Introduction
Lung cancer (LC) is an important etiology of
malignant mortality worldwide with global statistics indicating
>1,000,000 deaths each year (1). Non-small cell lung cancer (NSCLC)
accounts for 80–85% of LC (2). The
5-year survival rate is ~6.6% for advanced stage LC (stage III or
IV) in the US (3). Apart from
surgery, adjuvant chemotherapy with gefitinib, erlotinib, and
epidermal growth factor receptor tyrosine kinase inhibitors
(EGFR-TKIs), has been widely used to clinically treat NSCLC.
However, their efficacy is limited because of natural or acquired
resistance (4). Therefore, there
is a need to identify and develop potential anticancer drugs with
increased selectivity and reduced toxicity.
Glycyrrhetinic acid (GA) is a bioactive component of
glycyrrhiza (GL), which is often used in Chinese traditional
medicine to treat various diseases. GA is known to possess
anti-inflammatory, anti-viral and cytokine-inducing activity
(5–10). Recently, the antitumor activity of
GA has been extensively studied. GA has been reported to have
cytotoxic effects against human ovarian cancer, hepatocellular
carcinoma, breast cancer, pituitary adenoma and human bladder
cancer (11–16). However, no inhibitory activity on
the growth of NCSLC cell lines has been reported.
Endoplasmic reticulum (ER) stress responses are
mediated by the activation of several unfolded protein response
(UPR)-signaling pathways. In mammalian cells, the UPR signals
increase expression of ER chaperone proteins GRP78/Bip, GRP94, and
CHOP (17). The UPR coordinates
the induction of ER chaperones, which decreases protein synthesis
and results in growth arrest in G1 phase of the cell cycle.
Previous studies have demonstrated that ER stress triggers G1-phase
cell cycle arrest in various cancer cells (18). However, the molecular mechanism
underlying UPR-induced G1 cell cycle arrest remains largely
unknown.
In this study, we investigated the effect of GA on
survival and proliferation of human NSCLC cell lines (A549 and
NCI-H460), and found that GA could suppress the proliferation of
both cell lines, with A549 being more sensitive than NCI-H460. GA
arrested cells in G1 phase via inactivation of CDK4/6-cyclin-D1/D3
complex through p18/p16 activation, and inactivation of
CDK2-cyclin-E2 complex through p27/p21 activation. This resulted in
pRb dephosphorylation and inactivation of E2F transcription factor
1 (E2F-1) in both cell types. E2F-1 is an essential transcription
factor that regulates cell cycle progression and apoptosis.
Additionally, GA was found to increase the expression of Bip,
protein kinase-like ER kinase (PERK) and ERP72, which are linked to
ER stress.
Materials and methods
Reagents
GA was purchased from Nanjing Zelang Medical
Technology Co., Ltd. (Jiangsu, China), and dissolved in dimethyl
sulfoxide (DMSO) (Sigma, St. Louis, MO, USA) to make a stock
solution before use. For treatment of cells, it was diluted in
culture medium at the appropriate concentrations, and the final
concentration of DMSO was <0.01% (v/v). Cisplatin (Lot no.
H20030675; Nanjing Pharmaceutical Factory Co., Ltd., Jiangsu,
China), and insulin, propidium iodide (PI),
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
and alamarBlue were from Sigma. Alexa Fluor 488 Annexin V/Dead Cell
Apoptosis kit was from Invitrogen Life Technologies (Carlsbad, CA,
USA). Antibodies against caspase-3, -7 and -9, p18, p16, p27, p21,
cyclin-D1, -D3 and -E2, CDK6, 4 and 2, E2F-1, pRb, Bip, PERK,
ERP72, β-actin, and HRP-conjugated antibodies (anti-rabbit or mouse
immunoglobulin G) were obtained from Cell Signaling Technology,
Inc. (Danvers, MA, USA).
BCA protein estimation kit was from Sigma.
Nitrocellulose (NC) blotting membrane was from Pall Corporation (DF
Mexico, Mexico). Enhanced chemiluminescence (ECL) was from Bio-Rad
(Hercules, CA, USA).
Cell culture
Human NSCLC cell lines A549 and NCI-H460 were
purchased from the Type Culture Collection of the Chinese Academy
of Sciences (Shanghai, China). A549 cells were cultured in DMEM/F12
(Gibco-BRL, Carlsbad, CA, USA), supplemented with 10% fetal bovine
serum (FBS). NCI-H460 cells were grown in RPMI-1640 medium
(Gibco-BRL), supplemented with 10% FBS. All cells were cultured
under 5% CO2 at 37°C.
In vitro viability assay
The effect of GA on cell viability was measured
using the MTT assay. Cells were seeded in 96-well plates at
5×103 cells/well in 100 μl of culture medium, and
treated with drug the next day for 24, 48, and 72 h. The final
concentrations of GA used in the assays were 50, 25, 12.5, 6.25 and
3.125 μmol/l in triplicate, respectively. Treated cells were
incubated with 20 μl of MTT (5 mg/ml) for 4 h at 37°C in the dark.
Optical density of producer after incubation was measured using a
microplate reader (Bio-Rad) at a wavelength of 490 nm.
Cell cycle analysis
After treatment with various concentrations of GA
for different time, the cells were harvested with trypsin, washed
once with PBS, and then fixed in 70% ethanol overnight at 4°C.
Before flow cytometry analysis, the cells were then treated with 1
mg/ml of RNase for 30 min at 37°C, and then stained with 40 μg/ml
of PI for 30 min. A total of 1×104 cells/sample were
analyzed using a FACSCalibur flow cytometer (BD Biosciences,
Heidelberg, Germany). Data were evaluated using ModFit
software.
Western blot analysis
After treatment with different concentrations of GA,
the cells were lysed in RIPA buffer containing 50 mM Tris/HCl (pH
8.0), 150 mM NaCl, 1% (w/v) Nonidet P-40, 1% (w/v) sodium
deoxycholate, 0.1% (w/v) SDS, 0.1 mM DTT, 0.05 mM PMSF, 0.002 mg/ml
aprotinin, 0.002 mg/ml leupeptin, and 1 mM NaVO3. The
protein concentrations of the supernatants were determined by the
BCA Protein Assay kit. Equal amounts of the protein were loaded and
separated by 10 or 12% SDS-PAGE, and then transferred onto NC
membranes. The membranes were incubated overnight with primary
antibodies against caspase-3, -7 and -9, p18, p16, p27, p21,
cyclin-D1, -D3 and -E2, CDK6, 4 and 2, E2F-1, pRb, Bip, PERK,
ERP72, or β-actin at 4°C, and then incubated with HRP-conjugated
secondary antibodies (anti-rabbit or mouse immunoglobulin G) for 1
h at room temperature. Immunoreactivity was detected by ECL
(Bio-Rad). β-actin was used as a loading control. Immunoblot
experiments were repeated three times. Quantitative analysis was
performed using Image Lab™ Software (Bio-Rad).
Statistical analysis
All values were expressed as mean ± SD (n=3).
One-way analysis of variance (ANOVA) was used to determine
statistical significance, followed by post hoc multiple comparisons
(Dunn’s test) using SPSS 19.0. P<0.05 was considered to be
statistically significant.
Results
GA suppresses the proliferation of NSCLC
cells in vitro
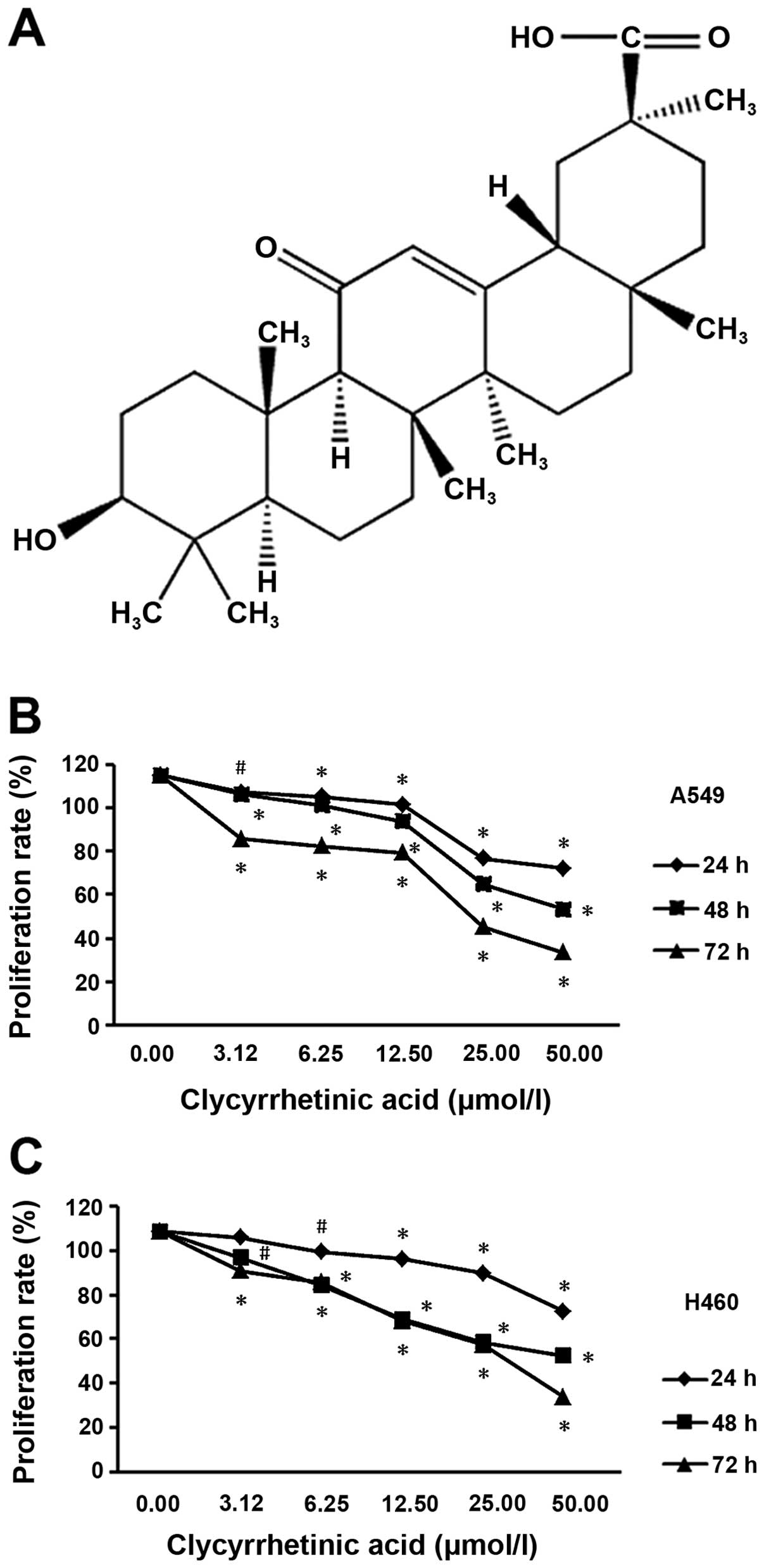
To determine the suppression effect of GA (structure
shown in Fig. 1A) on NSCLC cells,
we performed a cell viability assay using A549 and NCI-H460 cell
lines, respectively. After treatment for 24, 48 or 72 h, the
viability of the two cell lines significantly decreased in a dose-
and time-dependent manner (Fig. 1B and
C).
GA arrests the cell cycle in G0/G1
phase
The results of flow cytometric analysis showed that
the percentage of G0/G1 phase of both of A549 and H460 increased
after treated with different concentrations of GA for 24, 48 and 72
h (Fig. 2A), respectively. Cell
cycle distribution analysis showed that GA prevents the cell cycle
progression by arresting the cells in the G0/G1 phase in both cell
lines. In A549 cells, percentage of cells in G0/G1 phase increased
from 52.03±1.42% (control group) to 71.63±6.61% for cells treated
with 25 μmol/l of GA for 24 h. At 48 h, percentage of cells in
G0/G1 phase increased from 54.90±5.90% (control group) to
83.00±1.41% for cells treated with 25 μmol/l of GA. While at 72 h,
percentage of cells in G0/G1 phase increased from 69.70±5.38%
(control group) to 81.83±2.58% for cells treated with 25 μmol/l of
GA (Fig. 2A, upper panel). In
NCI-H460, percentage of cells in the G0/G1 phase increased from
66.10±0.99% (control group) to 80.95±1.91% for cells treated with
25 μmol/l of GA. At 48 h, percentage of cells in G0/G1 phase
increased from 84.60±2.40% (control group) to 88.95±2.19% for cells
treated with 25 μmol/l of GA. While at 72 h, percentage of cells in
the G0/G1 phase increased from 85.00±0.85% (control group) to
91.00±2.26% for cells treated with 25 μmol/l of GA (Fig. 2B, upper panel). No increase in S or
G2/M peak was observed in either cell line.
 | Figure 2Glycyrrhetinic acid (GA) induces
G1-phase cell cycle arrest in non-small cell lung cancer (NSCLC)
cells without induction of apoptosis. Effect of GA on cell cycle
was investigated using propidium iodide (PI) staining. Cells were
treated with 0–50 μmol/l of GA for 24, 48 and 72 h, and then
stained with PI. Green peak represents G0/G1 phase, red peak
represents S phase and blue peak represents G2 phase, respectively.
Upper panel shows representative of three independent experiments
with similar results, and lower panel represents the bar graphs of
cells in different phases. Bar graph represents mean ± SD from
three independent experiments. (A) Cells were strikingly
accumulated in the G1 phase after treatment with GA for 24, 48 and
72 h. (B) Representative bar graph for A549 and (C) for NCI-H460
cells. (D) Western blot analysis of caspase-3, -7 and -9 protein
expression after treatment with GA in a time (24 h)- and dose (0,
12.5, 25, 50 μmol/l)-dependent manner. β-actin was used as a
loading control. (E) Caspase-3, -7 and -9 activity in A549 and
NCI-H460 cells treated with GA in a time (24h)- and dose (0, 12.5,
25, 50 μmol/l)-dependent manner. Results shown are the mean of
three independent experiments; error bars represent SD. GA-mediated
G1-phase arrest is dependent on regulatory cyclin-dependent kinase
inhibitors (CKIs) p18, p16, p27, p21, and GA decreases the levels
of G1-phase regulatory CDKs and cyclins in both cell lines. |
Taken together, our data strongly suggested that GA
did not induce apoptosis but caused cell cycle arrest in G0/G1
phase in NSCLC cells. Annexin assay did not show any significant
changes in apoptotic/necrotic cell population for all
concentrations of GA as compared to the control group in either
cell line at 24, 48 and 72 h, respectively. To further validate the
above data, we checked the expression levels of caspase-3, -7 and
-9 in both cell lines after 24 h treatment with GA by western blot
analysis. Expression of caspase-3 decreased with increase in drug
concentration in NCI-H460 cells, but no significant changes in
caspase-7 and -9 protein levels or activity were observed in A549
or NCI-H460 cells (Fig. 2D and E),
suggesting that GA did not hinder the viability of cells.
GA downregulates the levels of cell cycle
regulatory proteins and retinoblastoma (Rb) phosphorylation
To investigate the causes of cell cycle arrest,
cyclin-dependent kinase inhibitors (CKIs), such as p27, p21, p18
and p16 that regulate G0/G1 phase of cell cycle progression were
examined by western blot analysis (19–21).
In A549 cells, the levels of p27, p21, p18 and p16 were
significantly increased after 24 h treatment with GA as compared to
the control cells (Fig. 3C). H460
cells also showed similar results (Fig. 3D). To further dissect the
biochemical events controlling the transition of cell cycle phases,
we examined the levels of several proteins, such as cyclin-D1, -D3
and -E2, CDK4, 6 and 2, which are involved in G0/G1-phase
progression, and found that GA significantly decreased the
expression of these proteins in both cell lines (Fig. 3E and F). GA also significantly
decreased the expression levels of E2F-1, the essential
transcription factor that regulates cell cycle progression and
apoptosis, and pRb (Fig. 3G and
H).
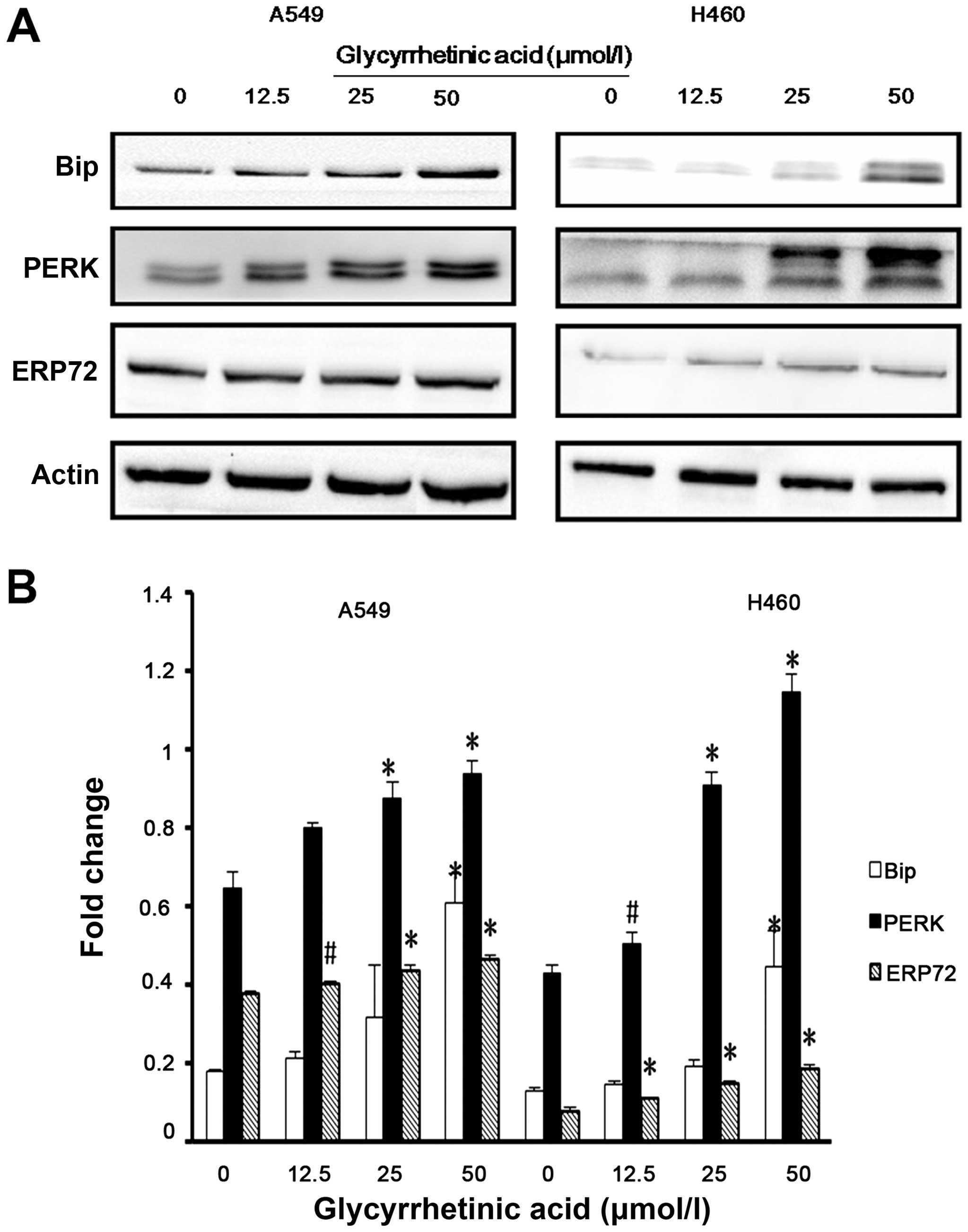
GA upregulated the levels of ER stress
regulatory proteins
Previous studies have demonstrated that ER stress
triggers G1-phase cell cycle arrest in various cancer cells
(22). Therefore, we examined
whether ER stress was induced by GA. Bip is the master regulator of
ER function. Phosphorylation of double-stranded RNA-activated PERK
is closely associated with Bip. Hence, we checked the expression of
Bip, PERK and ERP72 in both cell lines by western blot analysis,
and found that the expression levels of these proteins were
significantly upregulated after 24 h treatment with GA (Fig. 4A and B).
Discussion
GA is a natural active compound that is extracted
from the Chinese herbal medicine glycyrrhiza. GA was shown
to induce cell cycle arrest in G1 phase (13,15).
The role of GA in NSCLC, especially its relationship with ER stress
has not been reported. In the present study, we found that GA
induced G0/G1 arrest in A549 and NCI-H460 cell lines, which
provides a useful model system to characterize the cytotoxic
effects of therapeutic agents. Furthermore, GA could have
therapeutic potential in the treatment of NSCLC.
Our results have shown that GA successfully
inhibited proliferation of two NSCLC cell lines, A549 and NCI-H460.
Cell cycle analysis by flow cytometry showed that GA induced a
modest increase in G0/G1 phase in both cell lines. However, based
on expression levels of caspase-3, -7 and -9 by western blot
analysis, GA did not induce apoptosis in either cell line.
It is well known that eukaryotic cell cycle is
regulated by the coordinated activity of protein kinase complexes,
each consisting of a cyclin-dependent kinase (CDK) and cyclins. CDK
complexes are formed and activated at specific stages of the cell
cycle, and their activities are required for progression through
distinct cell cycle phases (23).
Progression through G1 phase requires the activities of
cyclin-D-dependent CDK4 or 6, followed by activation of the
cyclin-E- and cyclin-A-dependent kinase CDK2. The cyclin-CDK
complex formed during G1-phase catalyses phosphorylation of the
dominant inhibitor of G1/S-cell cycle progression, the Rb family of
tumor suppressor proteins, thereby blocking their inhibitory
activity allowing the cell to progress into S phase (24–27).
It is also known that these cyclin-CDK complexes often bind to CKIs
including p16, p18, p21 and p27, which inhibit their kinase
activities, and prevent cell cycle progression (28). E2F-1 is an essential transcription
factor that regulates cell cycle progression and cell
proliferation. E2F-1 activity is regulated by the Rb protein that
binds activator E2F proteins to inhibit transcription outside of
G1/S in animals (29).
Flow cytometric analysis of A549 and NCI-H460 cells
treated with GA showed that GA inhibits cell cycle progression by
blocking the transition from G1 to S phase. To further investigate
this result, western blot analysis was used to examine proteins
associated with the cell cycle, e.g., cyclin-D1/D3, which is
expressed in G1 phase and binds to CDK4 and 6 to activate them,
followed by activation of the cyclin-E-dependent kinase CDK2. These
protein kinase complexes were inhibited by GA. GA also
significantly decreased the expression levels of E2F-1 and pRb in
both cell lines. Our results indicated that GA induced growth
inhibition mainly via regulation of p16, p18, p21 and p27 status in
A549 and NCI-H460 cells.
In the present study, the analysis of DNA content
versus light scatter of the GA-treated A549 and NCI-H460 cells
indicated no apoptosis. Similarly, the expression of caspase-3, -7
and -9 measured by western blot analysis indicated that GA could
not induce apoptosis in these cells. However, GA induced expression
of ER proteins GRP78/Bip, PERK and ERP72, which are associated with
ER stress. This result suggested that GA inhibited proliferation of
A549 and NCI-H460 cells and caused G0/G1-phase cell cycle arrest
via ER stress rather than apoptosis.
GRP78/Bip is a major cellular target of the UPR, an
ER chaperone that not only binds to unfolded proteins but also
regulates the activation of ER stress transducers such as IRE1,
PERK, and ATF6 (30–32). GRP78/Bip is ubiquitously expressed
at very low levels in growing cells, but it is highly expressed in
response to numerous cellular stresses. ERP72, a member of the
protein disulfide isomerases (PDI) family, is localized in the ER,
and plays a major role in quality control and folding (33). Dysregulation of ER
chaperone/folding enzymes ERP72 and GRP78/Bip occurred early after
ablation of PERK function suggesting that changes in ER secretory
functions could reduce insulin gene expression and cell
proliferation (34,35).
Previous studies have found that CKIs and cyclins
play important roles in ER stress and cell cycle arrest. p27 was
reported to be a critical mediator of ER stress-induced G1 cell
cycle arrest in melanoma cells (36). p21 integrates the DNA damage
response with ER stress signaling, which then regulates
mitochondrial death pathways during chronic genotoxic stress
(37). Translational regulation of
cyclin-D1 in response to ER stress is a mechanism for checkpoint
control that prevents cell cycle progression (17). PERK has been shown to mediate cell
cycle arrest by blocking cyclin-D1 translation during UPR (17,38).
Similarly, our study has shown that induction of members of the
INK4 (p16, p18) or Kip/Cip (p21, p27) families of cell cycle kinase
inhibitors causes ER stress, and accumulation of unfolded proteins
in the ER triggers UPR, which is a stress signaling pathway. The
UPR coordinates the induction of ER chaperones with decreased
protein synthesis and growth arrest in G1 phase of the cell cycle.
Based on our results, we propose a model for the mechanism of
action of GA in NSCLC cells (Fig.
5).
 | Figure 5The proposed mechanism of action of
glycyrrhetinic acid (GA)-induced G1-phase arrest in non-small cell
lung cancer (NSCLC) cells. The G1/S cell cycle checkpoint controls
the passage of eukaryotic cells from the first phase (G1) into the
DNA synthesis phase (S). Two cell cycle kinases, CDK4/6-cyclin-D
and CDK2-cyclin-E, and the transcription complex that includes
retinoblastoma (Rb) and E2F are pivotal in controlling this
checkpoint. During G1 phase, the Rb-HDAC repressor complex binds to
the E2F-DP-1 transcription factors, inhibiting downstream
transcription. Phosphorylation of Rb by CDK4/6 and CDK2 dissociates
the Rb-repressor complex, permitting transcription of S-phase genes
encoding for proteins that amplify the G1- to S-phase switch, and
are required for DNA replication. Many different stimuli exert
checkpoint control including endoplasmic reticulum (ER) stress,
TGFβ, DNA damage, contact inhibition, replicative senescence and
growth factor withdrawal. ER stress is induced by members of the
INK4 (p16, p18) and Kip/Cip (p21, p27) families of cell cycle
kinase inhibitors. The accumulation of unfolded proteins in the ER
triggers the unfolded protein response (UPR), which is a stress
signaling pathway. The UPR coordinates the induction of ER
chaperones, decreases protein synthesis, and causes growth arrest
in G1 phase of the cell cycle. |
We have convincingly shown that GA inhibits
proliferation of NSCLC cell lines by causing cell cycle arrest in
G0/G1 phase in a time- and dose-dependent manner, without inducing
apoptosis. We have elucidated a new mechanism of action of GA
against NSCLC by inducing G1-phase cell cycle arrest through ER
stress pathway. Since GA synergizes the effect of anticancer drugs,
it provides new insight into the therapeutic index of NSCLC
treatment.
Acknowledgements
This study was supported by the National Science and
Technology Pillar Program in the 11th Five-year Plan of China
2006BAI11B08-01 (to H.F. and X.Z.), the U.S. National Institutes of
Health grants P01 CA116676 (to H.F.), the Priority Academic Program
Development (PAPD) of Jiangsu Higher Education Institutions (to
X.Z.), China and Europe taking care of healthcare solutions, CHETCH
Grant Agreement Number: PIRSES-GA-2013-612589 (to X.Z.) and the
Natural Science Foundation of Jiangsu Province (BK20131415) (to
M.C.).
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Siegel R, Ward E, Murray T, Xu J
and Thun MJ: Cancer statistics, 2007. CA Cancer J Clin. 57:43–66.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang T, Nelson RA, Bogardus A and Grannis
FW Jr: Five-year lung cancer survival: which advanced stage
nonsmall cell lung cancer patients attain long-term survival?
Cancer. 116:1518–1525. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carrera S, Buque A, Azkona E, et al:
Epidermal growth factor receptor tyrosine-kinase inhibitor
treatment resistance in non-small cell lung cancer: biological
basis and therapeutic strategies. Clin Transl Oncol. 16:339–350.
2014. View Article : Google Scholar
|
|
5
|
Agarwal MK, Iqbal M and Athar M:
Inhibitory effect of 18beta-glycyrrhetinic acid on
12-O-tetradecanoyl phorbol-13-acetate-induced cutaneous oxidative
stress and tumor promotion in mice. Redox Rep. 10:151–157. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jeong HG, You HJ, Park SJ, et al:
Hepatoprotective effects of 18beta-glycyrrhetinic acid on carbon
tetrachloride-induced liver injury: inhibition of cytochrome P450
2E1 expression. Pharmacol Res. 46:221–227. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matsui S, Matsumoto H, Sonoda Y, et al:
Glycyrrhizin and related compounds down-regulate production of
inflammatory chemokines IL-8 and eotaxin 1 in a human lung
fibroblast cell line. Int Immunopharmacol. 4:1633–1644. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Abe N, Ebina T and Ishida N: Interferon
induction by glycyrrhizin and glycyrrhetinic acid in mice.
Microbiol Immunol. 26:535–539. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dai JH, Iwatani Y, Ishida T, et al:
Glycyrrhizin enhances interleukin-12 production in peritoneal
macrophages. Immunology. 103:235–243. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ukil A, Biswas A, Das T and Das PK: 18
Beta-glycyrrhetinic acid triggers curative Th1 response and nitric
oxide up-regulation in experimental visceral leishmaniasis
associated with the activation of NF-kappa B. J Immunol.
175:1161–1169. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee CS, Yang JC, Kim YJ, Jang ER, Kim W
and Myung SC: 18β-glycyrrhetinic acid potentiates apoptotic effect
of trichostatin A on human epithelial ovarian carcinoma cell lines.
Eur J Pharmacol. 649:354–361. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kuang P, Zhao W, Su W, et al:
18β-glycyrrhetinic acid inhibits hepatocellular carcinoma
development by reversing hepatic stellate cell-mediated
immunosuppression in mice. Int J Cancer. 132:1831–1841. 2013.
View Article : Google Scholar
|
|
13
|
Satomi Y, Nishino H and Shibata S:
Glycyrrhetinic acid and related compounds induce G1 arrest and
apoptosis in human hepatocellular carcinoma HepG2. Anticancer Res.
25:4043–4047. 2005.PubMed/NCBI
|
|
14
|
Sharma G, Kar S, Palit S and Das PK:
18β-glycyrrhetinic acid induces apoptosis through modulation of
Akt/FOXO3a/Bim pathway in human breast cancer MCF-7 cells. J Cell
Physiol. 227:1923–1931. 2012. View Article : Google Scholar
|
|
15
|
Wang D, Wong HK, Feng YB and Zhang ZJ:
18Beta-glycyrrhetinic acid induces apoptosis in pituitary adenoma
cells via ROS/MAPKs-mediated pathway. J Neurooncol. 116:221–230.
2014. View Article : Google Scholar
|
|
16
|
Lin KW, Huang AM, Hour TC, Yang SC, Pu YS
and Lin CN: 18β-glycyrrhetinic acid derivatives induced
mitochondrial-mediated apoptosis through reactive oxygen
species-mediated p53 activation in NTUB1 cells. Bioorg Med Chem.
19:4274–4285. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brewer JW, Hendershot LM, Sherr CJ and
Diehl JA: Mammalian unfolded protein response inhibits cyclin D1
translation and cell-cycle progression. Proc Natl Acad Sci USA.
96:8505–8510. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Puthalakath H, O’Reilly LA, Gunn P, et al:
ER stress triggers apoptosis by activating BH3-only protein Bim.
Cell. 129:1337–1349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chu IM, Hengst L and Slingerland JM: The
Cdk inhibitor p27 in human cancer: prognostic potential and
relevance to anticancer therapy. Nat Rev Cancer. 8:253–267. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pérez-Sayáns M, Suárez-Peñaranda JM,
Gayoso-Diz P, Barros-Angueira F, Gándara-Rey JM and García-García
A: The role of p21Waf1/CIP1 as a Cip/Kip type cell-cycle regulator
in oral squamous cell carcinoma (Review). Med Oral Patol Oral Cir
Bucal. 18:e219–e225. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Drexler HG: Review of alterations of the
cyclin-dependent kinase inhibitor INK4 family genes p15, p16, p18
and p19 in human leukemia-lymphoma cells. Leukemia. 12:845–859.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu GY, Wong BC, Lu A, et al: Alkylphenols
from the roots of Ardisia brevicaulis induce G1 arrest and
apoptosis through endoplasmic reticulum stress pathway in human
non-small-cell lung cancer cells. Chem Pharm Bull (Tokyo).
60:1029–1036. 2012. View Article : Google Scholar
|
|
23
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sherr CJ: D-type cyclins. Trends Biochem
Sci. 20:187–190. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guadagno TM and Newport JW: Cdk2 kinase is
required for entry into mitosis as a positive regulator of
Cdc2-cyclin B kinase activity. Cell. 84:73–82. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Geng Y, Whoriskey W, Park MY, et al:
Rescue of cyclin D1 deficiency by knockin cyclin E. Cell.
97:767–777. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jin YH, Choi J, Shin S, Lee KY, Park JH
and Lee SK: Panaxadiol selectively inhibits cyclin A-associated
Cdk2 activity by elevating p21WAF1/CIP1 protein levels in mammalian
cells. Carcinogenesis. 24:1767–1772. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Grana X and Reddy EP: Cell cycle control
in mammalian cells: role of cyclins, cyclin dependent kinases
(CDKs), growth suppressor genes and cyclin-dependent kinase
inhibitors (CKIs). Oncogene. 11:211–219. 1995.PubMed/NCBI
|
|
29
|
Bertoli C, Skotheim JM and de Bruin RA:
Control of cell cycle transcription during G1 and S phases. Nat Rev
Mol Cell Biol. 14:518–528. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Harding HP, Zhang Y, Zeng H, et al: An
integrated stress response regulates amino acid metabolism and
resistance to oxidative stress. Mol Cell. 11:619–633. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shi K, Wang D, Cao X and Ge Y: Endoplasmic
reticulum stress signaling is involved in mitomycin C (MMC)-induced
apoptosis in human fibroblasts via PERK pathway. PLoS One.
8:e593302013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yoon JS, Kim HM, Yadunandam AK, et al:
Neferine isolated from Nelumbo nucifera enhances anti-cancer
activities in Hep3B cells: molecular mechanisms of cell cycle
arrest, ER stress induced apoptosis and anti-angiogenic response.
Phytomedicine. 20:1013–1022. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mazzarella RA, Srinivasan M, Haugejorden
SM and Green M: ERp72, an abundant luminal endoplasmic reticulum
protein, contains three copies of the active site sequences of
protein disulfide isomerase. J Biol Chem. 265:1094–1101.
1990.PubMed/NCBI
|
|
34
|
Brewer JW and Diehl JA: PERK mediates
cell-cycle exit during the mammalian unfolded protein response.
Proc Natl Acad Sci USA. 97:12625–12630. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Feng D, Wei J, Gupta S, McGrath BC and
Cavener DR: Acute ablation of PERK results in ER dysfunctions
followed by reduced insulin secretion and cell proliferation. BMC
Cell Biol. 10:612009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Han C, Jin L, Mei Y and Wu M: Endoplasmic
reticulum stress inhibits cell cycle progression via induction of
p27 in melanoma cells. Cell Signal. 25:144–149. 2013. View Article : Google Scholar
|
|
37
|
Vitiello PF, Wu YC, Staversky RJ and
O’Reilly MA: p21(Cip1) protects against oxidative stress by
suppressing ER-dependent activation of mitochondrial death
pathways. Free Radic Biol Med. 46:33–41. 2009. View Article : Google Scholar :
|
|
38
|
Hamanaka RB, Bennett BS, Cullinan SB and
Diehl JA: PERK and GCN2 contribute to eIF2alpha phosphorylation and
cell cycle arrest after activation of the unfolded protein response
pathway. Mol Biol Cell. 16:5493–5501. 2005. View Article : Google Scholar : PubMed/NCBI
|