Introduction
In Japan, dietary changes favoring more Western
eating habits have resulted in an increased rate of obesity
(1). The prevalence of obesity in
adult men has increased since the 1970s, according to the data of
the Japanese Ministry of Health, Labour and Welfare (Tokyo, Japan)
(2). Obesity has become a major
social and health issue. Obesity is related to an increased risk of
not only metabolic diseases including cardiovascular disease,
hypertension, and diabetes, but also of various cancers,
particularly those with a hormonal basis such as breast and
prostate cancer (PCa) (3). Indeed,
the incidence of PCa has been increasing annually since the early
1990s in the Japanese population. PCa is predicted to become the
second most common cancer among all cancers of Japanese men by 2020
(4). However, the relationship
between PCa and obesity remains controversial, with some studies
indicating that obesity is associated with a decreased incidence of
PCa, and other studies suggesting an increased incidence and a
worse survival among obese men (5–7).
Several reports, including the comment by Freedland (8), have suggested that obesity might be
more important as a risk factor for PCa progression rather than for
its incidence.
Leptin, a 16-kDa polypeptide adipokine that is
secreted predominantly by adipose tissue, is considered to be
involved in satiety regulation in humans and animals (9). In adults, circulating leptin levels
are closely related to the amount of adipose tissue (10), so that serum leptin level is
elevated in obese individuals compared to non-obese individuals.
Leptin acts as a regulatory signal for the central nervous system,
reflects adipose tissue stores, and influences energy homeostasis,
eating behavior, appetite regulation and energy expenditure
(11). Leptin also plays a role in
reproduction, hematogenesis, neovascularization and cancer cell
proliferation (12,13).
Several studies have investigated the relationship
between obesity, plasma leptin levels, and PCa. Chang et al
(14) showed that men with
elevated leptin levels had an increased risk of diagnosis with
high-volume disease. It has also been shown that high leptin levels
correlate with high stage and high grade of PCa (15). High expression levels of leptin and
leptin receptor (ObR), which is a member of the class I
cytokine-receptor family, have been reported in prostate tissues
(16); therefore, leptin may play
an important role in the process of carcinogenesis and progression
of prostate tumors.
Several recent in vitro studies have
indicated that leptin acts as a mitogenic factor for various cell
types including both normal and neoplastic cells (17,18).
ObRs have been identified in prostate cell membranes (19), and appear to function in activation
of mitogenic signaling pathways (20,21).
In those studies, the proliferative effects of leptin on the
androgen-sensitive (AS) PCa cell line, LNCaP, were less pronounced
than its effects on the androgen-resistant lines, DU145 and PC-3.
In contrast to these reported proliferative effects of leptin on
only androgen-insensitive (AI) cell lines, Deo et al have
suggested that leptin induces the proliferation of only AS cell
lines (22). They reported a
dose-dependent increase in proliferation when LNCaP cells were
cultured with leptin, but no changes in the growth of AI cells,
even if they were incubated with a high dose of leptin. Thus, the
effects of leptin on the growth of PCa cells remain controversial.
In addition, the exact mechanism by which leptin exerts
proliferative effects on cancer cells, including PCa cells, is not
fully understood.
In the present study, we investigated the effects of
leptin on the growth of both AS and AI PCa cells and on the
underlying mechanisms of leptin action. Because exposure to
elevated leptin levels for a long time is expected to result in
higher PCa risk or higher PCa tumor aggressiveness in humans, the
present study was performed using PCa cells that were cultured in
medium containing various concentrations of leptin for 28 days.
Materials and methods
Materials
RPMI-1640 and Ham’s F12 media for cell culture were
supplied by Wako Pure Chemical Industries, Ltd. (Osaka, Japan) and
Life Technologies (Carlsbad, CA, USA), respectively. Fetal bovine
serum (FBS) was obtained from Nichirei Biosciences, Inc. (Tokyo,
Japan). Monoclonal antibody against human β-actin and polyclonal
rabbit antibodies against human ObR, and phosphorylated forkhead
box O1 (FOXO1) (Ser256) were purchased from Sigma-Aldrich Japan
K.K. (Tokyo, Japan) and Abcam (Tokyo, Japan), respectively.
LY294002, used as an PI3K inhibitor, and polyclonal rabbit
antibodies against phosphorylated Akt, Akt, cyclin D1, p21Cip1 and
lamin A/C were obtained from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Polyclonal rabbit antibody against FOXO1 was
obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA
USA).
Cell culture and treatment
The AS human PCa cell line LNCaP and the AI human
PCa cell lines DU145 and PC-3 used in this study were purchased
from the American Type Culture Collection (ATCC) (Rockville, MD,
USA). LNCaP and DU145 cells were maintained in RPMI-1640 medium and
PC-3 cells were maintained in Ham’s F-12 medium. Media were
supplemented with 10% heat-inactivated FBS in the presence or
absence of human recombinant leptin (Sigma Aldrich) at the
indicated concentrations, in a humidified incubator at 37°C and 5%
CO2. The medium was changed every 4 days. Cells were
passaged when they were 50–70% confluent. Cells were transferred
into serum-free medium and cultured for 24 h under the same
conditions except for the absence of serum.
Western blots
Cells were lysed with ice-cold lysis buffer and
protease inhibitor and phosphatase inhibitor cocktails
(Sigma-Aldrich). Samples were centrifuged at 12,000 × g for 10 min
at 4°C, and supernatants were electrophoresed on sodium dodecyl
sulfate (SDS)-polyacrylamide gels and transferred to polyvinylidene
difluoride membranes (Millipore, Bedford, MA, USA). After blocking
with 5% skimmed milk, the membranes were probed with primary
antibodies overnight at 4°C, followed by horseradish
peroxidase-conjugated secondary antibody (GE Healthcare,
Buckinghamshire, UK) for 1 h at room temperature. The immune
complexes were visualized with the Enhanced Chemiluminescence Plus
detection system (GE Healthcare) according to the manufacturer’s
instructions.
Gene silencing with siRNA
For small interfering RNA (siRNA) experiments, siRNA
cocktails were obtained from, and functionally annotated by Cosmo
Bio Co., Ltd. (Tokyo, Japan) and Bioneer, Inc. (Alameda, CA, USA).
The siRNA cocktails are indicated in Table I. For the control, non-targeting
siRNA (cat. no. 4390844) was obtained from Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). Cells were transiently
transfected with each siRNA or control siRNA cocktail using the
Neon Transfection System (Life Technologies). After 48-h
incubation, cells were utilized for each assay.
 | Table IThe siRNA used in this study. |
Table I
The siRNA used in this study.
| siRNA | Sense (5′→3′) | Antisense
(5′→3′) |
|---|
| ObR |
GGAAGAUGUAUGAGGUUUATT |
UAAACCUCAUACAUCUUCCTT |
|
GGCUAGAUGGACUGGGAUATT |
UAUCCCAGUCCAUCUAGCCTT |
|
CCAGAGUGAUGCAGGUUUATT |
UAAACCUGCAUCACUCUGGTT |
| FOXO1 |
GCUGCUGUAGAUAAGGACU |
AGUCCUUAUCUACAGCAGC |
|
GUCCAAGACAUAGCUGGUU |
AACCAGCUAUGUCUUGGAC |
|
CUGCAUAGCAUCAAGUCUU |
AAGACUUGCUGCUAUGCAG |
Cell proliferation assay
The relative effect of leptin on the proliferation
of each cell line was assessed using WST-8 assays and a Cell
Counting kit-8 (Dojindo, Kumamoto, Japan). Cells that had been
cultured in the presence or absence of graded concentrations of
leptin for each day were re-seeded in 96-well plates at a density
of 3×103 cells/well and were cultured in the same graded
concentrations of leptin in at least three replicate wells at 37°C
for 48 h. The WST-8 reagent was added to each well, and the cells
were further incubated for another 3 h. Absorbance was determined
using a spectrophotometer (Thermo Scientific Multiskan FC; Thermo
Fisher Scientific, Inc.) at 450 nm. Absorbance values were
expressed as percentages relative to untreated controls.
Cell invasion assay
The cell invasion assay was performed using a
Matrigel Invasion Chamber from Becton-Dickinson (BD Biosciences,
Tokyo, Japan) according to the manufacturer’s instructions.
Approximately 3×104 cells that had been cultured in the
presence or absence of 100 ng/ml of leptin for 28 days were seeded
onto the top of chamber and were incubated at 37°C in a 5%
CO2 incubator. After 48-h incubation, the media were
aspirated, and the cells remaining on the upper surface of the
membrane and the Matrigel Matrix were removed with a sterile cotton
swab. Invasive cells on the bottom surface of the membrane that had
passed through the Matrigel were fixed in methanol and stained with
Diff-Quik stain (Sysmex Corp., Hy go, Japan). The number of invaded
cells on four representative sections of each membrane was counted
in high-power fields under a light microscope (BX63; Olympus Corp.,
Tokyo, Japan). Percent invasion, which was the percent of the
seeded cells that had passed through the Matrigel, was calculated.
The assay was repeated at least three times.
Scratch wound-healing migration
assay
Cells that had been cultured in the presence or
absence of 100 ng/ml of leptin for 28 days were seeded into 6-well
plates and cultured in at least three replicate wells at 37°C. When
cells were grown to 90% or above confluence, a crossing linear
scratch was made with a 200-μl pipette tip, and floating cells were
removed by washing with phosphate-buffered saline (PBS). These
plates were photographed at the identical location both before and
18 h after incubation. The average area of the open scratch area
was calculated using ImageJ ver 1.46r (NIH, Bethesda, MD, USA).
Immunofluorescence
Approximately 1×104 cells were seeded in
a Lab-Tek II Chamber Slide (Thermo Fisher Scientific, Inc.). Cells
were then further cultured in serum-free media for 48 h with the
same leptin concentration. For PI3K inhibition, cells were
pre-treated with LY294002 for 2 h. The cells were then fixed with
4% paraformaldehyde and permeabilized with PBS containing 0.2%
Triton X-100 followed by blocking with 1% bovine serum albumin.
Thereafter, the cells were incubated with polyclonal anti-FOXO1
antibody followed by incubation with fluorescein
isothiocyanate-conjugated secondary antibody (Jackson
ImmunoResearch Labs, West Grove, PA, USA). Trihydrochloride
(Hoechst 33342; Dojindo) staining was used for nuclear detection.
Fluorescence was photographed using an Olympus IX71 microscope
(Olympus Corp.).
Subcellular fractionation
Subcellular fractionation was carried out using
Nuclear/Cytosol Fractionation kits (BioVision, Inc., Milpitas, CA,
USA) as described by the manufacturer. Equal volumes of each
fraction were loaded onto an SDS-polyacrylamide gel, and western
blotting was performed as previously described.
Statistical analysis
All values are expressed as the means ± standard
deviation (SD) of at least three independent experiments.
Statistical analysis was performed using Student’s unpaired t-test.
P<0.05 was considered statistically significant.
Results
Long-term exposure to leptin enhances PCa
cell proliferation, invasion and migration
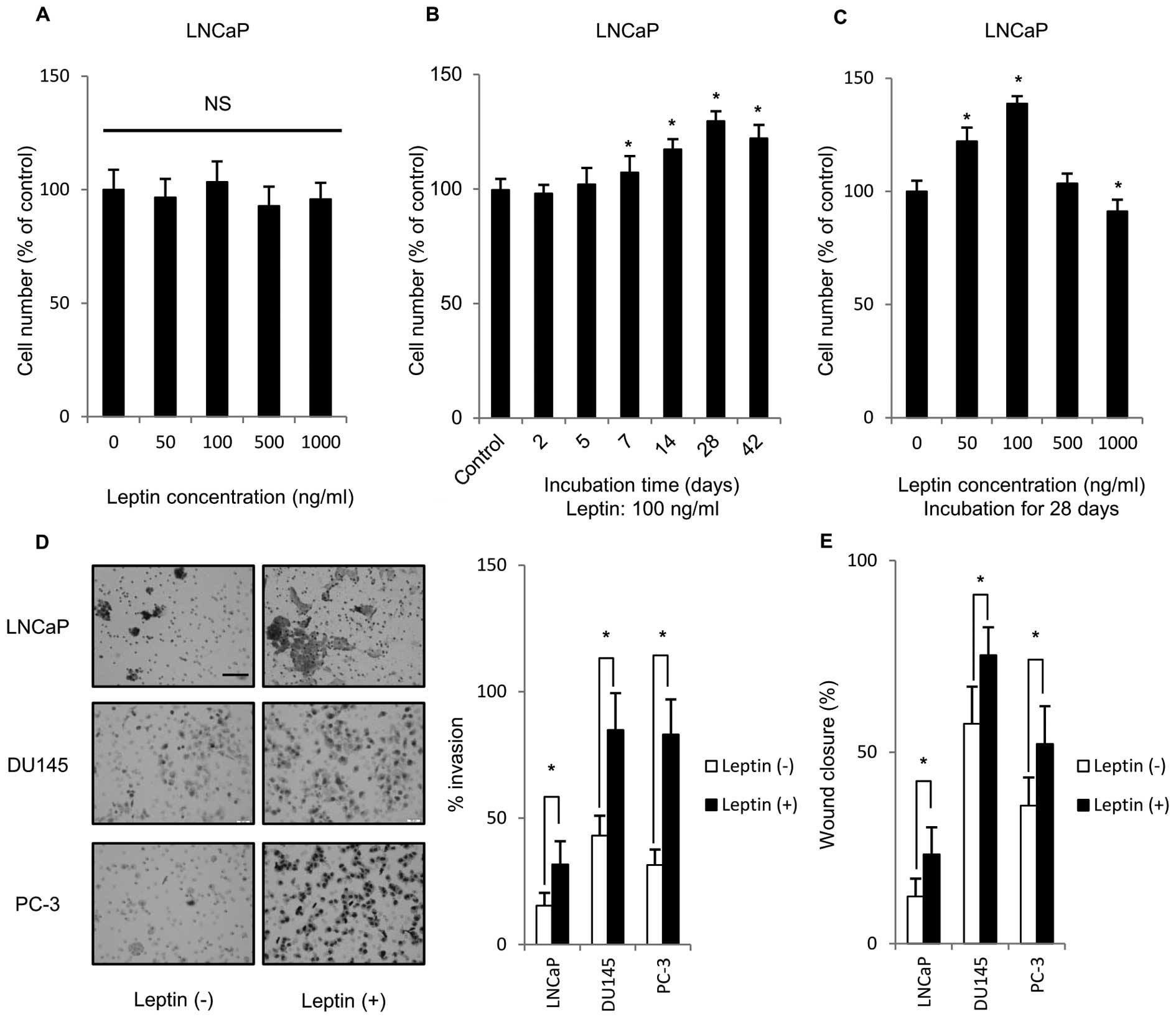
To clarify the effect of leptin on PCa cell
proliferation, LNCaP, DU145 and PC-3 were treated with various
concentrations of leptin for 48 h. Fig. 1A–C shows only results derived from
LNCaP cells, because the similar tendency was found in their
experiments using three PCa cells examined. The incubation for 48 h
did not affect the growth (Fig.
1A). Because short-term leptin exposure may not reflect the
in vivo situation, we tried to examine the effects of
long-term leptin exposure. Interestingly, time-course experiments
showed that cell growth reached maximum after 28-day incubation
with 100 ng/ml leptin (Fig. 1B).
The most effective concentration of leptin was at 100 ng/ml
(Fig. 1C), thus, 100 ng/ml was
selected as the leptin concetration for further experiments.
Maximum physiological concentration of serum leptin in human is
~100 ng/ml (10). These results
indicated that treatment with 100 ng/ml leptin for 28 days might be
the relevant condition for induction of proliferation of all PCa
cells. We tested the effect of long-term leptin treatment (1 month)
on the ability of PCa cells to invade through Matrigel. As shown in
Fig. 1D, PCa cells that were
treated long-term with leptin showed significantly higher transit
to the bottom surface of a Matrigel-coated membrane than
non-leptin-treated cells. In addition, the effect of long-term
leptin treatment on the migration ability of PCa cells was
estimated using a scratch wound-healing migration assay. As shown
in Fig. 1E, PCa cells treated long
term with leptin showed a significantly larger healed area after
scratching compared to non-treated cells. Thus, long-term exposure
to leptin enhanced the malignant potential of PCa cell lines.
Long-term exposure to leptin increases
ObR expression and Akt phosphorylation
Next, the effect of long-term leptin treatment on
leptin-induced intracellular signaling pathways in PCa cells was
investigated. We first determined if the ObR and intracellular Akt
phosphorylation played a role in the enhanced proliferation
observed in long-term leptin-treated cells. Akt phosphorylation was
assayed because several groups have previously demonstrated that
leptin enhances cell proliferation via the PI3K/Akt signaling
pathway. Expression of the ObR, as assessed by western blotting,
was significantly increased by long-term leptin treatment of all
PCa cell lines, compared to non-treated cells, as shown in Fig. 2A. Furthermore, we confirmed that
the level of Akt phosphorylation increased in all PCa cell lines
that were treated with leptin for 1 month compared to non-treated
cells.
ObR gene silencing and PI3K inhibitor
suppress cell proliferation
To confirm that ObR signaling played a role in
leptin-induced proliferation of PCa cells, ObR specific siRNA was
transfected into PCa cells that had been exposed to leptin for 1
month. After 48-h transfection, ObR protein expression was markedly
decreased in comparison to cells transfected with scrambled
non-specific siRNA (Fig. 2B). The
silencing of ObR significantly decreased the proliferation of all
PCa cell lines compared to control siRNA cells. This result
suggests that the ObR receptor mediates leptin effects on
proliferation.
To further analyze the role of Akt activation in
leptin- enhanced PCa cell proliferation, we used LY294002 to
inhibit Akt activation. Following long-term leptin culture,
LY294002 or control dimethyl sulfoxide (DMSO) was added for 2 h.
And cells were incubated in serum-free media for 48 h. As shown in
Fig. 2C, inhibition of Akt
phosphorylation with LY294002 decreased cell proliferation in the
absence of leptin and further suppressed leptin-induced
proliferation in LNCaP cells. We observed similar results in DU145
and PC-3 (data not shown). These results suggest that the PI3K/Akt
signaling pathway is important for leptin-induced cell
proliferation.
Long-term exposure to leptin induces
phosphorylation of FOXO1 via the PI3K/Akt signaling pathway
The PI3K/Akt signaling pathway has been reported to
phosphorylate FOXO1, which mediates some of its downstream
signaling. To determine if FOXO1 was involved in leptin-mediated
cellular effects, we first determined whether long-term exposure to
leptin induces phosphorylation of FOXO1 using western blotting. As
shown in Fig. 3A, long-term
exposure to leptin induced a higher level of FOXO1 phosphorylation
than that of non-treated cells. We further assayed if this
phosphorylation is mediated by PI3K/Akt by assaying the effect of
adding the specific PI3K inhibitor LY294002 (25 μM) for the last 2
h of long-term leptin culture of PCa cells on FOXO1
phosphorylation. Following LY294002 treatment FOXO1 phosphorylation
induced by leptin treatment was greatly reduced compared to control
cells (Fig. 3A). These results
suggested that long-term exposure to leptin induced phosphorylation
of FOXO1 via the PI3K/Akt signaling pathway.
FOXO1 gene silencing enhances PCa cell
proliferation
To clarify the role of FOXO1 in the proliferation of
these PCa cell lines, we transiently transfected leptin-untreated
cells with FOXO1 siRNA. After 48-h transfection, FOXO1 protein
expression was decreased in comparison to cells transfected with
scrambled non-specific siRNA (Fig.
3B). FOXO1 gene silencing significantly increased the
proliferation of these cells compared to control cells (Fig. 3B). These results indicated that
FOXO1 functions to suppress PCa cell proliferation.
Long-term exposure to leptin regulates
nuclear/cytoplasmic localization of FOXO1 via PI3K
One possible mechanism by which Akt-mediated
phosphorylation of FOXO1 might mediate FOXO1 effects on
leptin-induced proliferation is by regulation of FOXO1 cellular
localization. Thus, if FOXO1 is phosphorylated on specific residues
by Akt, FOXO1 can be excluded from the nucleus, thereby
inactivating its transcriptional activity. To confirm the effect of
long-term exposure to leptin on the intracellular localization of
FOXO1, immunocytochemistry was performed. As shown in Fig. 4A, FOXO1 was expressed in both the
nucleus and the cytoplasm in DU145 cells. In contrast, FOXO1
expression in the nucleus was strongly decreased by long-term
leptin treatment. The addition of LY294002 to the medium with
leptin restored the intra-nuclear expression of FOXO1 protein. We
observed similar results in LNCaP and PC-3 cells (data not shown).
To further confirm the intracellular distribution of the FOXO1
protein, we compared FOXO1 expression in isolated nuclear fractions
of leptin-treated and non-treated cells by western blotting.
Fig. 4B shows that FOXO1
expression in the nuclei of leptin-treated cells was significantly
decreased to ~50% of that in non-treated cells.
Long-term exposure to leptin regulates
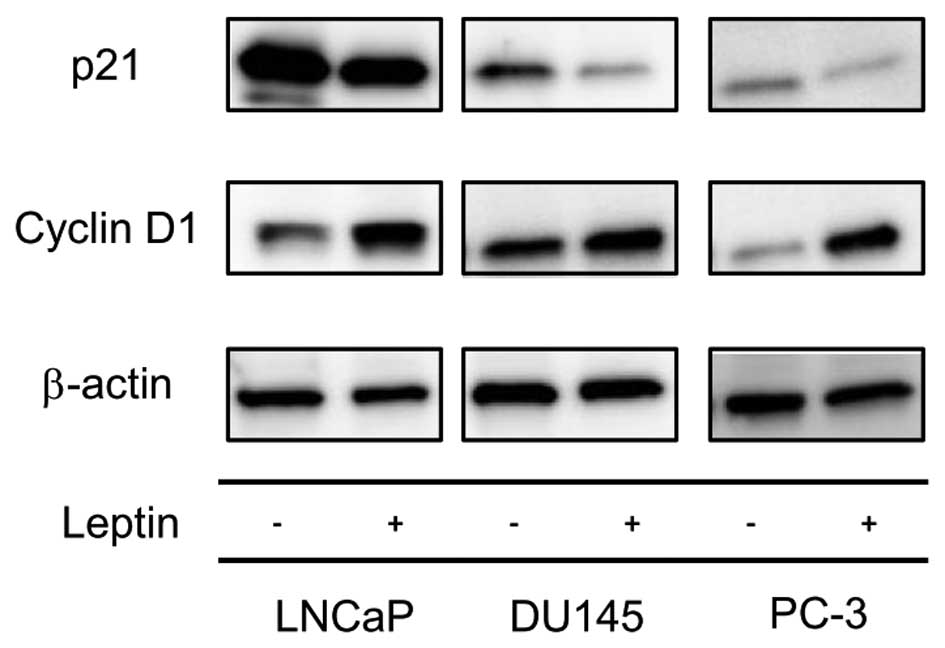
the expression of cell cycle-related proteins
As described above, long-term exposure of
leptin-induced PCa cell proliferation resulted from FOXO1 exclusion
out of a nuclear region. We therefore next determined if the
nuclear exclusion of FOXO1 that resulted from long-term exposure to
leptin might affect the expression of cell cycle-related proteins.
Increased cyclin D1 expression and decreased p21 expression was
shown by western blotting of cells following long-term leptin
treatment compared to non-treated cells (Fig. 5), suggesting that genes that
regulate the G1/S transition were modulated towards cell cycle
progression by leptin treatment.
Discussion
To our knowledge, this is the first report that
long-term exposure of leptin increases proliferation, migration and
invasion of PCa cells through inactivation of FOXO1.
Most in vitro studies of leptin have only
focused on analysis of the short-term effects of leptin exposure
over 24–72 h. In preliminary studies we analyzed the effect of
short-term leptin exposure on PCa cell proliferation and found that
treatment with leptin concentrations of 0–1,000 ng/ml for 48 h did
not modulate PCa cell proliferation (Fig. 1A). However, short-term leptin
exposure may not reflect the in vivo situation. Thus, obese
people are exposed to high levels of leptin over a long period. In
addition, in humans, higher PCa risk or higher tumor aggression
correlates with elevated leptin levels. For example, the study by
Wang et al showed that a low-fat diet decreased the tumor
growth of xenografted LNCaP cells (23). Another study demonstrated that a
high-fat diet induced a high level of serum leptin and high
argyrophilic nucleolar organizer region (AgNOR) counts in a
transgenic adenocarcinoma of mouse prostate (TRAMP) model (24). These studies suggested that chronic
hyperleptinemia might be related to PCa progression. The present
study was therefore performed to clarify whether long-time exposure
(28 days) to leptin affected the aggressiveness of three human PCa
cell lines; LNCaP, DU145 and PC-3, in vitro.
First of all, long-term exposure to leptin
significantly enhanced the aggressiveness of all PCa cells. Leptin
had a similar influence on both AS and AI PCa cells, although
previous studies have not shown common effects of leptin on AS and
AI PCa cells (20–22). In addition, we demonstrated that
long-term exposure to leptin enhanced expression of the ObR and Akt
phosphorylation. Overexpression of leptin and the expression of ObR
have been reported to be associated with tumor progression in
several types of cancers (25–28).
ObR gene silencing resulted in a significant decrease in long-term
leptin-induced cell proliferation. Moreover, the PI3K inhibitor
also suppressed cell proliferation. These results suggested that
the PI3K/Akt signaling pathway, via ObR, has an important role in
leptin-induced proliferation.
Furthermore, we showed that the Akt phosphorylation
induced FOXO1 phosphorylation. The PI3K/Akt signaling pathway is
known to be important for FOXO1 phosphorylation (29). The FOXO family is composed of four
members (FOXO1, 3a, 4, and 6) that are known to be important
factors in many cellular processes including apoptosis, repair of
damaged DNA, glucose metabolism and cell cycle arrest (29,30).
FOXO1 is known to function in the tumor suppression of several
malignant tumors (31–35). Also in the present study, FOXO1
silencing promoted the proliferation of all PCa cell lines
examined. Furthermore, FOXO1 has a role as a transcription factor
in the nucleus that regulates a large spectrum of tumor suppression
genes. The activities of FOXOs are controlled by post-translational
modifications such as phosphorylation and acetylation (29,30,36,37).
Unphosphorylated FOXO proteins usually reside only in the nucleus.
Once they are phosphorylated by intracellular environmental
changes, they are exported to the cytoplasm and inactivated
(37,38). FOXO1 promotes cell cycle arrest in
the nucleus via upregulation of cell cycle inhibitors or
downregulation of cell cycle regulators (29,30,39).
We demonstrated that PCa cell proliferation induced by long-term
exposure to leptin was a result of FOXO1 exclusion from the
nucleus. In addition, increased cyclin D1 expression and decreased
p21 expression was shown following long-term leptin treatment.
These data suggest that leptin treatment may release these cells
from cell cycle arrest, which FOXO1 regulates in the nucleus, by
exclusion of FOXO1 from the nucleus. These findings closely match
those of previous studies (40,41),
which indicate the induction of apoptosis through PI3K/FOXO1
signaling pathways. Lee et al have shown that MHY-449, a
novel dihydrobenzofuro[4,5-b] naphthyridin-6-one derivative,
induced apoptotic cell death in PC-3 cells through a reduction in
the phosphorylation of Akt and FOXO1 and induced subcellular
translocation of FOXO1 from the cytoplasm to the nucleus in these
cells (41).
PCa growth depends mainly on androgen signaling via
the androgen receptor (AR). Liu et al demonstrated that
FOXO1 represses AR signaling, and that the nuclear localization of
FOXO1 is necessary for its inhibition (42). Zhang et al also demonstrated
that FOXO1 silencing attenuates methylselenic acid suppression of
AR activity (33). Thus, in the
case of AS PCa cells, leptin may increase AR activity by exclusion
of FOXO1 from the nucleus.
In conclusion, our findings provide supporting
evidence for a signaling pathway between long-term leptin treatment
and FOXO1 function and contribute to an understanding of the
association between obesity and PCa progression.
Acknowledgements
We are grateful to the members of the Higashiyama
Laboratory (Ehime University Graduate School of Medicine, Ehime,
Japan) for helpful suggestions. We thank Ms. Kazumi Kanno, Ms.
Izumi Tanimoto, and Ms. Maria Mori for their excellent technical
assistance.
References
|
1
|
Kaneko G, Miyajima A, Yuge K, Hasegawa M,
Takeda T, Jinsaki M, Kikuchi E, Nakagawa K and Oya M: Periprostatic
fat area is an independent factor that prolonged operative time in
laparoscopic radical prostatectomy. Urology. 82:1304–1309. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Japanese Ministry of Health, Labour and
Welfare. Results of Kokumin-Kenkou-Eiyou Chousa (in Japanese).
http://www.mhlw.go.jp/.
|
|
3
|
Bray GA: The underlying basis for obesity:
relationship to cancer. J Nutr. 132(Suppl 11): 3451S–3455S.
2002.PubMed/NCBI
|
|
4
|
Utada M, Ohno Y, Soda M and Kamo K:
Estimation of cancer incidence in Japan with an age-period-cohort
mode. Asian Pac J Cancer Prev. 11:1235–1240. 2010.
|
|
5
|
Giovannucci E, Rimm EB, Liu Y, Leitzmann
M, Wu K, Stampfer MJ and Willett WC: Body mass index and risk of
prostate cancer in U.S. health professionals. J Natl Cancer Inst.
95:1240–1244. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Andersson SO, Wolk A, Bergström R, Adami
HO, Engholm G, Englund A and Nyrén O: Body size and prostate
cancer: a 20-year follow-up study among 135006 Swedish construction
workers. J Natl Cancer Inst. 89:385–389. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schuurman AG, Goldbohm RA, Dorant E and
van den Brandt PA: Anthropometry in relation to prostate cancer
risk in the Netherlands Cohort Study. Am J Epidemiol. 151:541–549.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Freedland SJ: Obesity and prostate cancer:
a growing problem. Clin Cancer Res. 11:6763–6766. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mistry AM, Swick AG and Romsos DR: Leptin
rapidly lowers food intake and elevates metabolic rates in lean and
ob/ob mice. J Nutr. 127:2065–2072. 1997.PubMed/NCBI
|
|
10
|
Liuzzi A, Savia G, Tagliaferri M,
Lucantoni R, Berselli ME, Petroni ML, De Medici C and Viberti GC:
Serum leptin concentration in moderate and severe obesity:
relationship with clinical, anthropometric and metabolic factors.
Int J Obes Relat Metab Disord. 23:1066–1073. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miller R, Tanofsky-Kraff M, Shomaker LB,
et al: Serum leptin and loss of control eating in children and
adolescents. Int J Obes (Lond). 38:397–403. 2014. View Article : Google Scholar
|
|
12
|
Sierra-Honigmann MR, Nath AK, Murakami C,
García-Cardeña G, et al: Biological action of leptin as an
angiogenic factor. Science. 281:1683–1686. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Somasundar P, Yu AK, Vona-Davis L and
McFadden DW: Differential effects of leptin on cancer in vitro. J
Sur Res. 113:50–55. 2003. View Article : Google Scholar
|
|
14
|
Chang S, Hursting SD, Contois JH, et al:
Leptin and prostate cancer. Prostate. 46:62–67. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tewari R, Rajender S, Natu SM, Goel A,
Dalela D, Goel MM and Tondon P: Significance of obesity markers and
adipocytokines in high grade and high stage prostate cancer in
North Indian men - a cross-sectional study. Cytokine. 63:130–134.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hoon Kim J, Lee SY, Myung SC, Kim YS, Kim
TH and Kim MK: Clinical significance of the leptin and leptin
receptor expressions in prostate tissues. Asian J Androl.
10:923–928. 2008. View Article : Google Scholar
|
|
17
|
Hardwick JC, Van Den Brink GR, Offerhaus
GJ, Van Deventer SJ and Peppelenbosch MP: Leptin is a growth factor
for colonic epithelial cells. Gastroenterology. 121:79–90. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hoda MR, Keely SJ, Bertelsen LS, Junger
WG, Dharmasena D and Barrett KE: Leptin acts as a mitogenic and
antiapoptotic factor for colonic cancer cells. Br J Surg.
94:346–354. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Somasundar P, Frankenberry KA, Skinner H,
et al: Prostate cancer cell proliferation is influenced by leptin.
J Surg Res. 118:71–82. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hoda MR and Popken G: Mitogenic and
anti-apoptotic actions of adipocyte-derived hormone leptin in
prostate cancer cells. BJU Int. 102:383–388. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Onuma M, Bub JD, Rummel TL and Iwamoto Y:
Prostate cancer cell-adipocyte interation: leptin mediates
androgen-independent prostate cancer cell proliferation through
c-Jun NH2-terminal kinase. J Biol Chem. 278:42660–42667. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Deo DD, Rao AP, Bose SS, et al:
Differential effects of leptin on the invasive potential of
androgen-dependent and -independent prostate carcinoma cells. J
Biomed Biotechnol. 2008:1639022008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Y, Corr JG, Thaler HT, Tao Y, Fair WR
and Heston WD: Decreased growth of established human prostate LNCaP
tumors in nude mice fed a low-fat diet. J Natl Cancer Inst.
87:1456–1462. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Park SH, Chang SN, Baek MW, Kim DJ, Na YR,
Seok SH, Lee BH, Kim KS and Park JH: Effects of dietary high fat on
prostate intraepithelial neoplasia in TRAMP mice. Lab Anim Res.
29:39–47. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Garofalo C, Koda M, Cascio S, Sulkowska M,
Kanczuga-Koda L, Golaszewska J, Russo A, Sulkowski S and Surmacz E:
Increased expression of leptin and the leptin receptor as a marker
of breast cancer progression: possible role of obesity-related
stimuli. Clin Cancer Res. 12:1447–1453. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ishikawa M, Kitayama J and Nagawa H:
Expression pattern of leptin and leptin receptor (OB-R) in human
gastric cancer. World J Gastroenterol. 12:5517–5522.
2006.PubMed/NCBI
|
|
27
|
Riolfi M, Ferla R, Del Valle L, et al:
Leptin and its receptor are overexpressed in brain tumors and
correlate with the degree of malignancy. Brain Pathol. 20:481–489.
2010. View Article : Google Scholar :
|
|
28
|
Howard JM, Beddy P, Ennis D, Keogan M,
Pidgeon GP and Reynolds JV: Associations between leptin and
adiponectin receptor upregulation, visceral obesity and tumour
stage in oesophageal and junctional adenocarcinoma. Br J Surg.
97:1020–2027. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang X, Tang N, Hadden TJ and Rishi AK:
Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta.
1813:1978–1986. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang H and Tindall DJ: Dynamic FoxO
transcription factors. J Cell Sci. 120:2479–2487. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li R, Erdamar S, Dai H, Wheeler TM, Frolov
A, Scardino PT, Thompson TC and Ayala GE: Forkhead protein FKHR and
its phosphorylated form p-FKHR in human prostate cancer. Hum
Pathol. 38:1501–1507. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang H, Pan Y, Zheng L, Choe C, Lindgren
B, Jensen ED, Westendorf JJ, Cheng L and Huang H: FOXO1 inhibits
Runx2 transcriptional activity and prostate cancer cell migration
and invasion. Cancer Res. 71:3257–3267. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang H, Fang J, Yao D, Wu Y, Ip C and
Dong Y: Activation of FOXO1 is critical for the anticancer effect
of methylseleninic acid in prostate cancer cells. Prostate.
70:1265–1273. 2010.PubMed/NCBI
|
|
34
|
Scodelaro Bilbao P and Boland R:
Extracellular ATP regulates FoxO family of transcription factors
and cell cycle progression through PI3K/Akt in MCF-7 cells. Biochim
Biophys Acta. 1830:4456–4469. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Berry E, Hardt JL, Clardy J, Lurain JR and
Kim JJ: Induction of apoptosis in endometrial cancer cells by
psammaplysene A involves FOXO1. Gynecol Oncol. 112:331–336. 2009.
View Article : Google Scholar :
|
|
36
|
Rena G, Guo S, Cichy SC, Unterman TG and
Cohen P: Phosphorylation of the transcription factor forkhead
family member FKHR by protein kinase B. J Biol Chem.
274:17179–17183. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kau TR, Schroeder F, Ramaswamy S,
Wojciechowski CL, Zhao JJ, Roberts TM, Clardy J, Sellers WR and
Silver PA: A chemical genetic screen identifies inhibitors of
regulated nuclear export of a Forkhead transcription factor in
PTEN-deficient tumor cells. Cancer Cell. 4:463–476. 2003.
View Article : Google Scholar
|
|
38
|
Wen Q, Duan X, Liao R, Little P, Gao G,
Jiang H, Lalit S, Quirion R and Zheng W: Characterization of
intracellular translocation of Forkhead transcription factor O
(FoxO) members induced by NGF in PC12 cells. Neurosci Lett.
498:31–36. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu Z, Sun H, Zeng W, He J and Mao X:
Upregulation of MircoRNA-370 induces proliferation in human
prostate cancer cells by downregulating the transcription factor
FOXO1. PLoS One. 7:e458252012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen Q, Ganapathy S, Singh KP, Shankar S
and Srivastava RK: Resveratrol induces growth arrest and apoptosis
through activation of FOXO transcription factors in prostate cancer
cells. PLoS One. 5:e152882010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lee SH, Kang YJ, Sung B, et al: MHY-449, a
novel dihydrobenzofuro[4,5-b][1,8] naphthyridin-6-one derivative,
induces apoptotic cell death through modulation of Akt/FoxO1 and
ERK signaling in PC3 human prostate cancer cells. Int J Oncol.
44:905–911. 2014.PubMed/NCBI
|
|
42
|
Liu P, Li S, Gan L, Kao TP and Huang H: A
transcription-independent function of FOXO1 in inhibition of
androgen-independent activation of the androgen receptor in
prostate cancer cells. Cancer Res. 68:10290–10299. 2008. View Article : Google Scholar : PubMed/NCBI
|