Introduction
Genistein (4′,5,7-trihydroxyisoflavone) is a common
type of isoflavone that is abundantly found in soy products, which
exerts diverse biological activities (1). Genistein is believed to have a potent
antitumor activity in various cancer cell lines (2–7). It
has attracted attention due of its extensive chemotherapeutic
functions for malignancies and its minimal side effects. Genistein
has inhibitory effects on the growth, survival, metastasis,
invasion and angiogenesis of cancer cells, by inhibiting cell
proliferation and inducing apoptosis (8–11).
In 1996, the pioneering study by Yamashita et al indicated
that genistein could intervene in OS cell lines (12). Subsequent reports from these
authors demonstrated that this flavonoid compound has antitumor
effects on human OS cell lines (13–16).
Additionally, it has been found that genistein decreased cell
invasion and motility potentials by inducing cell differentiation
in murine OS cell line LM8 (17).
Recently, there have been significant interests in exploring
malignancy specific receptors activated by genistein - including in
OS.
Genistein is known as a phytoestrogen that has the
ability to bind with an estrogen receptor (ER) (18). Previous studies on OS cells
suggested that genistein can affect multiple intracellular events
by binding different ER isoforms (13,19)
and that its inhibition becomes statistically significant from 1
nmol/l (20). The specific
inhibition of genistein on the epidermal growth factor receptor
protein tyrosine kinase (PTK) is a well-known mechanism (21). Genistein has also shown
anti-proliferative effects on OS cells, which is probably due to
its anti-PTK mechanism (5).
I was proposed that phytoestrogens might act through
nuclear receptors other than ERs, and genistein was reported to act
as a natural ligand of PPARγ (22), provoking PPARγ-dependent gene
transcription (23) and regulating
the PPARγ signaling pathway (24).
In addition, a high concentration genistein treatment (>1
μmol/l) can bind with PPARγ and downregulate osteogenesis, which is
consistent with the effects of inducing PPARγ (22).
PPARγ, an isotype of PPARs, is a ligand-dependent
transcription factor of the nuclear hormone receptor superfamily
(25). It regulates gene
transcription in multistep metabolic processes, such as lipid,
glucose homeostasis, inflammation (26,27)
and tumorigenesis (28). It has
aroused remarkable interests due to its antitumor effects against a
variety of malignancies (29–36).
Increasing evidence demonstrates that PPARγ activation may be
sought as a probable intervention in OS (37–41).
PPARγ agonists can promote osteoblastic differentiation of OS cells
(42) and suppress its
proliferation (43). Moreover,
several novel natural compounds, which have PPARγ agonistic
activities, possess potent antitumor activities. Therefore, one
future direction of this study is to develop a safe and effective
PPARγ agonist that can be used as a chemo-preventive agent for OS
patients.
The therapeutic potential of genistein as an
antitumor agent is clearly evident. However, it is not clear
whether genistein is involved with the antitumor activities of OS
cells by directly activating PPARγ. Thus, we explored the effects
of genistein on OS cell viability via PPARγ pathway and
investigated the potential underlying mechanisms.
Materials and methods
Reagents and antibodies
Genistein (4′,5,7-trihydroxyisoflavone),
2-chloro-5-nitrobenzanilide (GW9662), dimethyl sulfoxide (DMSO),
Triton X-100, RNase A and propidium iodide (PI) were all purchased
from Sigma-Aldrich Corp. (Sigma-Aldrich Corp., St. Louis, MO, USA).
Dissolving genistein and GW9662 in DMSO were recommended. According
to the product information, genistein was dissolved to prepare a 10
mg/ml stock solution and GW9662 was reconstituted as a 26 mg/ml
stock solution. The solutions were then diluted to appropriate
concentrations with a culture medium. Dulbecco’s modified Eagle’s
medium (DMEM), penicillin-streptomycin, trypsin-EDTA, trypsin and
fetal bovine serum (FBS) were all obtained from Gibco/Life
Technologies (Carlsbad, CA, USA). Rabbit polyclonal antibodies
specific for PPARγ, phosphatase and tensin homolog (PTEN),
P21WAF1/CIP1, Cyclin B1, B-cell lymphoma-2 (BCL-2),
survivin and mouse monoclonal antibody specific for β-actin were
purchased from Assay Biotechnology (Sunnyvale, CA, USA).
Horseradish peroxidase-conjugated goat anti-mouse and goat
anti-rabbit secondary antibodies were obtained from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA).
Cell and cell culture
The MG-63 OS cell line was purchased from the China
Center Type Culture Collection (CCTCC, Shanghai, China). The cells
have been maintained under standard culture conditions in DMEM,
supplemented with 10% (vol/vol) FBS and 1% pencillin-streptomycin
at 37°C, under a humidified atmosphere of 5% CO2 in our
laboratory. The medium was replaced every 3 days before full
confluency.
Cytotoxicity assay
Cells were seeded with a density of
2–4×103/well in 96-well plates, which was allowed to
attach overnight. The medium was replaced with a fresh medium
containing different concentrations of genistein (0–120 μmol/l or
GW9662 0–100 μmol/l), diluted from the stock solution. Since the
final DMSO concentration of genistein in the medium was >0.1%, a
DMSO (solvent) concentration of 0.32% (vol/vol) was used for
vehicle control. After 24, 48 and 72 h of incubation, 10 μl of Cell
Counting Kit-8 (CCK-8) (Dojindo, Tokyo, Japan) solution was added.
Absorbance at 450 nm was measured by the Microplate Reader (Thermo
Labsystems, Helsinki, Finland). The relative folds of drug
resistance were analyzed by comparing with IC50.
As stated above, cells were plated and allowed to
attach overnight; the medium was then removed and replaced with a
fresh medium containing various doses of genistein (40, 60 and 80
μmol/l), with 1 μmol/l of GW9662, for 48 h. The effect of GW9662 on
cell viability was examined by CCK-8 method, as described
above.
Cell proliferation assay
The proliferation assay of MG-63 cells was performed
using a Cell-Light™ 5-ethynyl-2′-deoxyuridine (EdU) DNA Cell
Proliferation kit (Ruibo Biotech, Guangzhou, China), based on the
manufacturer’s protocols (44–46).
MG-63 cells were cultured in 24-well plates at
2–3×104/well with different concentrations of genistein
(40, 60 and 80 μmol/l), with or without GW9662 (1 μmol/l), for 48
h. Then, the cells were incubated with 100 μmol/l of EdU for
another 2 h. Finally, the cells were fixed with 4% formaldehyde in
phosphate buffered solution (PBS) buffer solution, for 30 min.
After labeling, five random × 100 fields were photographed in each
well. The EdU-positive cells were counted, averaged and compared.
Assays were performed three times using triplicate wells.
Annexin V-FITC/PI double staining
To measure the apoptosis rate of the cells, we used
the Annexin V-FITC/PI doublefluorescence apoptosis detection kit
(Biouniquer Technology, Shanghai, China). Briefly, the cells were
seeded in 6-well plates (2×105 cells/ml) and exposed to
genistein (40, 60 and 80 μmol/l), with or without GW9662 (1
μmol/l), for 48 h. The cells were washed 2 times by PBS buffer
solution at 4°C, diluted with 500 μl of Annexin V binding liquid
and 5 μl of Annexin V-FITC was added at 4°C for 15 min in the dark.
Finally, 10 μl of PI was added at 4°C for 5 min in the dark.
Samples were analyzed using a FACScan flow cytometer
(Becton-Dickinson, San Jose, CA, USA) - within 1 h after
staining.
Cell cycle progression
MG-63 cells were plated in T25 tissue
culture flasks at 2×106/flask in a culture medium. After
24 h, cells were washed in PBS buffer and treated with a fresh
medium, with or without 1 μmol/l of GW9662 or various
concentrations of genistein (40, 60 and 80 μmol/l). Following 48 h
of genistein treatment, the cells were detached and trypsinized and
washed two times in PBS buffer solution at 4°C. Then, the cells
were fixed and permeabilized with 70% ethanol on ice for 2 h. Cells
were washed again in 0.5 ml of PBS buffer solution and stained for
30 min with 1 ml of DNA fluorochrome solution at normal
temperature, containing 200 μg of PI, 0.1% of Triton X-100 and 2 mg
of RNase A. Then 2×104 cells were counted and analyzed
using a FACScan flow cytometer (Becton-Dickinson).
Western blot analysis
The 2×106 cells that were treated for 48
h with genistein (40, 60 and 80 μmol/l) and/or GW9662 (1 μmol/l)
were washed twice in an ice-cold PBS buffer solution; and the cells
were resuspended in 200 μl of ice-cold lysis buffer solution (50
mmol/l Tris-HCl, 150 mmol/l NaCl, 0.02% NaN3 and 1%
NP40) for 30 min. The lysates were clarified by
centrifugation for 15 min at 14,000 rpm at 4°C. The protein
concentration of the supernatants was determined by using a
bicinchoninic acid protein assay (Pierce, Rockford, IL, USA). Equal
amounts of lysate protein were separated by 8–12%
SDS-polyacrylamide gel electrophoresis, which were then transferred
onto polyvinylidene fluoride membranes. After being blocked with
10% non-fat dry milk for 1 h at room temperature, the membranes
were washed three times with a PBS Tween-20 (PBST) buffer solution
and incubated with monoclonal antibodies or phosphorylated
antibodies overnight, at 4°C. Subsequently, the membranes were
washed three times with PBST buffer solution and incubated with
peroxidase-conjugated secondary anti-mouse or anti-rabbit
antibodies for one hour at room temperature. The bands were
visualized using enhanced chemiluminescence detection and exposure
to film.
Real-time quantitative RT-PCR
analysis
RNA extraction and cDNA conversion were performed.
MG-63 cells in the logarithmic growth phase were placed into 6-well
plates with a density of 2×105/ml; and the cells were
treated with different genistein concentrations (40, 60 and 80
μmol/l), with or without 1 μmol/l of GW9662, for two days. Total
RNA was isolated with a TRIzol reagent (Takara Biotech, Dalian,
China). cDNA was generated from total RNA by using a PrimeScript RT
Master Mix kit (Takara Biotech). Quantitative real-time PCR was
performed using LightCycler™ 480 Real-Time system (Roche Applied
Science, IN, USA) and SYBR Premix Ex Taq™ kit (Tli RNaseH Plus)
(Takara Biotech). The threshold cycle (Ct) value of each
gene was measured from each RT sample. The Ct value of
β-actin was used as an endogenous reference for normalization
purposes. Thus, the obtained Ct value was normalized to
negative control and was expressed as fold changes. The primers and
reaction conditions used for the amplification are listed in
Table I.
 | Table IPrimers used for real-time PCR. |
Table I
Primers used for real-time PCR.
| Gene | Priners | Sequences | GenBank | Amplicon (bp) |
|---|
| PPARγ | Forward |
5′-ATTCCATTCACAAGAACAGATCCAG-3′ | NM_005037.5 | 195 |
| Reverse |
5′-TTTATCTCCACAGACACGACATTCA-3′ | | |
| PTEN | Forward |
5′-GAGCGTGCAGATAATGACAAGGAAT-3′ | NM_000314.4 | 152 |
| Reverse |
5′-GGATTTGACGGCTCCTCTACTGTTT-3′ | | |
| β-actin | Forward |
5′-CATTGCCGACAGGATGCA-3′ | NM_ 001101.3 | 142 |
| Reverse |
5′-CATCTGCTGGAAGGTGGACAG-3′ | | |
Statistical analysis
Statistical analysis was performed using SPSS 13.0
software (SPSS Inc., Chicago, IL, USA). The data were expressed as
means ± SD and the statistical differences between multiple groups
were calculated by one-way ANOVA, followed by Dunnett’s test.
P<0.05 values were considered statistically significant. The
results from the in vitro studies are representative of at
least three independent experiments.
Results
The effects of genistein, GW9662 and
their combination on the viability of MG-63 cells
To determine the cytotoxicity of genistein on OS
cells, we tested the effects of various doses of genistein on the
viability of MG-63 cell line using CCK-8 assay. The cells were
treated for 24, 48 and 72 h; and cell inhibition was found to be
significant at 48 and 72 h (data at 24 h was not significant). As
shown in Fig. 1A, cell growth was
reproducibly inhibited by genistein treatment in a dose-dependent
manner. After 48-h treatment with 20, 40, 60, 80, 100 and 120
μmol/l of genistein, the cell survival rates were 103.4±2.7,
76.7±3.7, 54.6±6.6, 40.0±3.5, 33±4.8 and 11.1±0.5, respectively.
After 72 h of MG-63 cell treatments with different genistein
concentrations, the cell survival rates were 122.3±7.1, 87.2±5.6,
48.8±1.9, 26.7±2.9, 17.7±2.3 and 9.2±0.5, respectively, inhibiting
cell growth (Fig. 1A). Based on
the data, the conditions used in 48-h treatments of genistein were
selected for further studies.
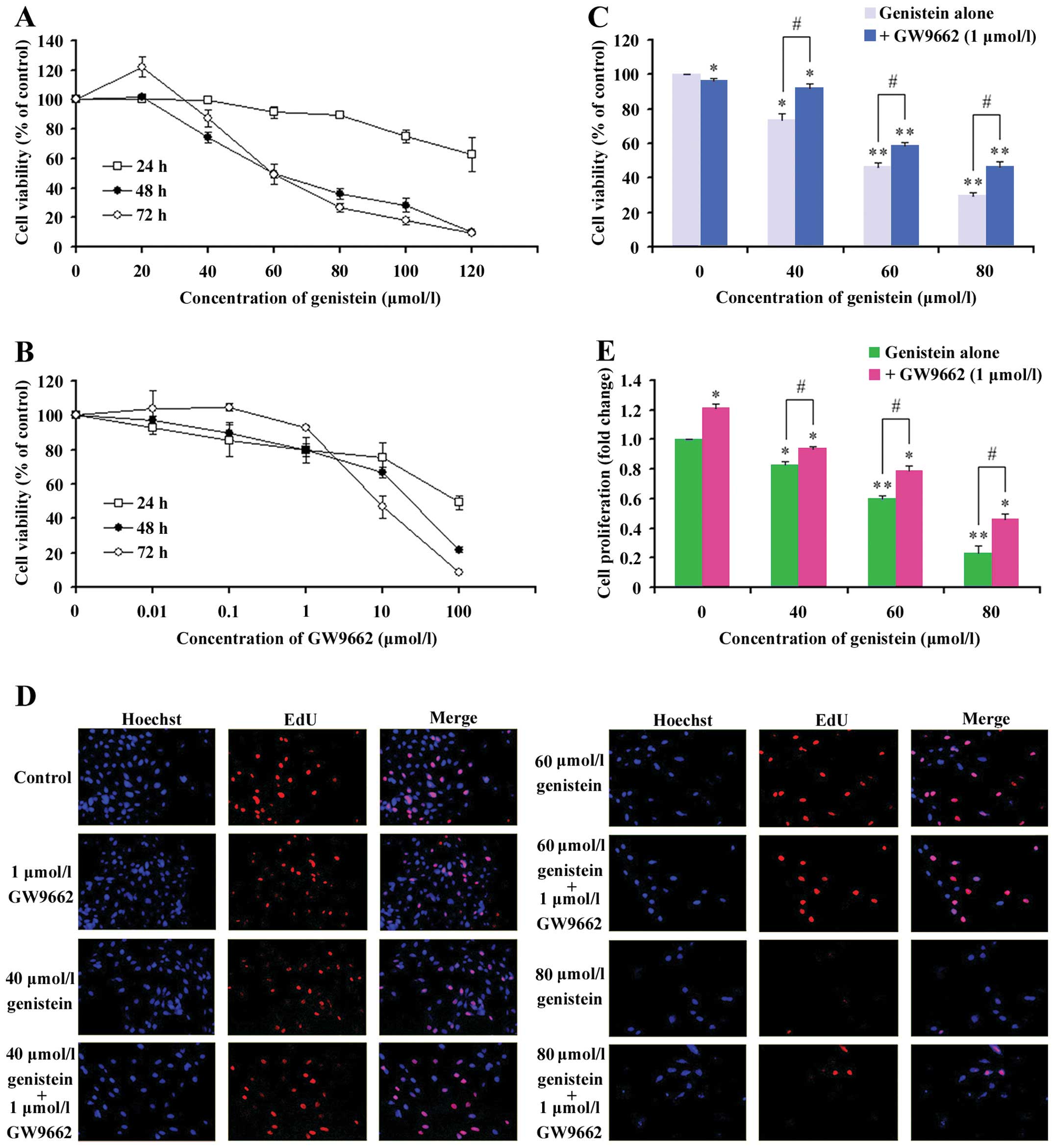 | Figure 1The human osteosarcoma MG-63 cell
growth inhibition by genistein, GW9662 and their combination: (A)
the effects of various doses of genistein on the viability of MG-63
cells after 24, 48 and 72 h; (B) the effects of various doses of
GW9662 on the viability of MG-63 cells after 24, 48 and 72 h; (C)
the effects of various doses of genistein with 1 μmol/l of GW9662
on the viability of MG-63 cells after 48 h. The total percentage of
viable cells was measured by CCK-8 assay. Data are expressed as
mean ± SD in three independent experiments. Bars marked with
asterisks are significant with respect to the controls at
*P<0.05 and **P<0.01. The number of
viable cells significantly increased, compared with the relative
genistein group (#P<0.05). EdU assay of relative
Hoechst stained cells and EdU add-in cells. (D) MG-63 cells were
treated with different concentrations of genistein, combination or
control. Forty-eight hours after treatment, EdU (100 μmol/l) was
added and the cells were cultured for 2 h. EdU and Hoechst staining
were performed, as described in Materials and methods. At least 200
cells were counted per well. (E) Data are expressed as mean ± SD in
the representative experiments performed in triplicate. The
proliferation rate of MG-63 cells, treated with different
concentrations of genistein or the combined group, significantly
decreased compared with the control (*P<0.05). The
proliferation rate of the combination group significantly
increased, compared with the relative genistein group
(#P<0.05). |
Subsequently, we evaluated the effects of GW9662 - a
potent specific antagonist of PPARγ. It was found that low
concentrations of GW9662 (0.01–1 μmol/l) played a weak role in
impairing the proliferation of MG-63 cells (Fig. 1B). Similarly, Seargent et al
(47) reported that GW9662 could
directly prevent tumor cells by the PPARγ-independent pathway. As a
single agent, these results indicated that GW9662 was a growth
inhibitor of OS cells. Therefore, 1 μmol/l of GW9662 was chosen for
our studies, due to its minimal effects on MG-63 cells.
To assess whether the inhibition of
genistein-induced cells are related to PPARγ activation (Fig. 1C), we investigated the effects of
combining genistein and GW9662 on cell viability by CCK-8 assay.
The cells were treated with different concentrations of genistein
(20, 40, 60, 80, 100 and 120 μmol/l), with or without GW9662 (1
μmol/l), for 48 h. MG-63 cells showed comparable cell growth
augmentation, when treated with genistein and GW9662. However,
genistein treatment alone caused a significant increase in cell
growth inhibition.
Therefore, to investigate the effects of PPARγ
activation by genistein, our subsequent studies primarily focused
on the MG-63 cell line, simulating different treatments for 48
h.
PPARγ inhibition attenuated
genistein-induced inhibition of MG-63 cell proliferation
Cell proliferation was evaluated by detecting
immunofluorescence for EdU. As shown in Fig. 1D and E, 48-h genistein treatments
(40, 60 and 80 μmol/l) potently decreased the number of
proliferating MG-63 cells. In contrast, combining different
concentrations of genistein (40, 60 and 80 μmol/l) and GW9662 (1
μmol/l) did not significantly affect the proliferation of MG-63
cells. Therefore, the inhibition of PPARγ with GW9662 significantly
reduced the genistein-induced anti-proliferation activites of MG-63
cells. These data suggest that the result of activating the PPARγ
mechanism partially caused genistein to lose its cell proliferation
ability.
The effects of GW9662 on
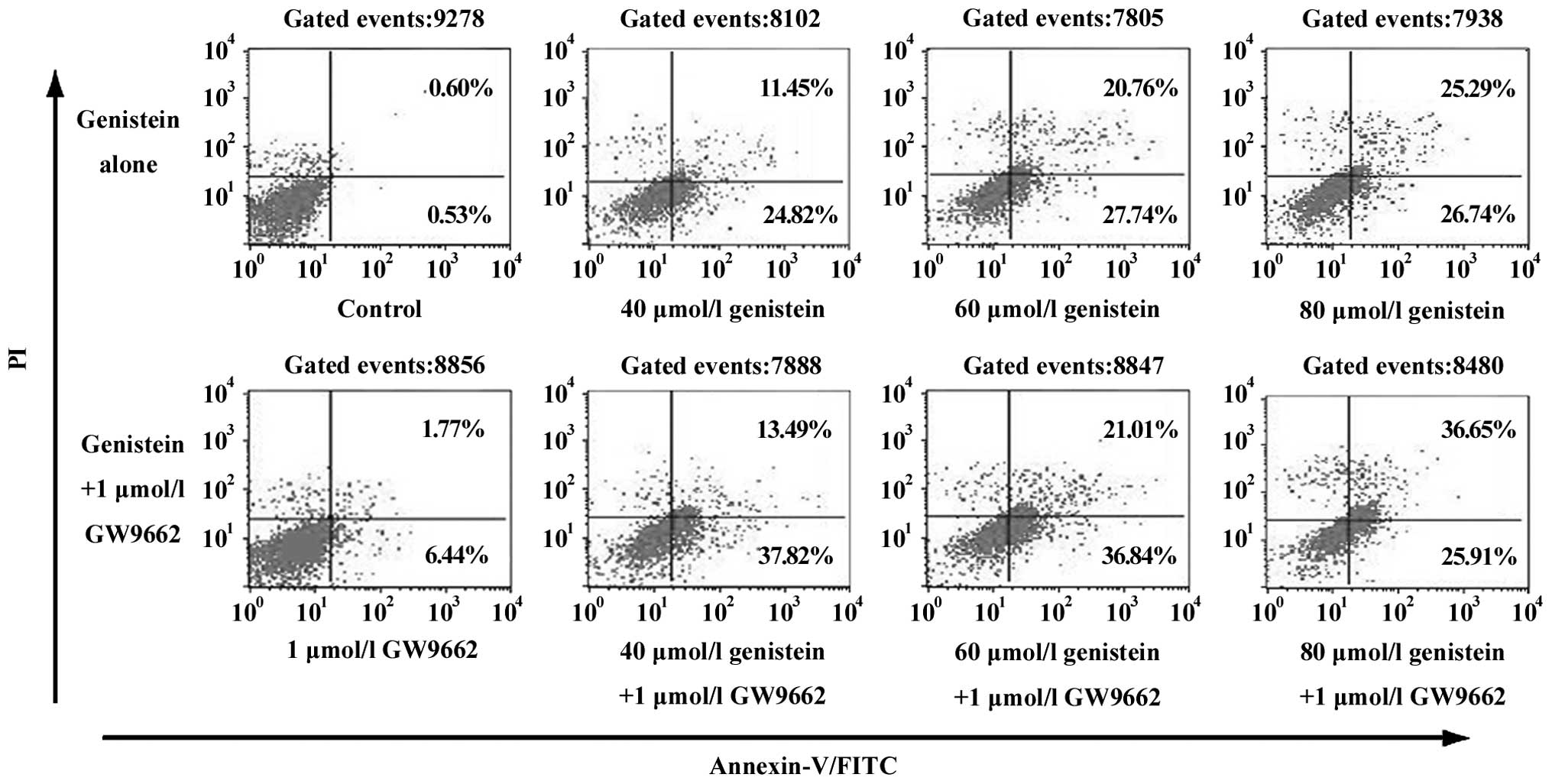
genistein-induced apoptosis in human OS MG-63 cells
Using fluorescence-activated cell sorting to
quantify apoptosis, we observed that genistein as a single agent
induced apoptosis in MG-63 cells dose-dependently (Fig. 2). DNA electrophoresis showed a more
significant DNA ladder in MG-63 cells that underwent a combined
treatment of genistein and GW9662. Flow cytometry experiments also
showed that GW9662 can enhance genistein-induced OS cell apoptosis.
The induction of genistein-induced apoptosis was dose-dependent and
was unrelated to PPARγ.
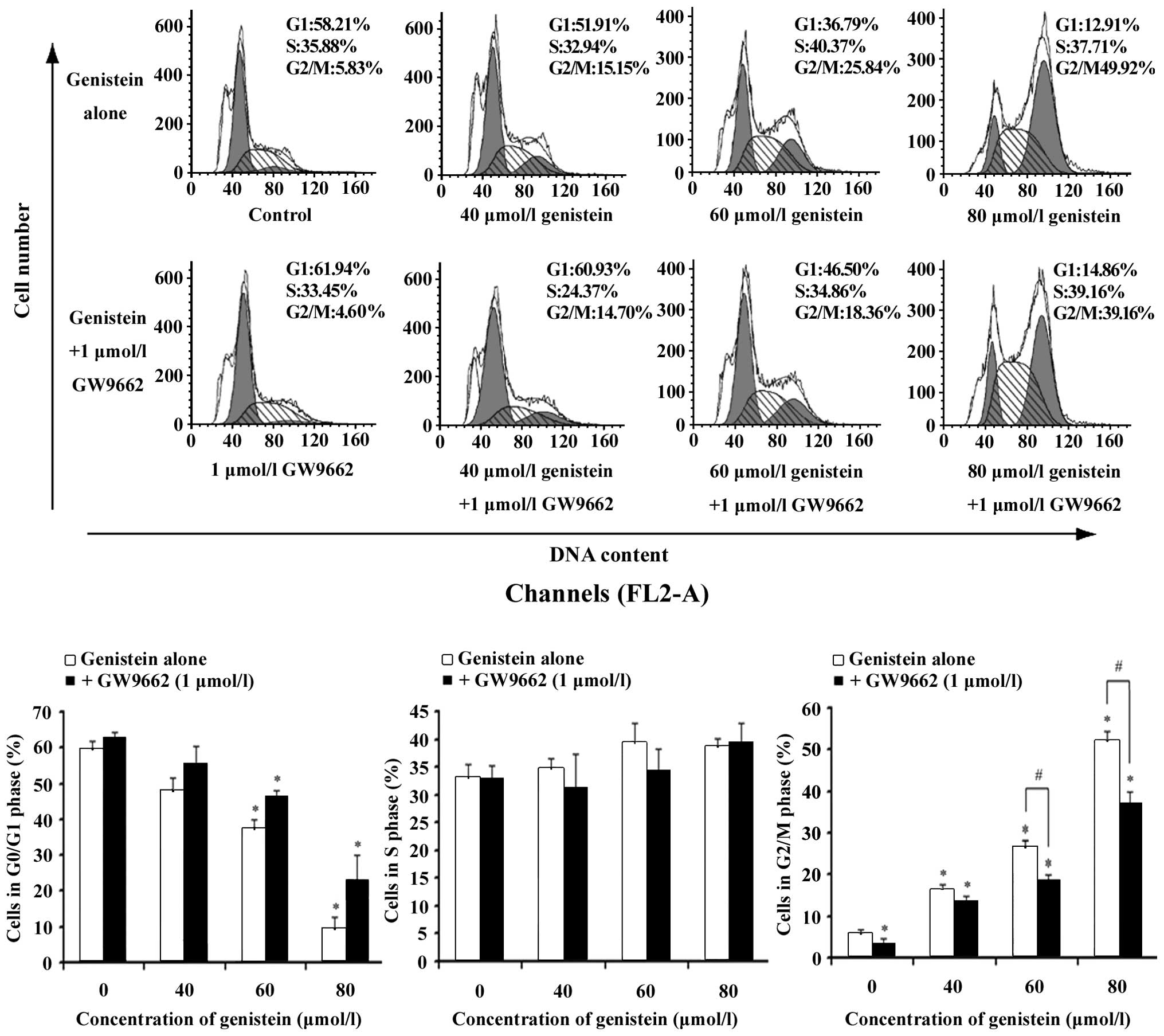
The effects of genistein and the
combination of genistein and GW9662 on cell cycle progression
To detect if the suppressive effect of genistein can
be caused by the specific perturbation of cell cycle-related
events, the DNA content of genistein-treated and
combination-treated MG-63 cells were measured using flow cytometry
after PI staining of the nuclei. The G1 and S cell
population significantly decreased in a dose-dependent manner and
after 48-h exposure to genistein (Fig.
3), while cells were arrested in the G2/M phase of
the cell cycle. Incubating with GW9662 reversed the effects of the
G2/M cell cycle arrest caused by genistein.
The inhibitive effects of genistein on
MG-63 cell growth via PPARγ pathway activation
PPARγ activation inhibits cell growth and causes
both differentiation and apoptosis in a variety of cancer cell
types (25). Therefore, in our
present study, we used GW9662, a PPARγ inhibitor that can block
PPARγ functions, to determine whether genistein prevents OS cell
growth via the PPARγ pathway. Although GW9662 (1 μmol/l) induced a
decrease in cell growth, GW9662 treatment partially reversed the
inhibitory effects of genistein based on the cell number; the
related MG-63 cell protein expression was also changed.
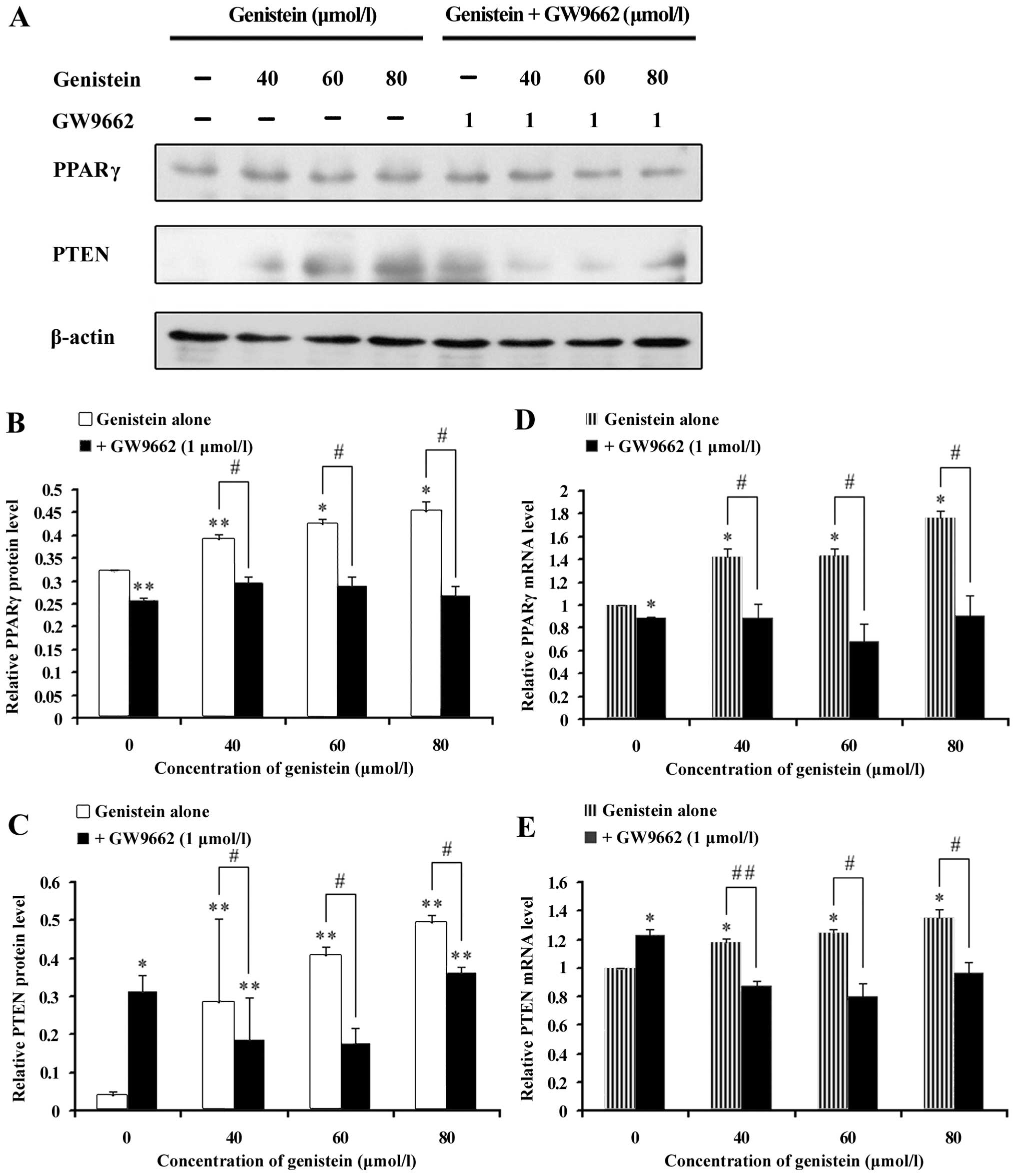
To elucidate the effect of genistein in OS cell
growth, proteins related to the PPARγ pathway were examined by
western blot analysis. As shown in Fig. 4A and B, a stable level of PPARγ
protein was observed in MG-63 cells after exposure to different
genistein concentrations; however, the PPARγ protein expression was
not altered by the GW9662 inhibitor. PTEN is a downstream target
protein that plays a notable role in malignancy growth and causes
the activation of the phosphoinositide-3 kinase/AKT (PI3K/Akt)
signaling pathway (48). Fig. 4A and C shows that genistein (40, 60
and 80 μmol/l) promoted the expression of the PPARγ target protein,
PTEN, in a concentration-dependent manner. GW9662 treatment
significantly decreased the PTEN expression caused by genistein;
suggesting possible PPARγ pathway implications on the mechanisms
responsible for the effects of genistein on OS cell growth.
Fig. 4D and E shows
the real-time quantitative RT-PCR results of PPARγ-related genes
(PPARγ and PTEN). There was a prominent increase in PPARγ gene
expression levels, as well as in the PTEN genes of MG-63 cells
treated with genistein. GW9662 significantly inhibited the
expression of these two genes induced by genistein. With β-actin
constitutively expressed, its levels were not affected by genistein
or the combined treatment. Thus, the real-time quantitative RT-PCR
results support the previous observations of our western blot
assay.
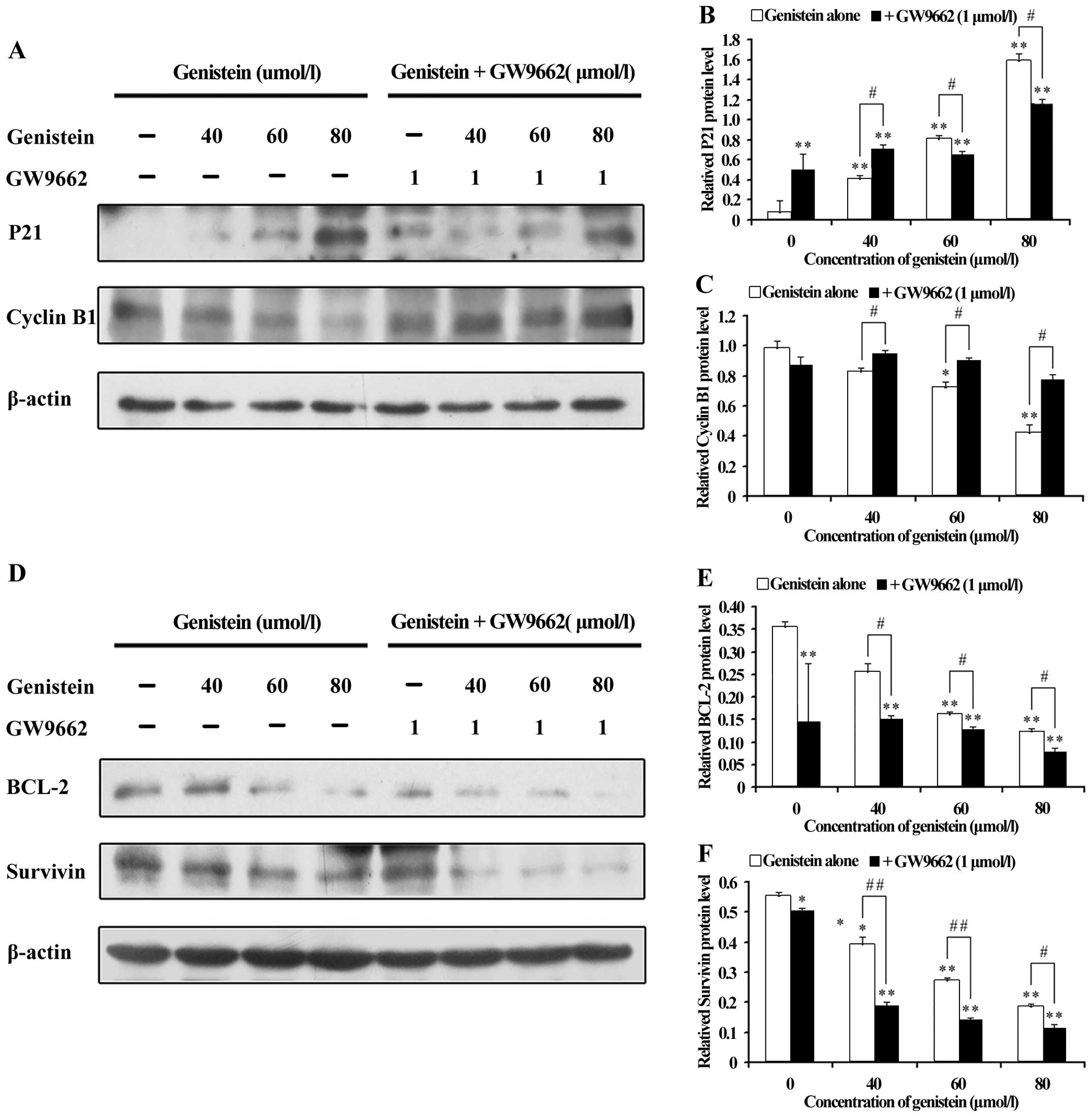
To further understand the downstream mechanism of
the PPARγ pathway by genistein treatment, G2/M
regulatory molecules were analyzed. Since genistein treatment
perturbed the G2/M phase of the cell cycle, as assessed
by flow cytometry, we examined the intracellular protein expression
of the cell cycle regulating components at the G2/M
boundary, such as P21 WAF1/CIP1 and Cyclin B1. As shown
in Fig. 5A and B, a dramatic
dose-dependent increase in the P21WAF1/CIP1 protein
levels were observed with genistein, compared with the
co-treatments. In contrast, Cyclin B1 levels (Fig. 5A and C) were reduced by either the
reagent or the combined treatment, the former causing a more
dramatic effect in a dose-dependent manner.
Several lines of evidence indicate that protein
BCL-2 and survivin, related to the protein of PTEN (49,50),
were generally considered to be apoptosis inhibitors. These were
frequently overexpressed in several types of human cancers,
including OS. Parallel to the above observations, BCL-2 and
survivin levels by genistein were decreased, as indicated by the
significantly decreased levels of BCL-2 and survivin, even with the
presence of GW9662 (Fig. 5D–F).
While the inhibition of PPARγ repressed apoptosis-related protein
levels, GW9662 did not reverse the genistein-induced effects on the
promoted apoptosis, which was consistent with earlier findings.
Taken together, the data suggest that the PPARγ
pathway was associated with the effects of genistein. Furthermore,
genistein could significantly induce OS cell cycle arrest by
activating PPARγ, while it is probable for genistein-induced
apoptosis to be independent from the PPARγ pathway.
Discussion
Genistein is one of the most important
phytoestrogens; and its natural plant-derived compound is
structurally similar to 17β-estradiol. Genistein is potentially
regarded as an ideal chemotherapy agent for OS, due to its natural
and safe applications, as well as its minimal side effects and
relatively low cost. Numerous studies have indicated that genistein
induced antitumor effects; through promoting cell death via
G2/M cell cycle arrest (51,52)
and inducing apoptosis in various types of cancer cell lines
(8–11). In human OS cell lines, genistein
has shown its ability to inhibit tyrosine kinase and to affect cell
growth dose-dependently inhibiting hyaluronan and proteoglycan
synthesis (13). Although some
studies reported mechanisms involving ERs, some antitumor effects
of estrogen-like compounds appear to be independent in their
ability to bind with these receptors (13,18,19).
However, there are still underlying mechanisms pertaining to the
antitumor effects of genistein on OS that are unclear.
PPARγ is a ligand-dependent nuclear transcription
factor that belongs to the nuclear hormone receptor superfamily;
which is involved in gene expression related metabolic processes,
such as lipid and glucose homeostasis and important
anti-inflammatory effects. Previous investigations revealed that
PPARγ agonists have antitumoral properties in OS (37–51).
PPARγ has been proposed as a possible target of genistein (22,23).
No previous studies were done on whether genistein could inhibit OS
cell growth by activating PPARγ.
In this study, phytoestrogen genistein inhibited
cell survival dose-dependently and induce G2/M cell
cycle arrest and apoptosis in the MG-63 OS cell line. With regards
to the underlying mechanisms, the cell growth alternation of
genistein and GW9662 and its inhibitory effects on MG-63 cells were
observed to last for up to 48 h, compared with genistein treatment
alone. The data in our study demonstrated that the antitumor effect
of genistein on MG-63 cells is, at least, partly dependent on PPARγ
activation - since it was inhibited by the selective PPARγ
antagonist, GW9662. Moreover, inhibiting PPARγ by GW9662 reversed
the repressing effects of genistein on cell proliferation, but did
not affect the genistein-induced cell apoptosis process.
To further explore the molecular mechanisms of the
above results, we performed western blot and real-time quantitative
RT-PCR analysis. PPARγ plays an critical role in tumorigenesis and
differentiation (53). We found
that PPARγ is highly expressed in genistein-treated MG-63 cells,
which can be reversed by inhibiting statistically significant
PPARγ. We then investigated the downstream signaling effects of
genistein and its co-application on MG-63 cells. PTEN, a well-known
tumor suppressor, potently regulates the PI3K/Akt pathway, which is
involved in cell proliferation and survival. Any altered expression
of PTEN may contribute to the growth of osteosarcoma (54,55).
There is evidence that genistein exposure elevates PTEN levels via
PPARγ activation (56). Our data
confirmed that genistein treatment caused a significant increase of
PTEN mRNA and protein expression and the accumulation of PPARγ
proteins with the corresponding cell alterations. These results
suggested that the ability of genistein to induce the PTEN to
increase was dependent on PPARγ activation.
We then compared the gene and protein expressions in
cells treated with genistein and cells treated with genistein and
GW9662, to confirm our hypothesis that S/G2/M cell cycle
arrest and apoptosis stimulation were triggered by PPARγ-mediated
epigenetic events, caused by genistein. This hypothesis was based
on current reports, where PTEN reactivates the transcription of
P21WAF1/CIP1, Cyclin B1 is dominantly regulated by
P21WAF1/CIP1 and that the reduced BCL-2 and Survivin
levels are regulated by the PPARγ-mediated PTEN activation. Cell
cycle progression is regulated through several different
cyclin-dependent kinase (CDK) regulatory mechanisms, while other
investigators demonstrated the ability of genistein to promote
G2/M cell cycle arrest in human prostate carcinoma cells
(51,57). As a member of the Cip/Kip family of
CDK inhibitors (CDKIs), P21WAF1/CIP1 is a cell cycle
regulatory molecule involved in G2/M arrest. This
molecule also downregulates the intracellular protein levels of
Cyclin B1, which plays an essential role as a positive regulator in
cell cycle progression, during the G2/M transition stage
(57). By blocking PI3K/Akt
signaling, PTEN promotes the nuclear movement of CDKIs and enhances
the cell cycle inhibitory activities of P21WAF1/CIP1
(58). This hypothesis was clearly
supported by the fact that GW9662 exhibits the capability of
reversing the genistein-induced increase of P21WAF1/CIP1
and downregulating Cyclin B1, as determined by western blot and
real-time quantitative RT-PCR analysis. On the other hand, BCL-2 is
a key member of the BCL-2 family of apoptosis regulator proteins
that have anti-apoptotic effects; and that the BCL-2 protein
expression can be upregulated by genistein (59). Survivin also acts as an apoptosis
inhibitor, which has been implicated in both apoptosis inhibition
and mitosis regulation (60).
BCL-2 and Survivin are frequently over-expressed in numerous types
of cancer (61). Additionally, Ma
and Wang (62) advocated that
Survivin protein was directly correlated with Bcl-2 in colorectal
cancer; and both proteins reacted on different apoptosis stages to
jointly promote cancer development synergistically. For the past
decade, many cancer research communities argued that PTEN decreased
the expressions of BCL-2 and Survivin to induce apoptosis (49,63).
In the MG-63 cells, we found that genistein dramatically decreased
the mRNA and protein expressions of BCL-2 and Survivin in a
dose-dependent manner. Interestingly, GW9662 enhanced the antitumor
effects of genistein in inhibiting BCL-2 and Survivin, rather than
reversing genistein-induced results, by inactivating PPARγ. The
present study provides evidence that the activation of PPARγ could
attenuate the degradation of BCL-2 and survivin by diverse
mechanisms (64–66). In addition, we found that 1 μmol/l
of GW9662 could slightly decrease the survival rate of the MG-63
cell line. Therefore, genistein-induced OS cell apoptosis may be
independent of PPARγ.
Overall, our results showed that genistein has
PPARγ-dependent inhibitive actions in the MG-63 cells, which was
mainly presented by promoting G2/M arrest although this
compound did not show any ability to increase apoptosis by PPARγ
activation. Genistein-induced effects were associated to increased
PPARγ transcriptional activity, concomitantly to a modulation of
downstream target PTEN, to gradually fulfill growth inhibition that
probably relates to PI3K/Akt signaling; thus, providing a novel
mechanism for the post-translational modification of PPARγ, which
may have therapeutic implications for treating OS.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 81270052).
References
|
1
|
Yang Z, Kulkarni K, Zhu W, et al:
Bioavailability and pharmacokinetics of genistein: mechanistic
studies on its ADME. Anticancer Agents Med Chem. 10:1264–1280.
2012. View Article : Google Scholar
|
|
2
|
Banerjee S, Li Y, Wang Z, et al:
Multi-targeted therapy of cancer by genistein. Cancer Lett.
2:226–242. 2008. View Article : Google Scholar
|
|
3
|
Zhang Z, Wang CZ, Du GJ, et al: Genistein
induces G2/M cell cycle arrest and apoptosis via atm/p53-dependent
pathway in human colon cancer cell. Int J Oncol. 1:289–296.
2013.
|
|
4
|
Ullah MF, Ahmad A, Zubair H, et al: Soy
isoflavone genistein induces cell death in breast cancer cells
through mobilization of endogenous copper ions and generation of
reactive oxygen species. Mol Nutr Food Res. 4:553–559. 2011.
View Article : Google Scholar
|
|
5
|
Mizushina Y, Shiomi K, Kuriyama I, et al:
Inhibitory effects of a major soy isoflavone, genistein, on human
DNA topoisomerase II activity and cancer cell proliferation. Int J
Oncol. 4:1117–1124. 2013.
|
|
6
|
Hwang KA, Kang NH, Yi BR, et al:
Genistein, a soy phytoestrogen, prevents the growth of BG-1 ovarian
cancer cells induced by 17beta-estradiol or bisphenol A via the
inhibition of cell cycle progression. Int J Oncol. 2:733–740.
2013.
|
|
7
|
Xia J, Cheng L, Mei C, et al: Genistein
inhibits cell growth and invasion through regulation of mir-27a in
pancreatic cancer cells. Curr Pharm Des. 33:5348–5353. 2014.
View Article : Google Scholar
|
|
8
|
Hilakivi-Clarke L, Onojafe I, Raygada M,
et al: Prepubertal exposure to zearalenone or genistein reduces
mammary tumorigenesis. Br J Cancer. 11:16821999. View Article : Google Scholar
|
|
9
|
Zhou JR, Mukherjee P, Gugger ET, et al:
Inhibition of murine bladder tumorigenesis by soy isoflavones via
alterations in the cell cycle, apoptosis, and angiogenesis. Cancer
Res. 22:5231–5238. 1998.
|
|
10
|
Li C, Teng RH, Tsai YC, et al: H-Ras
oncogene counteracts the growth-inhibitory effect of genistein in
T24 bladder carcinoma cells. Br J Cancer. 1:80–88. 2004.
|
|
11
|
Yang CH, Murti A, Pfeffer SR, et al:
Interferon α/β promotes cell survival by activating nuclear factor
κB through phosphatidylinositol 3-kinase and Akt. J Biol Chem.
17:13756–13761. 2001.
|
|
12
|
Yamashita K, Suzuki M, Iwata H, et al:
Tyrosine phosphorylation is crucial for growth signaling by tissue
inhibitors of metalloproteinases. FEBS Lett. 1:103–107. 1996.
View Article : Google Scholar
|
|
13
|
Nikitovic D, Tsatsakis AM, Karamanos NK,
et al: The effects of genistein on the synthesis and distribution
of glycosaminoglycans/proteoglycans by two osteosarcoma cell lines
depends on tyrosine kinase and the estrogen receptor density.
Anticancer Res. 23(1A): 459–464. 2003.PubMed/NCBI
|
|
14
|
Morris C, Thorpe J, Ambrosio L, et al: The
soybean isoflavone genistein induces differentiation of MG63 human
osteosarcoma osteoblasts. J Nutr. 5:1166–1170. 2006.
|
|
15
|
Zhang B, Shi ZL, Liu B, et al: Enhanced
anticancer effect of gemcitabine by genistein in osteosarcoma: the
role of Akt and nuclear factor-κB. Anticancer Drugs. 3:288–296.
2010. View Article : Google Scholar
|
|
16
|
Liang C, Li H, Shen C, et al: Genistein
potentiates the anti-cancer effects of gemcitabine in human
osteosarcoma via the downregulation of Akt and nuclear
factor-kappaB pathway. Anticancer Agents Med Chem. 5:554–563. 2012.
View Article : Google Scholar
|
|
17
|
Nakamura A, Aizawa J, Sakayama K, et al:
Genistein inhibits cell invasion and motility by inducing cell
differentiation in murine osteosarcoma cell line LM8. BMC Cell
Biol. 1:242012. View Article : Google Scholar
|
|
18
|
Rickard DJ, Monroe DG, Ruesink TJ, et al:
Phytoestrogen genistein acts as an estrogen agonist on human
osteoblastic cells through estrogen receptors alpha and beta. J
Cell Biochem. 3:633–646. 2003. View Article : Google Scholar
|
|
19
|
Salvatori L, Caporuscio F, Coroniti G, et
al: Down-regulation of epidermal growth factor receptor induced by
estrogens and phytoestrogens promotes the differentiation of U2OS
human osteosarcoma cells. J Cell Physiol. 1:35–44. 2009. View Article : Google Scholar
|
|
20
|
Djiogue S, Njamen D, Halabalaki M, et al:
Estrogenic properties of naturally occurring prenylated isoflavones
in U2OS human osteosarcoma cells: Structure-activity relationships.
J Steroid Biochem Mol Biol. 4–5:184–191. 2010. View Article : Google Scholar
|
|
21
|
Nakamura H, Wang Y, Kurita T, et al:
Genistein increases epidermal growth factor receptor signaling and
promotes tumor progression in advanced human prostate cancer. PLoS
One. 5:e200342011. View Article : Google Scholar
|
|
22
|
Dang ZC, Audinot V, Papapoulos SE, et al:
Peroxisome proliferator-activated receptor gamma as a molecular
target for the soy phytoestrogen genistein. J Biol Chem. 2:962–967.
2003. View Article : Google Scholar
|
|
23
|
Mezei O, Banz WJ, Steger RW, et al: Soy
isoflavones exert anti-diabetic and hypolipidemic effects through
the PPAR pathways in obese Zucker rats and murine RAW 264.7 cells.
J Nutr. 5:1238–1243. 2003.
|
|
24
|
Xiang Q, Lin G, Fu X, et al: The role of
peroxisome proliferator-activated receptor-γ and estrogen receptors
in genistein-induced regulation of vascular tone in female rat
aortas. Pharmacology. 2:117–124. 2010. View Article : Google Scholar
|
|
25
|
Youssef J and Badr M: Peroxisome
proliferator-activated receptors and cancer: challenges and
opportunities. Br J Pharmacol. 1:68–82. 2011. View Article : Google Scholar
|
|
26
|
Willson TM, Lambert MH and Kliewer SA:
Peroxisome proliferator-activated receptor gamma and metabolic
disease. Annu Rev Biochem. 70:341–367. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Berger J and Moller DE: The mechanisms of
action of PPARs. Annu Rev Med. 53:409–435. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bundscherer A, Reichle A, Hafner C, et al:
Targeting the tumor stroma with peroxisome proliferator activated
receptor agonists. Anticancer Agents Med Chem. 7:816–821. 2009.
View Article : Google Scholar
|
|
29
|
Rumi MA, Ishihara S, Kazumori H, et al:
Can PPAR gamma ligands be used in cancer therapy? Curr Med Chem
Anticancer Agents. 6:465–477. 2004. View Article : Google Scholar
|
|
30
|
Tontonoz P, Singer S, Forman BM, et al:
Terminal differentiation of human liposarcoma cells induced by
ligands for peroxisome proliferator-activated receptor gamma and
the retinoid X receptor. Proc Natl Acad Sci USA. 1:237–241. 1997.
View Article : Google Scholar
|
|
31
|
Kubota T, Koshizuka K, Williamson EA, et
al: Ligand for peroxisome proliferator-activated receptor gamma has
potent antitumor effect against human prostate cancer both in vitro
and in vivo. Cancer Res. 15:3344–3352. 1998.
|
|
32
|
Asou H, Verbeek W, Williamson E, et al:
Growth inhibition of myeloid leukemia cells by troglitazone, a
ligand for peroxisome proliferator activated receptor gamma, and
retinoids. Int J Oncol. 5:1027–1031. 1999.
|
|
33
|
Kitamura S, Miyazaki Y, Shinomura Y, et
al: Peroxisome proliferator-activated receptor gamma induces growth
arrest and differentiation markers of human colon cancer cells. Jpn
J Cancer Res. 1:75–80. 1999. View Article : Google Scholar
|
|
34
|
Elstner E, Muller C, Koshizuka K, et al:
Ligands for peroxisome proliferator-activated receptorgamma and
retinoic acid receptor inhibit growth and induce apoptosis of human
breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci
USA. 15:8806–8811. 1998. View Article : Google Scholar
|
|
35
|
Mueller E, Sarraf P, Tontonoz P, et al:
Terminal differentiation of human breast cancer through PPAR gamma.
Mol Cell. 3:465–470. 1998. View Article : Google Scholar
|
|
36
|
Chang TH and Szabo E: Induction of
differentiation and apoptosis by ligands of peroxisome
proliferator-activated receptor gamma in non-small cell lung
cancer. Cancer Res. 4:1129–1138. 2000.
|
|
37
|
Ali AA, Weinstein RS, Stewart SA, et al:
Rosiglitazone causes bone loss in mice by suppressing osteoblast
differentiation and bone formation. Endocrinology. 3:1226–1235.
2005. View Article : Google Scholar
|
|
38
|
Rzonca SO, Suva LJ, Gaddy D, et al: Bone
is a target for the antidiabetic compound rosiglitazone.
Endocrinology. 1:401–406. 2004. View Article : Google Scholar
|
|
39
|
Rajkumar T and Yamuna M: Multiple pathways
are involved in drug resistance to doxorubicin in an osteosarcoma
cell line. Anticancer Drugs. 3:257–265. 2008. View Article : Google Scholar
|
|
40
|
Yamaguchi K, Whitlock NC, Liggett JL, et
al: Molecular characterisation of canine nonsteroidal
anti-inflammatory drug-activated gene. Vet J. 1:89–95. 2008.
View Article : Google Scholar
|
|
41
|
He BC, Chen L, Zuo GW, et al: Synergistic
antitumor effect of the activated PPARgamma and retinoid receptors
on human osteosarcoma. Clin Cancer Res. 8:2235–2245. 2010.
View Article : Google Scholar
|
|
42
|
Haydon RC, Luu HH and He TC: Osteosarcoma
and osteoblastic differentiation: a new perspective on oncogenesis.
Clin Orthop Relat Res. 454:237–246. 2007. View Article : Google Scholar
|
|
43
|
Haydon RC, Zhou L, Feng T, et al: Nuclear
receptor agonists as potential differentiation therapy agents for
human osteosarcoma. Clin Cancer Res. 5:1288–1294. 2002.
|
|
44
|
Yu LX, Yan HX, Liu Q, et al: Endotoxin
accumulation prevents carcinogen-induced apoptosis and promotes
liver tumorigenesis in rodents. Hepatology. 4:1322–1333. 2010.
View Article : Google Scholar
|
|
45
|
Lv L, Xiao XY, Gu ZH, et al: Silencing
USP22 by asymmetric structure of interfering RNA inhibits
proliferation and induces cell cycle arrest in bladder cancer
cells. Mol Cell Biochem. 1–2:11–21. 2011. View Article : Google Scholar
|
|
46
|
Lu Q, Lu S, Gao X, et al: Norisoboldine,
an alkaloid compound isolated from Radix Linderae, inhibits
synovial angiogenesis in adjuvant-induced arthritis rats by
moderating Notch1 pathway-related endothelial tip cell phenotype.
Exp Biol Med. 8:919–932. 2012. View Article : Google Scholar
|
|
47
|
Seargent JM, Yates EA and Gill JH: GW9662,
a potent antagonist of PPARgamma, inhibits growth of breast tumour
cells and promotes the anticancer effects of the PPARgamma agonist
rosiglitazone, independently of PPARgamma activation. Br J
Pharmacol. 8:933–937. 2004. View Article : Google Scholar
|
|
48
|
Patel L, Pass I, Coxon P, et al: Tumor
suppressor and anti-inflammatory actions of PPARgamma agonists are
mediated via upregulation of PTEN. Curr Biol. 10:764–768. 2001.
View Article : Google Scholar
|
|
49
|
Wu ZX, Song TB, Li DM, et al:
Overexpression of PTEN suppresses growth and induces apoptosis by
inhibiting the expression of survivin in bladder cancer cells.
Tumour Biol. 1:9–15. 2007. View Article : Google Scholar
|
|
50
|
Mikhail M, Velazquez E, Shapiro R, et al:
PTEN expression in melanoma: relationship with patient survival,
Bcl-2 expression, and proliferation. Clin Cancer Res. 14:5153–5157.
2005. View Article : Google Scholar
|
|
51
|
Raffoul JJ, Wang Y, Kucuk O, et al:
Genistein inhibits radiation-induced activation of NF-kappaB in
prostate cancer cells promoting apoptosis and G2/M cell cycle
arrest. BMC Cancer. 6:1072006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ouyang G, Yao L, Ruan K, et al: Genistein
induces G2/M cell cycle arrest and apoptosis of human ovarian
cancer cells via activation of DNA damage checkpoint pathways. Cell
Biol Int. 12:1237–1244. 2009. View Article : Google Scholar
|
|
53
|
Wagner ER, He BC, Chen L, et al:
Therapeutic implications of PPARgamma in human osteosarcoma. PPAR
Res. 9564272010.PubMed/NCBI
|
|
54
|
Miao J, Wu S, Peng Z, et al: MicroRNAs in
osteosarcoma: diagnostic and therapeutic aspects. Tumour Biol.
4:2093–2098. 2013. View Article : Google Scholar
|
|
55
|
Weng L, Brown J and Eng C: PTEN induces
apoptosis and cell cycle arrest through
phosphoinositol-3-kinase/Akt-dependent and -independent pathways.
Hum Mol Genet. 3:237–242. 2001. View Article : Google Scholar
|
|
56
|
Waite KA, Sinden MR and Eng C:
Phytoestrogen exposure elevates PTEN levels. Hum Mol Genet.
11:1457–1463. 2005. View Article : Google Scholar
|
|
57
|
Choi YH, Lee WH, Park KY, et al:
p53-independent induction of p21, reduction of Cyclin B1 and G2/M
arrest by the isoflavone genistein in human prostate carcinoma
cells. Jpn J Cancer Res. 2:164–173. 2000. View Article : Google Scholar
|
|
58
|
Mayo LD and Donner DB: The PTEN, Mdm2, p53
tumor suppressor-oncoprotein network. Trends Biochem Sci.
9:462–467. 2002. View Article : Google Scholar
|
|
59
|
Zhang T, Wang F, Xu H-X, et al: Activation
of nuclear factor erythroid 2-related factor 2 and PPARγ plays a
role in the genis-tein-mediated attenuation of oxidative
stress-induced endothelial cell injury. Br J Nutr. 2:223–235. 2013.
View Article : Google Scholar
|
|
60
|
Halasova E, Adamkov M, Matakova T, et al:
Expression of Ki-67, Bcl-2, survivin and p53 proteins in patients
with pulmonary carcinoma. Adv Exp Med Biol. 756:15–21. 2013.
View Article : Google Scholar
|
|
61
|
Gao Q, Yang S and Kang MQ: Influence of
survivin and Bcl-2 expression on the biological behavior of
non-small cell lung cancer. Mol Med Rep. 6:1409–1414. 2012.
|
|
62
|
Ma Y and Wang HS: Correlations of Bcl-2
and survivin gene protein expressions in colorectal cancer. Applied
Mech Mater. 423–426:362–365. 2013. View Article : Google Scholar
|
|
63
|
Kumar P, Miller AI and Polverini PJ: p38
MAPK mediates gamma-irradiation-induced endothelial cell apoptosis,
and vascular endothelial growth factor protects endothelial cells
through the phosphoinositide 3-kinase-Akt-Bcl-2 pathway. J Biol
Chem. 41:43352–43360. 2004. View Article : Google Scholar
|
|
64
|
Fong WH, Tsai HD, Chen YC, et al:
Anti-apoptotic actions of PPAR-gamma against ischemic stroke. Mol
Neurobiol. 2–3:180–186. 2010. View Article : Google Scholar
|
|
65
|
Ren Y, Sun C, Sun Y, et al: PPAR gamma
protects cardiomyocytes against oxidative stress and apoptosis via
Bcl-2 upregulation. Vascul Pharmacol. 2–3:169–174. 2009. View Article : Google Scholar
|
|
66
|
Kim YJ, Park KJ, Song JK, et al: The
PPARgamma agonist protects cardiomyocytes from oxidative stress and
apoptosis via thioredoxin overexpression. Biosci Biotechnol
Biochem. 12:2181–2187. 2012. View Article : Google Scholar
|