Introduction
Endometrial carcinoma, comprising of several types
of malignancies arise from the endometrium or lining of the uterus,
is the most common gynecologic malignancy among women in the United
States, with an estimated 52,630 new case and 8,590 deaths in 2014
(1). Endometrial carcinoma cases
in Chinese women increased in the last decade. Based on clinical
features and pathogenesis endometrial carcinomas have been
classified into two types (2).
Type I endometrial carcinomas occur commonly in perimenopausal
women, with low grade, related to obesity and estrogen exposure.
Type II endometrial carcinomas are more common in older women,
unrelated to hormone excess, with a worse outcome than Type I.
Previous studies have reported that various gene mutations, and
expression deregulation are related to endometrial carcinoma
genesis. PTEN and K-ras mutations occur in Type I endometrial
carcinomas, often with Wnt, AKT and PI3KCA deregulation. TP53 and
PPP2R1A mutations are frequent in Type II endometrial carcinomas,
with HER2/neu overexpression and p16 inactivation (3,4).
LncRNAs are a spectrum of RNAs transcripted by RNA
polymerase II, but not translated into proteins, with more than 200
nucleotides in length. LncRNAs which have previously been
identified as transcription noise are proved to be involved in the
process of multiple gene expression regulation (5,6).
Increasing evidence suggests that the deregulation of lncRNAs is
linked to disease, especially carcinoma genesis. UCA1 (7–10)
upregulated in bladder carcinoma, is a functional lncRNA molecule
involved in cell growth, cell cycle, cell invasion and
tumorigenesis. LncRNA AFAP1-AS1 (11) is hypomethylated and upregulated in
Barrett’s esophagus (BE), esophageal adenocarcinoma (EAC) tissues
and cell lines. Inhibition of its expression in EAC cells was able
to diminish cell growth, migration, and invasion, as well as
increase apoptosis. MALAT1 (12)
is first discovered in non-small cell lung cancer and its
over-expression associates with high metastatic potential and poor
patient prognosis in variety of cancer. A recent report showns that
the promoter hypermethylation silencing of tumor suppressor PCDH10
contributed to MALAT1 upregulation in endometrioid endometrial
cancer (EEC) (13).
The mechanism of endometrial carcinoma genesis is
complex and related with multiple gene mutation and deregulation.
The exploration of gene expression profile, especially the
potential functional lncRNA will help to gain better knowledge and
to discover new therapeutic candidates for endometrial carcinoma.
We performed a microarray analysis to study the expression of
lncRNAs and coding genes in 3 endometrial carcinomas and their
paired adjacent non-tumor tissues. Our result demonstrated that the
expression profile of lncRNAs is significantly different between
endometrial carcinomas and non-tumor endometrium. The novel
ASLNC04080 lncRNA is upregulated in endometrial carcinomas and
HEC-1-B cell line. Additionally the deregulation of coding genes
was also detected in endometrial carcinomas, and these deregulated
coding transcripts were involved in multiple biological processes,
cellular components, molecular functions and pathways which were
related to carcinogenesis and cancer progression. We constructed a
co-expression network of lncRNAs and coding gene transcripts based
on the expression level relation between lncRNAs and coding gene
transcripts. According to the co-expression network ASLNC04080’s
expression was correlated to 19 coding transcripts, showing a
potential co-regulation function of lncRNA ASLNC04080. Taken
together, these results suggest the altered expression levels of
lncRNAs may contribute to endometrial carcinomas genesis and
multiple molecular processes. In addition, ASLNC04080 could be a
functional lncRNA molecule with potential use as biomarker or
therapeutic target.
Materials and methods
Patient samples
All human endometrial carcinomas and their paired
adjacent nontumor tissues were obtained from patients of the First
Affiliated Hospital, School of Medicine of Xi’an Jiaotong
University. Tissue samples were collected with informed consent
from patients, as approved by the Hospital Ethics Committees. All
tissue samples were pathologically confirmed. Three pairs of these
patient samples were randomly selected for human lncRNA microarray
analysis.
DNA microarray
RNA quality was assessed by Nanodrop ND-1000 and RNA
integrity was assessed using standard denaturing agarose gel
electrophoresis. The RNA extraction was performed by KangChen
Bio-tech, Shanghai, China.
The Human 12×135 k Long Non-coding RNA Array was
manufactured by Roche NimbleGen. Each array represents all long
transcripts, both protein coding mRNAs and lncRNAs (long non-coding
RNAs) in the human genome. More than 23000 lncRNAs are collected
from the authoritative data sources including NCBI RefSeq, UCSC,
RNAdb, lncRNAs from literature and UCRs. The microarray analysis
was performed by KangChen Bio-tech.
RNA labeling and array hybridization
Double-strand cDNA (ds-cDNA) was synthesized from 5
μg of total RNA using an Invitrogen SuperScript ds-cDNA synthesis
kit in the presence of 100 pmol oligo dT primers. ds-cDNA was
cleaned and labeled in accordance with the Nimblegen Gene
Expression Analysis protocol (Nimblegen Systems, Inc., Madison, WI,
USA). Briefly, ds-cDNA was incubated with 4 μg RNase A at 37°C for
10 min and cleaned using phenol:chloroform:isoamyl alcohol,
followed by ice-cold absolute ethanol precipitation. The purified
cDNA was quantified using a nanodrop ND-1000. For Cy3 labeling of
cDNA, the Nimblegen One-Color DNA labeling kit was used according
to the manufacturer’s guideline detailed in the Gene Expression
Analysis protocol (Nimblegen Systems, Inc.). ds-cDNA (1 μg) was
incubated for 10 min at 98°C with 1 OD of Cy3-9mer primer. Then,
100 pmol of deoxynucleoside triphosphates and 100 units of the
Klenow fragment (New England Biolabs, Ipswich, MA, USA) were added
and the mix incubated at 37°C for 2 h. The reaction was stopped by
adding 0.1 volume of 0.5 M EDTA, and the labeled ds-cDNA was
purified by isopropanol/ethanol precipitation. Microarrays were
hybridized at 42°C for 16–20 h with 4 μg of Cy3 labelled ds-cDNA in
Nimblegen hybridization buffer/hybridization component A in a
hybridization chamber (Hybridization System - Nimblegen Systems,
Inc.). Following hybridization, washing was performed using the
Nimblegen Wash Buffer kit (Nimblegen Systems, Inc.). After being
washed in an ozone-free environment, the slides were scanned using
the Axon GenePix 4000B microarray scanner. The microarray analysis
was performed by KangChen Bio-tech.
Data analysis
Slides were scanned at 5 μm/pixel resolution using
an Axon GenePix 4000 B scanner (Molecular Devices Corp.) piloted by
GenePix Pro 6.0 software (Axon). Scanned images (TIFF format) were
then imported into NimbleScan software (version 2.5) for grid
alignment and expression data analysis. Expression data were
normalized through quantile normalization and the Robust Multichip
Average (RMA) algorithm included in the NimbleScan software. The
Probe level (*_norm_RMA.pair) files and mRNA level (*_RMA.calls)
files were generated after normalization. All mRNA level files were
imported into Agilent GeneSpring Software (version 11.0) for
further analysis. Differentially expressed lncRNAs and mRNAs were
identified through Fold Change filtering. Hierarchical clustering
was performed using the Agilent GeneSpring GX software (version
11.0). GO analysis and Pathway analysis was performed using the
standard enrichment computation method. The analysis was performed
by KangChen Bio-tech.
GO analysis
The Gene Ontology project provides a controlled
vocabulary to describe gene and gene product attributes in any
organism (http://www.geneontology.org). The
ontology covers three domains: Biological Process, Cellular
Component and Molecular Function. Fisher’s exact test is used to
find if there is more overlap between the DE list and the GO
annotation list than would be expected by chance. The p-value
denotes the significance of GO terms enrichment in the DE genes.
The lower the p-value, the more significant the GO term (p≥0.05 is
recommended).
Pathway analysis
Pathway analysis is a functional analysis mapping
genes to KEGG pathways. The p-value (EASE-score, Fisher p-value or
Hypergeometric-p-value) denotes the significance of the Pathway
correlated to the conditions. The lower the p-value, the more
significant the pathway is (the recommend p-value cut-off is
0.05).
Co-expression network
We constructed a coding-noncoding gene co-expression
network to investigate the relation between lncRNAs and their
coding genes, 6 lncRNAs up- or down-regulated in endometrial
carcinoma tissues were selected to draw the network. i) The data
were preprocessed by using the median gene expression value of all
transcripts expressed from the same coding gene, without special
treatment of the lncRNA expression value. ii) Then the data were
screened for differentially expressed lncRNAs and mRNAs and removed
from the dataset. iii) The R-value was used to calculate the
correlation coefficient of the PCC between lncRNA and coding genes
(only lncRNA-coding PCC, not including lncRNA-lncRNA or
coding-coding PCC). iv) Based on Pearson’s correlation coefficient
selecting PCC ≥0.95 as meaningful related pair v) The co-expression
network was drawn using Cytoscape. In the network, a round node
represents the coding gene, a box node represents the lncRNA, a red
node represents an upregulated lncRNA/mRNA and a green node
represents an under-regulated lncRNA/mRNA. A red solid line
indicates a positive correlation, and a blue dashed line indicates
a negative correlation.
RNA extraction and cell culture
Total RNA from tissue samples, whole blood and cells
was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA).
RNA concentration and integrity were determined by
spectrophotometry and standard RNA gel electrophoresis.
The human endometrial carcinoma (HEC-1-B); cervical
carcinoma (Siha HeLa) and ovarian cancer (3AO SKOV3) cell lines
were cultured in RPMI-1640 medium (Gibco-BRL, Gaithersburg, MD,
USA) supplemented with 10% bovine calf serum. Cultures were
maintained at 37°C in a humidified atmosphere with 5%
CO2.
Expression analysis of ASLNC04080 by
RT-PCR and qRT-PCR
Total RNA isolated from tissue samples, whole blood
and cells was reverse transcribed by using PrimeScript™ RT Reagent
kit with gDNA Eraser (Takara, Dalian, China). Takara Taq™ (Takara)
was used for 32 PCR cycles, annealing temperature was 65°C.
SYBR® Premix Ex Taq™ was used for real-time PCR,
annealing temperature was 61°C. Primer sequences were as follows:
ASLNC04080: forward primer, 5′-CGCTATGTGTGGTGCCTGGGGTG-3′ and
reverse primer, 5′-CAGCGCCTGAGTGGGTTTCGG-3′; 18S: forward primer,
5′-GCTCAGCGTGTGCCTACCCTAC-3′ and reverse primer,
5′-GTAGTAGCGACGGGCGGTGTGTA-3′.
Rapid amplification of cDNA ends (5′- and
3′-RACE)
One microgram of total RNA of whole blood was
purified further by treating with RNase-Free DNaseI (Takara), then
reverse transcribed with the SMART RACE cDNA Amplification kit
(Clontech, Mountain View, CA) according to manufacturer’s
instructions. Specific 5′- and 3′-RACE cDNA ends were amplified
with the universal primer mix provided in the kit and gene specific
primers (GSPs) with the advantage 2 PCR polymerase mix (Clontech).
The PCR products were subcloned into pGEM-T Easy vector (Promega,
Madison, WI, USA) and several recombinant clones were isolated for
sequencing. The GSP sequences: 5′RACE Gene-Specific Primers (Out
Primer 5′-AGCGCCTGAGTGGGTTTCGG-3′; Inner Primer
5′-GGGGGTCATACTCCCCGAAAGG-3′). 3′RACE Gene-Specific Primers (Out
Primer 5′-TTAATAGATTAGGCACAGGATGGGT-3′; Inner Primer
5′-CAGGATGGGTGTTTAATTCTCGGCAA-3′).
Amplification of truncated full length
cDNA sequence
Total RNA isolated from whole blood and cells was
reverse transcribed by using PrimeScript II 1st Strand cDNA
Synthesis kit (Takara). The Takara LA Taq® (Takara) was
used for 30 cycles PCR amplification, annealing temperature was
62°C. The truncated full length cDNA sequence primers: forward
primer 5′-ACCGCACCCGGCAGTAGTAC-3′, reverse primer
5′-ATTGATCACCTCTGAAGTTCAGTAGCA-3′. The PCR products were subcloned
into pGEM-T Easy vector (Promega) and several recombinant clones
were isolated for sequencing.
Small interfering RNA transfection
Three different small interfering RNAs (siRNA)
targeted ASLNC04080 (si-RNA616, si-RNA1315, si-RNA1535; (Gene
Pharma, Shanghai, China) and nonsense siRNA control were transected
to HEC-1-B cells by X-tremeGENE siRNA Transfection Reagent (Roche,
Mannheim, Germany). The sequences of the 3 siRNAs were as follows:
si-RAN616: 5′-CAGGGUUCAUUUCCAGACU-3′; si-RNA1315:
5′-GGCGUACUUAAGCAGAUGA-3′; si-RNA1535: 5′-GGAGUUGGGACAAUCUCUA-3′.
The knockdown effect of siRNA was detected by qRT-PCR.
Cell proliferation assays
HEC-1-B cells were plated 3000 cells per well onto
96-well plates, after 24 h siRNA1535 and nonsense siRNA control
were transfected. Every 24 h cell proliferation reagent CCK8
(Dojindo, Kumamoto, Japan) was added 10 μl per well and then
incubated at 37°C for 2 h. Optical density was measured at 450 nm
using a microplate reader (EnSpire, USA), and the proliferation
activity curve was drawn.
Cell apoptosis assays
HEC-1-B cells were plated at 15×104 cells
per well into 6-well plates. After 48 h of treatment with
siRNA-1535 and nonsense siRNA control, HEC-1-B cells were harvested
and stained with Annexin V and PI using Annexin V-FITC/PI apoptosis
detection kits (Beyotime, Haimen, China) and then examined by flow
cytometry (BD FACSCantoII, BD Biosciences, San Jose, CA, USA).
Cellular proteins were extracted 48 h after siRNA transfection.
Caspase-3 (Cell Signaling Technology, Danvers, MA) expression was
detected by western blotting.
Cell cycle analysis
HEC-1-B cells were plated at 15×104 cells
per well into 6-well plates. After 48 h of treatment with siRNA1535
and nonsense siRNA control, HEC-1-B cells were harvested, washed
with ice-cold phosphate-buffered saline, fixed with 70% ethanol
overnight, and pretreated with 5 mg/ml ribonuclease for 30 min at
37°C and then stained with PI (100 μg/ml). Cell cycle profile was
determined by flow cytometry (BD FACSCanto II, BD Biosciences)
analysis of DNA content of cell nuclei.
Results
ASLNC04080 is upregulated in endometrial
carcinoma and cell lines
We performed a microarray analysis of lncRNA in 3
paired endometrial carcinoma and adjacent non-tumor tissues. In the
lncRNA expression profiling data, a total of 23,837 lncRNAs
expressed in endometrial carcinoma were detected. A comparison of
lncRNA expression level between endometrial carcinoma and adjacent
non-tumor tissues identified 53 lncRNAs which were significantly
differentially expressed (fold change ≥2.0, p≤0.05) (Fig. 1A and C and Table I). ASLNC04080 was the most
upregulated lncRNA (fold change =3.38, p=0.038), and CD109474 was
the most downregulated lncRNA (fold change =4.72, p=0.034) in the
endometrial carcinoma group.
 | Table IDifferentially expressed lncRNAs with
>2-fold change in 3 paired endometrial carcinoma tissues (C) vs.
adjacent non-tumor tissues (N). |
Table I
Differentially expressed lncRNAs with
>2-fold change in 3 paired endometrial carcinoma tissues (C) vs.
adjacent non-tumor tissues (N).
| SEQ_ID | Log2 fold change
C/N | Regulation |
|---|
| ASLNC00294 | 2.5359635 | Up |
| ASLNC02239 | 2.0272071 | Up |
| ASLNC04080 | 3.3796694 | Up |
| ASLNC06245 | 2.5081081 | Up |
| ASLNC06537 | 2.5098338 | Up |
| ASLNC08884 | 2.236056 | Up |
| ASLNC09130 | 2.2931793 | Up |
| ASLNC09507 | 2.4517279 | Up |
| ASLNC10247 | 2.355146 | Up |
| ASLNC12496 | 2.2451406 | Up |
| ASLNC14462 | 2.097873 | Up |
| ASLNC14832 | 2.020886 | Up |
| ASLNC15527 | 2.5785253 | Up |
| ASLNC17047 | 2.3866124 | Up |
| ASLNC17464 | 2.1540515 | Up |
| ASLNC23065 | 2.1494098 | Up |
| ASLNC23333 | 2.0054722 | Up |
| ASLNC23414 | 2.4249542 | Up |
| ASLNC23671 | 2.4036572 | Up |
| AV732045 | 2.1128297 | Up |
| BC146594 | 2.3366451 | Up |
| BF448178 | 2.5298557 | Up |
| BF958740 | 2.079252 | Up |
| BI046482 | 2.8131595 | Up |
| BI520265 | 2.009207 | Up |
| BX642924 | 3.1237326 | Up |
| BX648912 | 2.0242465 | Up |
| DA195606 | 2.1546383 | Up |
| DB269443 | 2.336296 | Up |
| DB335254 | 2.1803222 | Up |
| DB341724 | 2.82081 | Up |
| DB349701 | 2.6501746 | Up |
| H44233 | 2.1850893 | Up |
| exon2439 | 2.012377 | Up |
| exon2817 | 2.0926008 | Up |
| exon3497 | 2.1918285 | Up |
| exon418 | 2.4487195 | Up |
| exon4652 | 2.4781675 | Up |
| exon844 | 2.45636 | Up |
| ASLNC00038 | 2.559887 | Down |
| ASLNC00087 | 2.1803436 | Down |
| ASLNC04117 | 2.09159 | Down |
| ASLNC05193 | 2.5719202 | Down |
| ASLNC05485 | 2.8765132 | Down |
| ASLNC09591 | 2.8102934 | Down |
| ASLNC10223 | 2.6993935 | Down |
| ASLNC10915 | 2.2386236 | Down |
| ASLNC14186 | 2.162685 | Down |
| ASLNC18394 | 2.2218604 | Down |
| ASLNC21513 | 2.0064554 | Down |
| BF507708 | 2.3711028 | Down |
| CD109474 | 4.7178984 | Down |
| HMlincRNA1454 | 2.032924 | Down |
The RT-PCR result showed ASLNC04080 expression
difference in 3 paired endometrial carcinoma and adjacent non-tumor
tissues which we used for microarray analysis (Fig. 2B). We evaluated the expression
level of lncRNA ASLNC04080 in 24 paired endometrial carcinoma and
adjacent non-tumor tissues as well as gynecological cancer cell
lines. In our result ASLNC04080 expression level in endometrial
carcinoma tissues was upregulated in 22 out of 24 paired samples
(Fig. 2A). The ASLNC04080
transcripts were also detected in endometrial carcinoma (HEC-1-B),
cervical carcinoma (Siha HeLa) and ovarian cancer (3AO SKOV3) cell
lines (Fig. 2C). Additionally,
according to UCSC information, ASLNC04080 was expressed in a series
of lymphocytes (Fig. 2D). We
detected the expression of ASLNC04080 in whole blood from both
endometrial carcinoma patients as well as healthy people (Fig. 2E).
Sequence structure of ASLNC04080
The 5′RACE and 3′RACE were performed to acquire
5′-end and 3′-end cDNA sequence of ASLNC04080 (Fig. 3A). According to the overlapping
region, we spliced 5′-end (744 bp) and 3′-end (1488 bp) sequence
forming the full length cDNA sequence (1867 bp) (Fig. 3B). The sequence message was
submitted to NCBI, GeneBank, Accession no. KJ782215. The ASLNC04080
cDNA sequence identity is 99.5% of the part human chromosome 1
cosmid (chr1: -28905061 - -28909492) sequence, by mapping with Blat
search program from UCSC. The full length ASLNC04080 cDNA contained
6 exons mapped with the 1 p35.3 (Fig.
3C), and the identity is 99% (Query cover 39%) of the
SNHG12.
The truncated full length cDNA sequence (184-1838,
1655 bp) was acquired from human whole blood and endometrial
carcinoma cell line HEC-1-B (Fig.
3D), and identity of 99% (Query cover 88%) of the full length
ASLNC04080 cDNA (KJ782215).
Coding gene expression profile in
endometrial carcinoma
The microarray analysis also included information on
the coding gene. A total of 18,738 coding transcripts (mRNA) could
be detected in the 3 paired tissue samples. Compared the mRNA
expression level between endometrial carcinoma and adjacent
non-tumor tissues, there were 46 mRNA differentially expressed; of
those 26 were upregulated and 20 were downregulated in the
endometrial carcinoma group (fold change ≥2.0, p≤0.05) (Fig. 1B and D; and Table II).
| Table IIDifferentially expressed mRNAs with
>2-fold change in 3 paired endometrial carcinoma tissues (C) vs.
adjacent non-tumor tissues (N). |
Table II
Differentially expressed mRNAs with
>2-fold change in 3 paired endometrial carcinoma tissues (C) vs.
adjacent non-tumor tissues (N).
| Gene symbol | Log2 fold change
C/N | Regulation |
|---|
| COL1A2 | 2.1063402 | Up |
| PLA2G1B | 2.4415922 | Up |
| OR1S1 | 2.391917 | Up |
| GEMIN7 | 2.4388766 | Up |
| C10orf114 | 2.1364949 | Up |
| FIGNL2 | 3.4194834 | Up |
| IL31 | 2.0702891 | Up |
| FAM23A | 2.0272226 | Up |
| ACOX3 | 2.8613539 | Up |
| NKPD1 | 2.0944095 | Up |
| FAM148C | 2.1054876 | Up |
| AKR1C1 | 2.4084988 | Up |
| TLE2 | 2.0259166 | Up |
| CDH7 | 2.0604987 | Up |
| HOXC6 | 2.1476102 | Up |
| C9orf61 | 2.0307186 | Up |
| KIR2DL1 | 2.1328676 | Up |
| TINAG | 2.2404068 | Up |
| MGRN1 | 2.1519182 | Up |
| CCR10 | 3.6094117 | Up |
| EMID2 | 2.6075573 | Up |
| BAX | 2.5370033 | Up |
| DAND5 | 2.2088263 | Up |
| HOXC6 | 2.1748328 | Up |
| ZBTB12 | 2.3850377 | Up |
| SCN1B | 2.1365902 | Up |
| GCH1 | 2.2663758 | Down |
| H2BFWT | 2.1060047 | Down |
| DDAH1 | 2.0587702 | Down |
| LOC120376 | 2.1094232 | Down |
| CRYM | 3.8512769 | Down |
| RDH5 | 2.0616512 | Down |
| SLC4A4 | 2.5983744 | Down |
| EGR3 | 2.0082827 | Down |
| TSPAN8 | 2.9661324 | Down |
| APBA3 | 2.10935 | Down |
| XBP1 | 2.010461 | Down |
| SERPINB3 | 2.4469104 | Down |
| KRT23 | 2.4278355 | Down |
| SERTAD4 | 2.100912 | Down |
| KIAA1324 | 3.5217378 | Down |
| SLC38A5 | 2.3809822 | Down |
| PARP15 | 2.4587605 | Down |
| CACNB2 | 3.3419204 | Down |
| ZNF320 | 2.033858 | Down |
| PRSS12 | 2.3177733 | Down |
With Pathway and Gene Ontology analysis we found
that these deregulated coding genes were involved in multiple
pathways and gene ontology. Pathway analysis indicated that 12
pathways corresponded to upregulated transcripts (Table III), and 12 pathways corresponded
to downregulated transcripts (Table
IV). Ras signaling pathway corresponded to 15 transcripts from
both up- and down-regulated data (Tables III and IV).
| Table IIIUpregulated coding gene transcripts
corresponding to 12 pathways. |
Table III
Upregulated coding gene transcripts
corresponding to 12 pathways.
| Pathway ID | Definition | Fisher -
P-value | Genes |
|---|
| hsa05032 | Morphine addiction
- Homo sapiens (human) | 0.004254111 |
ADORA1//GABRB1//GABRQ//PDE3A//PRKCG |
| hsa00512 | Mucin type O-Glycan
biosynthesis - Homo sapiens (human) | 0.00528077 |
GALNT13//GALNTL6//ST3GAL2 |
| hsa04972 | Pancreatic
secretion - Homo sapiens (human) | 0.005316287 |
ATP2A1//ATP2B3//PLA2G1B//PRKCG//PRSS2 |
| hsa04974 | Protein digestion
and absorption - Homo sapiens (human) | 0.01848372 |
COL12A1//COL1A2//COL4A6//PRSS2 |
| hsa04672 | Intestinal immune
network for IgA production - Homo sapiens (human) | 0.01969566 |
CCR10//CCR9//CD86 |
| hsa00140 | Steroid hormone
biosynthesis - Homo sapiens (human) | 0.02776873 |
AKR1C1//CYP19A1//UGT2B28 |
| hsa04510 | Focal adhesion -
Homo sapiens (human) | 0.03121468 |
COL1A2//COL4A6//MYLPF//PAK4//PRKCG//VEGFC |
| hsa00592 | α-Linolenic acid
metabolism - Homo sapiens (human) | 0.03328378 | ACOX3//PLA2G1B |
| hsa04950 | Maturity onset
diabetes of the young - Homo sapiens (human) | 0.03328378 | HNF4A//NKX2-2 |
| hsa05211 | Renal cell
carcinoma - Homo sapiens (human) | 0.04036226 |
GAB1//PAK4//TCEB2 |
| hsa00514 | Other types of
O-glycan biosynthesis - Homo sapiens (human) | 0.04651509 | GLT25D2//RFNG |
| hsa04014 | Ras signaling
pathway - Homo sapiens (human) | 0.0466148 |
ANGPT4//GAB1//PAK4//PLA2G1B//PRKCG//VEGFC |
| Table IVDownregulated coding gene transcripts
corresponding to 12 pathways. |
Table IV
Downregulated coding gene transcripts
corresponding to 12 pathways.
| Pathway ID | Definition | Fisher -
P-value | Genes |
|---|
| hsa04015 | Rap1 signaling
pathway - Homo sapiens (human) | 0.002010251 |
CSF1//FGF13//GNAQ//HGF//ITGAM//ITGB1//KITLG//MLLT4//PIK3CB |
| hsa05152 | Tuberculosis -
Homo sapiens (human) | 0.00262324 |
ARHGEF12//ATP6V0A2//CYP27B1//HLA-DOA//IL10//ITGAM//NFYC//PIK3C3 |
| hsa04014 | Ras signaling
pathway - Homo sapiens (human) | 0.003095239 |
CSF1//ETS1//FGF13//HGF//KITLG//MLLT4//PIK3CB//PLA2G4A//SHC1 |
| hsa05146 | Amoebiasis -
Homo sapiens (human) | 0.003198103 |
COL5A2//GNAQ//IL10//ITGAM//PIK3CB//SERPINB3 |
| hsa00380 | Tryptophan
metabolism - Homo sapiens (human) | 0.0158507 |
ACMSD//KYNU//OGDHL |
| hsa05140 | Leishmaniasis -
Homo sapiens (human) | 0.01747982 |
HLA-DOA//IL10//ITGAM//ITGB1 |
| hsa04270 | Vascular smooth
muscle contraction - Homo sapiens (human) | 0.03014367 |
ARHGEF12//GNAQ//GUCY1A3//KCNU1//PLA2G4A |
| hsa04970 | Salivary secretion
- Homo sapiens (human) | 0.03164653 |
ATP1A2//GNAQ//GUCY1A3//STATH |
| hsa04912 | GnRH signaling
pathway - Homo sapiens (human) | 0.03392592 |
GNAQ//GNRH1//MAP3K3//PLA2G4A |
| hsa04964 | Proximal tubule
bicarbonate reclamation - Homo sapiens (human) | 0.03706998 | ATP1A2//SLC4A4 |
| hsa05150 | Staphylococcus
aureus infection - Homo sapiens (human) | 0.04175273 |
HLA-DOA//IL10//ITGAM |
| hsa04730 | Long-term
depression - Homo sapiens (human) | 0.04544242 |
GNAQ//GUCY1A3//PLA2G4A |
Gene ontology (GO) analysis was preformed to show
that the differently expressed mRNA transcripts were associated
with biological processes (BP), cellular components (CC) and
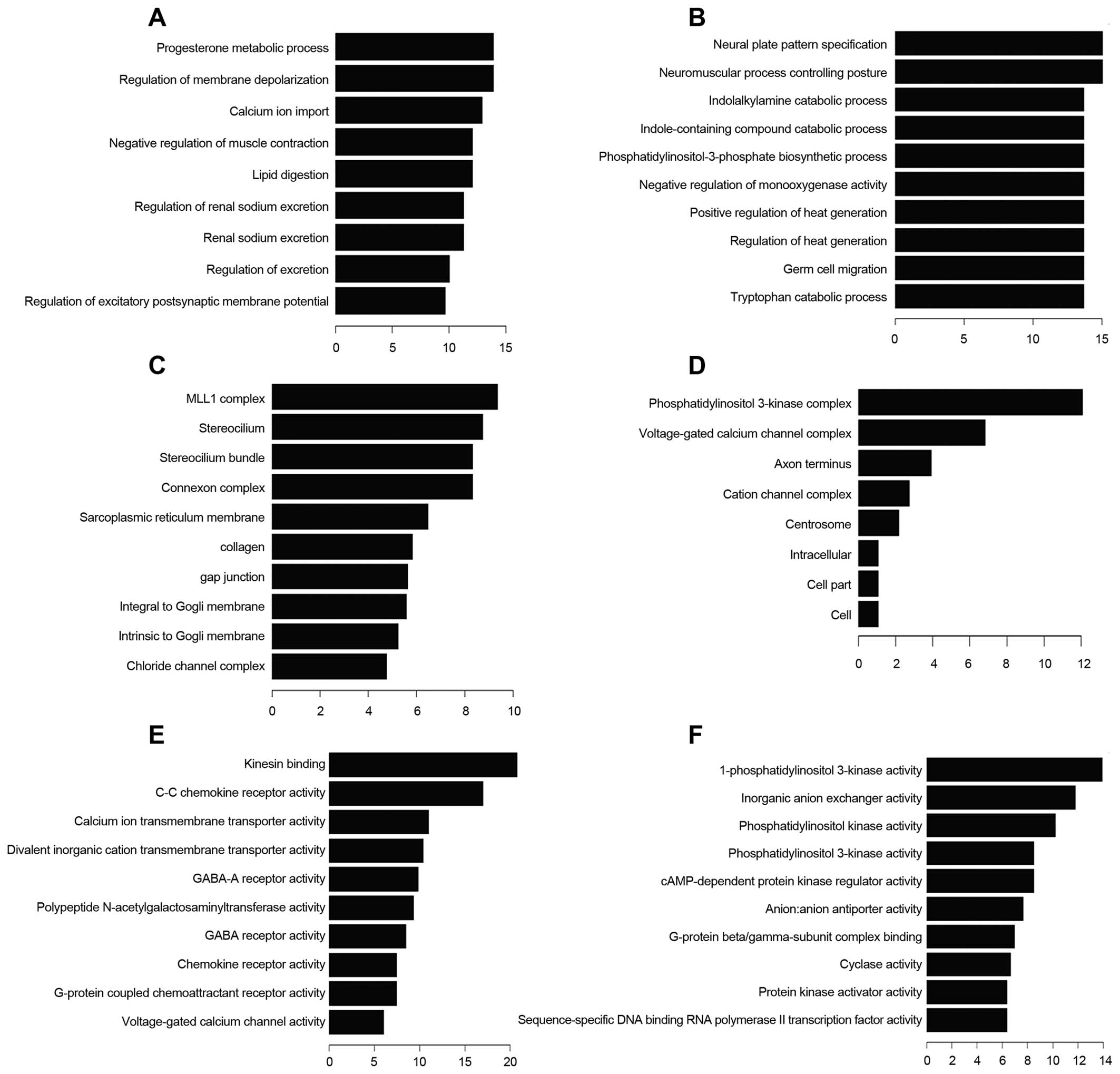
molecular function (MF) (Fig. 4).
Progesterone metabolic process is one of the most frequent fold
enrichment biological processes. There were 13 upregulated and 18
downregulated transcripts involved in biological process of wound
healing. Deregulated transcripts were involved in the molecular
function voltage-gated channel activity, including voltage-gated
ion channel activity, voltage-gated channel activity, voltage-gated
cation channel activity and voltage-gated calcium channel
activity
Expression of ASLNC04080 correlates with
the coding genes
Based on the correlation analysis between
differently expressed lncRNAs and mRNAs, we constructed a
coding-non-coding gene co-expression network. Four upregulated and
2 downregulated lncRNAs in endometrial carcinoma tissues were
selected to draw the network. The expression of 289 mRNAs was
related (Pearson’s correlation coefficients: PCC ≥0.95) to these 6
lncRNAs (Fig. 5).
ASLNC04080 expression level was correlated with 19
coding gene transcripts (Fig. 5).
Besides, these coding genes were involved in multiple pathways and
gene ontology. For example CCR10 was upregulated in endometrial
carcinoma and positively correlated with ASLNC04080 expression (PCC
>0.95), and participated in intestinal immune network for IgA
production. In addition, SYCP2 negatively correlated with
ASLNC04080 expression (PCC >0.95), involved in the pathway of
the cell cycle. These results implicated that ASLNC04080 has an
inter-regulation relation with the coding gene, and could be a
potential functional molecule in endometrial carcinoma genesis and
progression.
Inhibition of ASLNC04080 expression
influences HEC-1-B cell proliferation, apoptosis and cell
cycle
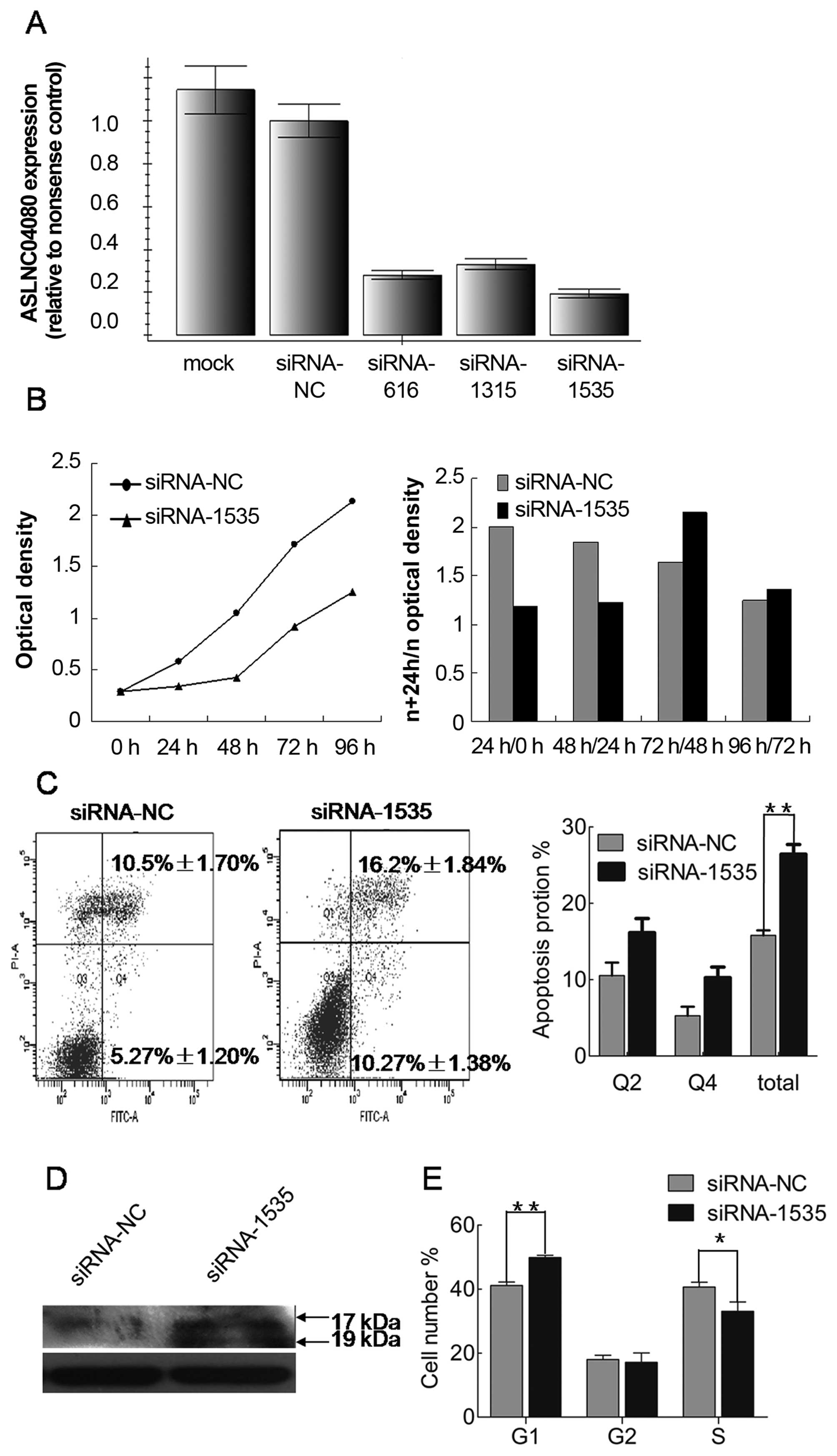
We used 3 different siRNAs to inhibit ASLNC04080
expression in human endometrial carcinoma cell line HEC-1-B. All 3
siRNAs were tested to have >50% reduction efficiency of
ASLNC04080 expression in HEC-1-B, and siRNA-1535 could reduce by
>80% the ASLNC04080 expression (Fig. 6A). Therefore, we performed several
in vitro assays to determine the functional consequences of
ASLNC04080 by inhibiting ASLNC04080 expression via siRNA-1535.
Compared with cells transfected with negative
control siRNA, HEC-1-B cell proliferation was significantly
suppressed within 48 h (Fig. 6B)
by inhibiting ASLNC04080 expression. The cell apoptosis assays were
examined by flow cytometry after HEC-1-B transfection with
ASLNC04080 and negative control siRNA. Knockdown of ASLNC04080
expression increased apoptosis in HEC-1-B cells (NC vs. siRNA-1535:
15.9±0.66% vs. 26.9±1.27%, T-test P<0.05) (Fig. 6C). Moreover, caspase-3 protein
cleavage was detected only in siRNA-1535 transfected cells
(Fig. 6D). We performed cell cycle
assays after siRNA transfection 48 h. Knockdown of ASLNC04080
expression induced G1 phase arrest (NC vs. siRNA-1535: 41.14±1.10%
vs. 49.85±0.77%, t-test P<0.05) (Fig. 6E).
These findings suggest that the ASLNC04080 lncRNA
could regulate endometrial carcinoma cell HEC-1-B proliferation,
apoptosis and cell cycle.
Discussion
Molecular alterations in endometrial carcinoma have
been studied for many years (14,15).
Microsatellite instability (MI), and gene mutations (PTEN, K-RAS
and PIK3CA) have been shown involved in type I endometrial
carcinoma. Type II endometrial carcinoma exhibits mutations of p53,
loss of heterozygosity (LOH), and molecular alterations (STK1 5,
p16, and c-erb-B2). Recently a comprehensive, multiplatform
analysis of 373 endometrial carcinomas identified new hotspot
mutations in POLE, and based on the genomic characterization they
classified endometrial cancers into four categories: POLE
ultramutated, microsatellite instability hypermutated, copy-number
low, and copy-number high (16).
The efforts on the integrated analysis of molecules not only help
us to construct new tumor classifications, but also affect
treatment recommendations for patients, provides opportunities for
genome-guided clinical trials and drug development. Besides the
known coding genes, noncoding RNAs act as a new hallmark for
endometrial carcinoma diagnosis and therapy, have attracted wide
attention. A large quantity of miRNAs target important genes in
tumor development and progression has been identified in
endometrial carcinoma (17).
Hiroki et al (18),
identified 120 miRNAs deregulated in endometrial serous carcinoma.
Moreover, their results showed that microRNA expression is
associated with clinical pathology and prognosis of patients with
endometrial serous adenocarcinoma.
LncRNA has been confirmed to regulate gene
expression in histone modification (19), regulation of transcription
(20) and splicing, and plays an
essential role in cellular proliferation, development and
metabolism (21). The deregulation
of lncRNA is associated with physiological disorders and disease
development (22). Increased
lncRNAs correlated with carcinogenesis and tumor progression have
been discovered accompanied with sequencing and microarray
technological development. A series of differentially expressed
lncRNAs (lncRNA-HEIH, lncRNA-MVIH, lncRNA-LALR1, lncRNA-LET and
lncRNA-Dreh) have been identified between HBV-related HCC and
paired peritumoral tissues by microarray (23–25).
With further studies, these lncRNAs were verified to be functional
molecules contributing to HCC tumor growth, angiogenesis,
metastasis and serving as a predictor for HCC patients’ poor
recurrence-free survival after hepatectomy. Although a systematic
study of lncRNA in endometrial carcinoma is lacking, a few
functional lncRNAs have been shown deregulated in endometrial
carcinoma. LncRNA NCT25 mutations were observed in endometrial
tumor specimens (in 23 of 48 of the samples), and could be a
mutational target specifically in endometrial cancer. MALAT1
upregulation is the result of tumor suppressor PCDH10 silencing in
endometrioid cancer (13).
Therefore, more functional lncRNA could be related to endometrial
carcinoma genesis and progression, and act as potential hallmark
for endometrial carcinoma diagnosis and therapy.
To investigate the lncRNA expression profile of
endometrial carcinoma, we performed a microarray analysis
containing lncRNA and coding gene information in 3 paired
endometrial carcinoma and adjacent non-tumor tissues. The
microarray data showed a significant difference of lncRNA
expression pattern between endometrial carcinoma and adjacent
non-tumor tissues. A total of 53 lncRNAs (C vs. N: 39 up-, 14
down-regulation, fold change >2.0, p<0.05) were deregulated
in endometrial carcinoma. ASLNC04080 is the most upregulated lncRNA
in endometrial carcinoma (fold change: 3.379, p=0.03778). The
expression level of ASLNC04080 is higher in endometrial carcinoma
tissues (22/24) compared with non-tumor tissues. ASLNC04080
expression could also be detected in endometrial carcinoma cell
line HEC-1-B as well as other gynecological cancer cell lines
(Siha, HeLa, 3AO and SKOV3). ASLNC04080 is transcribed from the
chr1: -28905061 - -28909492 loci (1 p35.3), consisting 6 exons
1,867nt in length.
According to the coding gene microarray data 46
mRNAs (C vs. N: 26 up-, 20 down-regulation, fold change >2.0,
p<0.05) exhibited a different expression between paired
endometrial carcinoma and adjacent non-tumor tissues. Previous
studies demonstrated that PTEN mutations occur early in endometrial
carcinogenesis and co-exist frequently with other deregulated
molecules targeting AKT-PI3K-mTOR pathway. Through pathway
analysis, we have found 6 up- and 9 down-regulated coding gene
transcripts in endometrial carcinoma targeting Ras signaling
pathway. Estrogen and progesterone accession and metabolism are
related to endometrial carcinoma genesis and progression (26). In our data progesterone metabolic
process is one of the most frequent fold enrichment biological
processes. Two endometrial carcinoma upregulated coding gene
transcripts CYP19A1 and AKR1C1 participated in this processes, and
correlated with endometrial carcinoma genesis (27,28).
Besides, 31 deregulated coding transcripts target the wound healing
process. Growth evidence shows that voltage-gated channel activity
alteration of carcinoma cell membrane is a characteristic of
carcinoma cells (29–31). In our results, the molecules target
voltage-gated ion channel activity, voltage-gated channel activity,
voltage-gated cation channel activity and voltage-gated calcium
channel activity, displaying deregulation in endometrial
carcinoma.
We constructed a coding-non-coding gene
co-expression network (CNC), by analyzing the correlation of
lncRNAs and the expression level of the coding gene transcripts.
The expression of six selected lncRNAs were showed to be related to
289 coding gene transcripts. The BX642924 expression was correlated
with 177 coding gene transcripts (Fig.
5), especially positively correlated to PRKCG (upregulated in
endometrial cacionoma, PCC >0.98) and negatively correlated to
KITLG (downregulated in endometrial cacionoma, PCC >0.0.95)
expression. Both PRKCG and KITLG were involved in Ras signaling
pathway, and have been showed related to tumor genesis and
progression (32,33). According to these analyses we
speculate that BX642924 may correlate with the Ras signaling
pathway. The expression of three coding gene transcripts (ZNF275,
PCDHGA8, PRSS21) was correlated with ASLNC02239 (upregulated in
endometrial carcinoma) and ASLNC10223 (downregulated in endometrial
carcinoma), showing a possible relation among these genes. More
work is needed to confirm the relations and the underlying
regulation mechanism of these coding and non-coding genes.
ASLNC04080 expression is correlated with 19 coding
gene transcripts. KLK3 is the most positively related transcript
with ASLNC04080 (PCC >0.97). Increasing evidence indicates that
KLK3 (kallikrein-related peptidase 3) is implicated in
carcinogenesis and acts as a biomarker or a diagnosis target in
multiple types of cancer (34).
Moreover, SYCP2 negatively correlates with ASLNC04080 expression
(PCC >0.95), and have been shown to participate in the cell
cycle pathway (35). Inhibition of
ASLNC04080 expression in HEC-1-B cells, resulted in decreased cell
proliferation, increased cell apoptosis and G1 phase arrest. Taking
together, these findings suggest that ASLNC04080 is a functional
lncRNA in human endometrial carcinoma and may contribute to
carcinogenesis via interaction with other coding genes. More
exploration of the function mechanism of ASLNC04080 is
required.
To our knowledge, this is the first systematic
research project of lncRNA expression profile in endometrial
carcinoma. We found several lncRNAs expressed in endometrial
carcinoma and correlated with multiple Gene Ontology and pathways
involved in carcinogenesis. These findings could help us enrich the
knowledge on the mechanism of endometrial carcinogenesis and find
new diagnostic or therapeutic targets for endometrial
carcinoma.
References
|
1
|
Siegel R, Ma JM, Zou ZH and Jemal A:
Cancer Statistics, 2014. CA Cancer J Clin. 64:9–29. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bokhman JV: Two pathogenetic types of
endometrial carcinoma. Gynecol Oncol. 15:10–17. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu FS: Molecular carcinogenesis of
endometrial cancer. Taiwan J Obstet Gynecol. 46:26–32. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Matias-Guiu X and Prat J: Molecular
pathology of endometrial carcinoma. Histopathology. 62:111–123.
2013. View Article : Google Scholar
|
|
5
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: insights into functions. Nat Rev Genet.
10:155–159. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ponting CP, Oliver PL and Reik W:
Evolution and functions of long noncoding RNAs. Cell. 136:629–641.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang F, Li X, Xie X, Zhao L and Chen W:
UCA1, a non-protein-coding RNA up-regulated in bladder carcinoma
and embryo, influencing cell growth and promoting invasion. FEBS
Lett. 582:1919–1927. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang C, Li X, Wang Y, Zhao L and Chen W:
Long non-coding RNA UCA1 regulated cell cycle distribution via CREB
through PI3-K dependent pathway in bladder carcinoma cells. Gene.
496:8–16. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Y, Chen W, Yang C, et al: Long
non-coding RNA UCA1a (CUDR) promotes proliferation and
tumorigenesis of bladder cancer. Int J Oncol. 41:276–284.
2012.PubMed/NCBI
|
|
10
|
Li Z, Li X, Wu S, Xue M and Chen W: Long
non-coding RNA UCA1 promotes glycolysis by upregulating hexokinase
2 through the mTOR-STAT3/microRNA143 pathway. Cancer Sci.
105:951–955. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu W, Bhagat TD, Yang X, et al:
Hypomethylation of noncoding DNA regions and overexpression of the
long noncoding RNA, AFAP1-AS1, in Barrett’s esophagus and
esophageal adenocarcinoma. Gastroenterology. 144:956–966. e42013.
View Article : Google Scholar
|
|
12
|
Weber DG, Johnen G, Casjens S, et al:
Evaluation of long noncoding RNA MALAT1 as a candidate blood-based
biomarker for the diagnosis of non-small cell lung cancer. BMC Res
Notes. 6:5182013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao Y, Yang Y, Trovik J, et al: A novel
wnt regulatory axis in endometrioid endometrial cancer. Cancer Res.
74:5103–5117. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bansal N, Yendluri V and Wenham RM: The
molecular biology of endometrial cancers and the implications for
pathogenesis, classification, and targeted therapies. Cancer
Control. 16:8–13. 2009.
|
|
15
|
Yeramian A, Moreno-Bueno G, Dolcet X, et
al: Endometrial carcinoma: molecular alterations involved in tumor
development and progression. Oncogene. 32:403–413. 2013. View Article : Google Scholar
|
|
16
|
Getz G, Gabriel SB, Cibulskis K, et al:
Integrated genomic characterization of endometrial carcinoma.
Nature. 497:67–73. 2013. View Article : Google Scholar
|
|
17
|
Devor EJ, Hovey AM, Goodheart MJ,
Ramachandran S and Leslie KK: microRNA expression profiling of
endometrial endometrioid adenocarcinomas and serous adenocarcinomas
reveals profiles containing shared, unique and differentiating
groups of microRNAs. Oncol Rep. 26:995–1002. 2011.PubMed/NCBI
|
|
18
|
Hiroki E, Akahira J, Suzuki F, et al:
Changes in microRNA expression levels correlate with
clinicopathological features and prognoses in endometrial serous
adenocarcinomas. Cancer Sci. 101:241–249. 2010. View Article : Google Scholar
|
|
19
|
Joh RI, Palmieri CM, Hill IT and Motamedi
M: Regulation of histone methylation by noncoding RNAs. Biochim
Biophys Acta. 1839:1385–1394. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bonasio R and Shiekhattar R: Regulation of
transcription by long noncoding RNAs. Annu Rev Genet. 48:433–455.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kornfeld JW and Brüning JC: Regulation of
metabolism by long, non-coding RNAs. Front Genet. 5:572014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schonrock N, Harvey RP and Mattick JS:
Long noncoding RNAs in cardiac development and pathophysiology.
Circ Res. 111:1349–1362. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang F, Zhang L, Huo XS, et al: Long
noncoding RNA high expression in hepatocellular carcinoma
facilitates tumor growth through enhancer of zeste homolog 2 in
humans. Hepatology. 54:1679–1689. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu D, Yang F, Yuan JH, et al: Long
noncoding RNAs associated with liver regeneration 1 accelerates
hepatocyte proliferation during liver regeneration by activating
Wnt/β-catenin signaling. Hepatology. 58:739–751. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang F, Huo XS, Yuan SX, et al: Repression
of the long noncoding RNA-LET by histone deacetylase 3 contributes
to hypoxia-mediated metastasis. Mol Cell. 49:1083–1096. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ito K, Utsunomiya H, Yaegashi N and Sasano
H: Biological roles of estrogen and progesterone in human
endometrial carcinoma - new developments in potential endocrine
therapy for endometrial cancer. Endocr J. 54:667–679. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Berstein LM, Imyanitov EN, Suspitsin EN,
et al: CYP19 gene polymorphism in endometrial cancer patients. J
Cancer Res Clin Oncol. 127:135–138. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rizner TL, Smuc T, Rupreht R, Sinkovec J
and Penning TM: AKR1C1 and AKR1C3 may determine progesterone and
estrogen ratios in endometrial cancer. Mol Cell Endocrinol.
248:126–135. 2006. View Article : Google Scholar
|
|
29
|
Roger S, Potier M, Vandier C, Besson P and
Le Guennec JY: Voltage-gated sodium channels: new targets in cancer
therapy? Curr Pharm Des. 12:3681–3695. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fraser SP, Ozerlat-Gunduz I, Brackenbury
WJ, et al: Regulation of voltage-gated sodium channel expression in
cancer: hormones, growth factors and auto-regulation. Philos Trans
R Soc Lond B Biol Sci. 369:201301052014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brackenbury WJ: Voltage-gated sodium
channels and metastatic disease. Channels. 6:352–361. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang Y, Hu X, Wang HK, et al:
Single-nucleotide polymorphisms of the PRKCG gene and osteosarcoma
susceptibility. Tumour Biol. 35:12671–12677. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kanetsky PA, Mitra N, Vardhanabhuti S, et
al: Common variation in KITLG and at 5q31.3 predisposes to
testicular germ cell cancer. Nat Genet. 41:811–815. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cicek MS, Liu X, Casey G and Witte JS:
Role of androgen metabolism genes CYP1B1, PSA/KLK3, and CYP11alpha
in prostate cancer risk and aggressiveness. Cancer Epidemiol
Biomarkers Prev. 14:2173–2177. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kouznetsova A, Novak I, Jessberger R and
Hoog C: SYCP2 and SYCP3 are required for cohesin core integrity at
diplotene but not for centromere cohesion at the first meiotic
division. J Cell Sci. 118:2271–2278. 2005. View Article : Google Scholar : PubMed/NCBI
|