Introduction
Colorectal cancer (CRC) is the second cause of
cancer deaths in Europe and the third in the United States, with
>1 million new cases estimated every year. Death occurs because
of metastasis to other organs (1).
Epithelial cells from gastrointestinal trait acquire
sequential genetic and epigenetic mutations in specific oncogene
and/or tumor suppressor genes, during colorectal adenocarcinoma
development, conferring them a selective advantage on proliferation
and self-renewal (2). Thus, normal
epithelium becomes hyperproliferative mucosa and subsequently gives
rise to a benign adenoma that progressively evolves into carcinoma
and, subsequently, generates metastasis (3–5).
Loss of genomic integrity facilitates the
accumulation of multiple mutations during the development of CRC.
Chromosomal instability (CIN), microsatellite instability (MIN),
aberrant DNA methylation and DNA repair defects are all mechanisms
involved in colorectal epithelial cell transformation (6).
At molecular level CRCs are a very heterogeneous
group of diseases showing several alterated molecular signaling
pathways, such as Wnt/APC/β-catenin, PI3K/Akt/glycogen synthase
kinase 3β (GSK3β), TGFβ/SMAD, NF-κB or mismatch repair genes (MMR).
These alterations confer individual susceptibility to cancer, and
are responsible for responsiveness or resistance to antitumor
agents (7).
Wnt/β-catenin pathway is the most frequently
dysfunctional signaling in sporadic CRC. When Wnt ligand, a
secreted glycoprotein, binds to its frizzled receptors, the
multifunctional kinase GSK3β is inactivated and β-catenin, that
acts both as E-cadherin cell-cell adhesion protein and as a
transcriptional activator, is stabilized, accumulated in the
cytoplasm and finally translocates into nucleus. Here it interacts
with members of the lymphoid enhancer factor (LEF)/T-cell factor
(TCF) and activates specific target genes. In the absence of Wnt
signal, casein kinase 1 (CK1) and the APC/Axin/GSK3β complex,
target β-catenin for ubiquitination and proteasomal degradation by
its phosphorylation (8).
GSK3β is a multifunctional serine/threonine kinase
and an important regulator of cell survival that may act as anti-
or pro-apoptotic, in a cell-specific manner. Its activity is
regulated by site-specific phosphorylation. Full activity of GSK3β
generally requires phosphorylation at tyrosine 216 (Tyr216),
whereas phosphorylation at serine 9 (Ser9) inhibits its activity.
It has been demonstrated that activation of PI3K and PKC inhibits
GSK3β activity by stimulating its Ser9 phosphorylation (9).
GSK3β also regulates NF-κB activity, a transcription
factor that plays a role in many physiological and
pathophysiological processes, including immune responses,
inflammation, cell proliferation, survival and differentiation. In
colon and pancreatic cancer cells, NF-κB is positively regulated by
GSK3β and its activation confers a selective growth advantage on
these cells, so acting as a tumor promoter. The molecular
mechanisms underlying GSK3β/NF-κB interaction remain to be further
investigated (10,11). More than 100 proteins, playing a
role in a wide spectrum of cellular processes, are substrates of
GSK3β, among which, β-catenin and NF-κB inhibitor IκB are the most
well-known. Genes upregulated by β-catenin/TCF/LEF and/or NF-κB
include proto-oncogenes, such as c-Myc and cyclin-D1,
and genes regulating cell invasion/migration, such as Snail,
CD44 and MMP-7 (12).
Recently, it has been suggested that
epithelial-to-mesenchymal transition (EMT) could be a common
biological mechanism that could represent a good target for
therapeutic intervention. EMT consists in an essential phenotypic
conversion of epithelial cells into cells with mesenchymal
phenotype. It is a reversible process that often occurs during
embryonic development and tissue remodeling and also plays a
critical role in early events occurring in invasion and metastasis
of many types of cancer, including CRC (13). EMT regulation is orchestrated by a
group of transcription factors, including Snail, Slug, ZEB1 and
Twist, but tumor microenvironment also plays a part into phenotypic
conversion through different signals, such as TGFβ, EGF, Wnt and
Notch (14,16).
During EMT epithelial cells lose their E-cadherin
expression, that specifically guarantees the epithelial phenotype,
destroy their intercellular adhesion, acquire mesenchymal
characteristics and increase migratory and invasive properties.
Furthermore, the EMT program induces stem cell-specific gene
expression, thus promoting self-renewal capability (14–16).
One of the main problems in cancer treatment is drug
resistance, responsible for relapses in many tumors and the failure
of medical treatments in metastatic disease. Probably, both
chemotherapy and radiation therapy too often miss the opportunity
to kill a part of a tumor cell subpopulation, such as CSC and
CSC-like cells.
We aimed to realize a tissue biobank from patients
affected by CRC and to establish primary cell cultures with the
main purpose of studying EMT and its reverting mechanism, the
mesenchymal-to-epithelial transition (MET) in CRC, in vitro.
We also investigated GSK3β inhibition as therapeutic target of
CRC.
Materials and methods
Patients
Blood samples, normal colorectal mucosa and CRC
tissues were obtained from patients with sporadic colon cancer
operated at the Istituto Nazionale dei Tumori in Naples (Italy) and
tissues were frozen in liquid nitrogen. Primary cell cultures were
also established from some of these CRC patients.
Samples from all subjects who participated in the
study were collected after being granted authorization from the
Comitato Etico per le Attività Biomediche ‘Carlo Romano’ of the
University of Naples Federico II, with protocol no. 120/10. Such
authorization is given only once the study has received ethics
approval, and the participants informed and written consent has
been obtained.
Cell cultures
Samples of CRC from CRC patients were washed
overnight at 4°C in PBS containing 300 U/ml penicillin, 300 μg/ml
streptomycin, and 2.5 μg/ml amphotericin B (all from Gibco-BRL,
Karlsruhe, Germany), finely minced with scissors (tissue pieces of
~30 mm3) and digested in 2 ml 0.1% collagenase II
(Boehringer Mannheim, Mannheim, Germany) in DMEM/FBS-10% for 1 h at
37°C, 5% CO2. The cell suspension was then collected by
centrifugation, washed twice with serum-free DMEM medium, and
subsequently cultured in DMEM/F12-10% FBS medium (1:1), 100 U/ml
penicillin, 100 μg/ml streptomycin, and 2.5 μg/ml amphotericin B
(all from Gibco-BRL). CRC cells were selected by differential
sedimentation, cultured on plates, and incubated with LiCl (30
mmol/l) for 1 and 24 h as well as 10 days. For in vitro
differentiation, DMEM/F12-5% FBS medium 30 mM LiCl with was used.
These primary colon cancer cells were then cultured as spheres in
serum-free stem cell medium and low-adhesion plates, as described
by Kreso and O’Brien (17), for
~60 days, disgregated six times every 10 days.
Cytogenetic analysis
Metaphase chromosome analysis was performed on cell
cultures from CRC patients, using high resolution G-banding (550
bands) according to standard procedures. Multicolor-FISH (M-FISH)
was carried out using MetaSystems’ 24XCyte color kit (MetaSystems
GmbH, Altlussheim, Germany).
FISH analysis was performed using whole chromosome
painting (WCP) probes for chromosomes 20 and 22 and locus-specific
DiGeorge probe mixture (MetaSystems GmbH) that contains a
SpectrumOrange probe located at 22q11.2 and a SpectrumGreen LSI
probe that maps at 22q13.3 region and subtelomeric probes for the p
(green) and q (red) arms of chromosomes 20.
Multicolor chromosome banding (MCB) was performed
using the multicolor banding DNA probe kit based on
micro-dissection derived region-specific libraries for chromosome
22 (MetaSystems GmbH) according to standard protocols (18).
FISH experiments were performed on metaphase spreads
and fluorescent images were analysed using a fluorescence
microscope (Axio Imager.Z1 mot; Carl Zeiss Microscopy, LLC,
Thornwood, NY, USA) with ISIS software imaging system (MetaSystems
GmbH) for image capturing and processing.
RER assay
The MSI status was confirmed with a fluorescent
multiplex system including six mononucleotide repeats (BAT-25,
BAT-26, BAT-40, NR21, NR24, and TGFβRII) and four dinucleotide
repeats (D2S123, D5S346, D17S250, and D18S58) as described earlier
(19), using the CC-MSI kit (AB
Analitica s.r.l., Padova, Italy), according to the manufacturer’s
instructions. PCR products were analysed by capillary
electrophoresis analysis using an ABI Prism 3130 Genetic Analyzer
(Applied Biosystems, Inc., Foster City, CA, USA).
RT-PCR analysis
Total RNA was extracted from colorectal tissues of
CRC patients, using QIAzol Reagent (Qiagen, Hilden, Germany), after
homogenization and cDNA was synthesized with 1 μg of total RNA, 500
ng of random hexamers, and 1 μl SuperScript III Reverse
Transcriptase (Invitrogen Life Technologies, Carlsbad, CA, USA), in
the presence of 4 μl 5X RT buffer, 1 μl DTT (0.1 M) and 1 mM dNTPs,
after DNase incubation (Invitrogen Life Technologies). The reaction
was run for 50 min at 42°C in a 20 μl reaction volume, heated to
70°C for 15 min and quickly chilled on ice. One microliter of the
cDNA was amplified by RT-PCR for CTK20 and 18 as well as E-cadherin
messengers, using primer pairs shown in Table I.
 | Table IOligonucleotide sequences. |
Table I
Oligonucleotide sequences.
|
Oligonucleotides | Primers | Accession nos. |
|---|
| Cytokeratin 18 | F:
5′-AGACTGGAGCCATTACTTC-3′ | [NM_199187.1; start
+441] |
| R:
5′-GCTCTGTCTCATACTTGACTC-3′ | [NM_199187.1; start
+563] |
| Cytokeratin 20 | F:
5′-CTGCAAATTGATAATGCTAA-3′ | [XM_005277792.1;
start +485] |
| R:
5′-GGTCATCAAAGACCTTATTC-3′ | [XM_005277792.1;
start +586] |
| B-Raf | F:
5′-TGCTTGCTCTGATAGGAAAATGAGA-3′ | [NC_018918.2; start
+140387483] |
| R:
5′-CTCAGCAGCATCTCAGGGCC-3′ | [NC_018918.2; start
+140387250] |
| E-cadherin | F:
5′-TCCTGGGCAGAGTGAATTTT-3′ | [NM_004360.3; start
+276] |
| R:
5′-CCGTAGAGGCCTTTGACTG-3′ | [NM_004360.3; start
+381] |
| Snail 1 | F:
5′-GCGAGCTGCAGGACTCTAAT-3′ | [NM_005985.3; start
+143] |
| R:
5′-TCCCAGATGAGCATTGGCAG-3′ | [NM_005985.3; start
+269] |
| Twist1 | F:
5′-CCTTCTCGGTCTGGAGGAT-3′ | [NM_000474; start
+908] |
| R:
5′-TCCTTCTCTGGAAACAATGACA-3′ | [NM_000474; start
+1004] |
| Vimentin | F:
5′-TCTGGATTCACTCCCTCTGG-3′ | [NM_003380; start
+1694] |
| R:
5′-GGTCATCGTGATGCTGAGAA-3′ | [NM_003380; start
+178 |
| GUS | F:
5′-GAAAATATGTGGTTGGAGAGCTCATT-3′ | [XR_242233.2; start
+1695] |
| R:
5′-CCGAGTGAAGATCCCCTTTTTA-3′ | [XR_242233.2; start
+1774] |
All oligonucleotides were obtained with Primer-BLAST
Software (http://www.ncbi.nlm.nih.gov/tools/primer-blast/).
Real-time PCR quantification
analysis
Real-time PCR quantification analysis was performed
for Twist1, Snail and cyclooxygenase-2 (COX2) messengers using 0.5
μl of the cDNA and primer pairs shown in Table I.
Relative expression was calculated with the
comparative Ct method and normalized against the Ct of
glucuronidase (GUS) mRNA. The quantitative real-time assays were
performed using the Bio-Rad iCycler iQ Real-Time PCR Detection
System (Bio-Rad Laboratories, Inc., Hercules, CA, USA) as
previously described (20,21), using healthy mucosa (HM) as a
calibrator to measure the relative expression.
Western blot assay
Total proteins were extracted from HM of colon and
colon tumor tissue (TT) of affected patients and from primary colon
cancer cultures, using QIAzol Reagent (Qiagen) following the
manufacturer’s instructions. Concentrations were determined by
using a protein assay kit adopting bovine serum albumin standards,
according to the manufacturer’s instructions (Bio-Rad Laboratories,
Inc.). A total of 30 μg of proteins were separated by
SDS-polyacrylamide gel electrophoresis and blots were prepared on a
Amersham Hybond-ECL nitrocellulose membrane (Amersham Pharmacia
Biotech, Inc./GE Healthcare Bio-Sciences Corp., Piscataway, NJ,
USA). The primary antibody against β-catenin, E-cadherin
(monoclonal, rabbit anti-human; no. 3195), CD44 (monoclonal, mouse
anti-human; no. 5640), Snail (monoclonal, rabbit anti-human; no.
3879) and vimentin (monoclonal, rabbit anti-human; no. 5741) was
from Cell Signaling Technology, Inc. (Beverly, MA, USA). The
antibody against actin (polyclonal, rabbit anti-human; no. sc-1615)
was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The
membrane was probed with a secondary antibody against
peroxidase-conjugated rabbit or goat immunoglobulin G, and
immunoreactive bands were detected using the enhanced
chemiluminescence Immobilon Western HRP Substrate (Millipore,
Billerica, MA, USA).
Immunofluorescence
The primary cancer cells were seeded and grown on
glass coverslips 24 h prior to the experiments. After fixation with
4% paraformaldehyde (PFA)-PBS for 10 min, cells were permeabilized
in 0.1% Triton X-100 in PBS and sequentially blocked with 10%
bovine serum albumin for 45 min. Following the overnight incubation
with primary antibody specific for cytokeratin (pan) (no. 18-0059,
1:100 dilution; Zymed Laboratories, Inc., San Francisco, CA, USA),
E-cadherin (no. 3195, 1:100 dilution), Nanog (polyclonal, rabbit
anti-human, no. 3580, 1:100 dilution), Sox2 (monoclonal, mouse
anti-human, no. 4900, 1:100 dilution), Oct4 (monoclonal, rabbit
anti-human, no. 2840, 1:100 dilution) and Snail (no. 3879, 1:100
dilution), and/or 2 h incubation with primary antibody specific for
vimentin (no. 5741, 1:200 dilution) and CD44 (no. 5640, 1:800
dilution) (all from Cell Signaling Technology, Inc.). Cells were
further incubated with appropriate secondary antibodies and DAPI
for nuclear labeling. The negative controls without primary
antibodies were also included, while no obvious staining was
observed. Immunofluorescence was visualized under a fluorescence
microscope and image-captured.
Results
Stabilisation of primary colon cancer
cell cultures and tumor status validation
We realized a tissue biobank by sampling pairs of
healthy colorectal mucosa and its matched CRC tissues from patients
with sporadic and hereditary CRC, frozen in liquid nitrogen. From
the same CRC tissues we established primary CRC cell cultures. For
molecular investigation, we chose two different cultures: one
mesenchymal-like (T88) (Fig. 1A)
and the other more epithelial-like (T93) (Fig. 1B). According to TNM staging,
classification of these two tumors was T3N1 and T4N2, for tumor 88
and 93, respectively. First of all, we validated the cancer status
of our cultures using molecular biology and cytogenetic
tecniques.
Karyotyping was performed on both cell cultures and
cytogenetic analyses were interpreted at a resolution level of 550
bands. Only the T93 cells showed a female karyotype with a
reciprocal translocation between the q arm of chromosome 20 and the
q arm of chromosome 22, but no numerical aberrations were found
(Fig. 1C). The high resolution MCB
image of the normal chromosome 22 compared to the rearranged
chromosome 22 shows that the 22q13.2qter region of chromosome 22
was translocated to chromosome 20, while the region of chromosome
20 translocated includes the region 20q13 to 20qter (Fig. 1E). FISH with subtelomeric probes
for the p (green) and q (red) arms of chromosomes 20 shows that
region 20q13 was translocated into chromosome 22 (Fig. 1D).
The complete chromosomal characterisation according
to ISCN 2013 (22) was as follows:
46,XX,t(20;22)(q13;q13)(20pter→20q13.2::22q13.2→22qter:22pter→22q13.1::20q13.3→20qter).
MSI analysis was performed on DNA extracted from TT
and pheripheral blood sample of patient no. 88, resulted negative
for CIN phenotype, who was found to have an MSI-high (MSI-H)
status, with instability at mononucleotide markers NR21, BAT-40 and
NR24 (Fig. 1F and G).
Cell cultures express mesenchymal and
epithelial markers, EMT transcription factors and stemness
markers
We have characterized T88 and T93 primary colon
cancer cell cultures for epithelial and mesenchymal markers, by
using RT-PCR, real-time PCR, western blot and immunofluorescent
analysis.
As shown in Fig. 2A and
B, Twist1, Snail and COX2 messengers were mainly upregulated in
both cell cultures when investigated by real-time RT-PCR. GUS mRNA
was used as calibrator in all real-time quantification assays and
each HM of colon was adopted as target sample for relative
quantification.
Furthermore, E-cadherin protein expression was
downregulated in T88 and upregulated in T93 TTs, while completely
absent in cell cultures. On the other hand, vimentin and Snail were
upregulated in TTs, compared to normal colon mucosa of each patient
but highly expressed in cell cultures, when analyzed by western
blot assay. CD44, instead, was upregulated in both cell cultures,
furthermore it was upregulated in TT from patient no. 88 but it
gave no hybridization signal in tissues from patient no. 93, in our
experimental conditions (Fig. 2C and
D).
Moreover, HM of colon, colon cancer tissues and cell
cultures expressed cytokeratin 18, whereas only healthy and cancer
mucosa expressed cytokeratin 20, when analyzed by RT-PCR (Fig. 2E and F).
To clarify if all the cells of these cultures
expressed mesenchymal markers, EMT-TF and epithelial markers or,
alternatively, were a heterogeneous cell population, we performed
an immunofluorescence assay using CD44, Snail, vimentin,
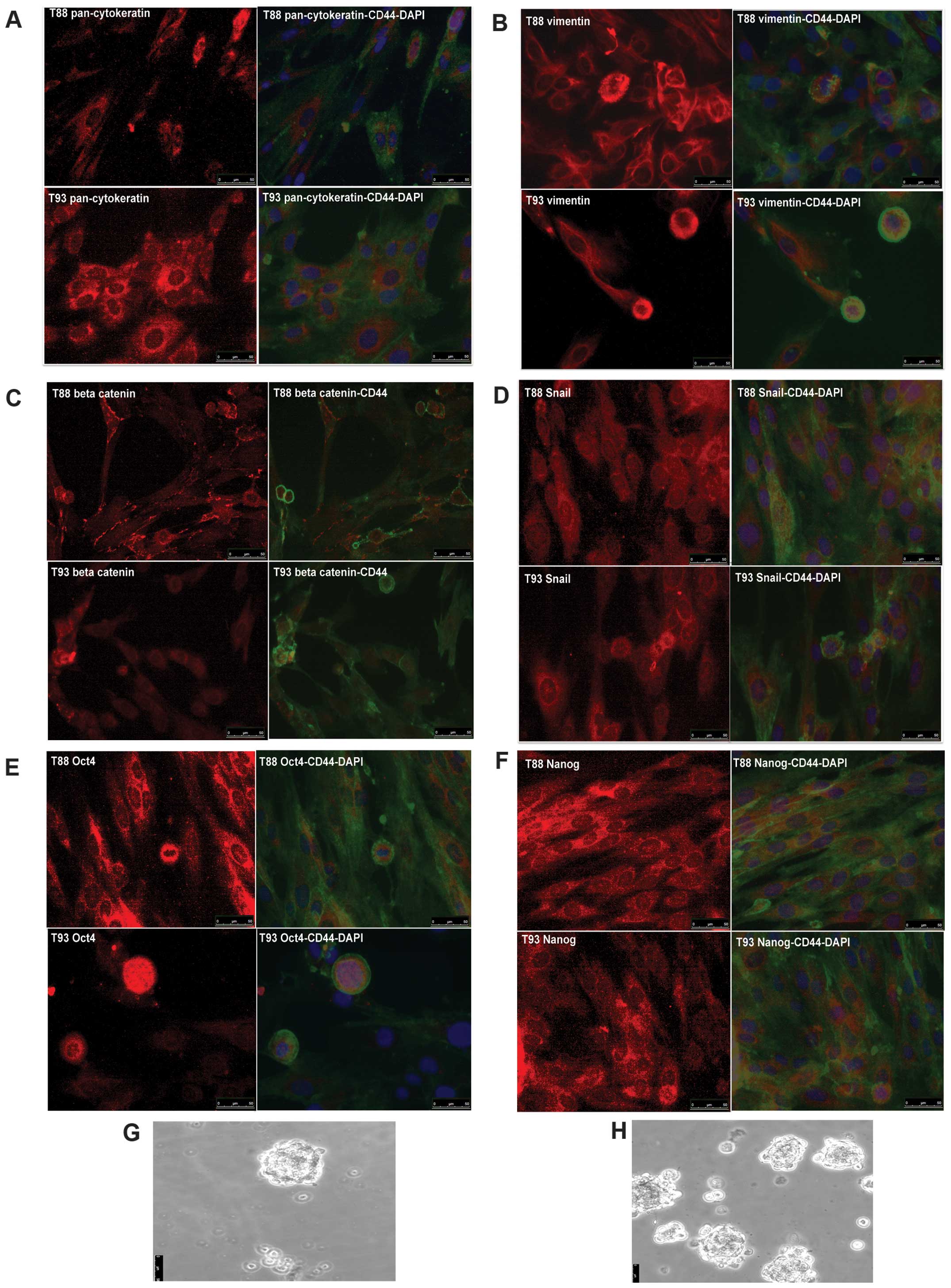
pan-cytokeratin, Oct4, Nanog and Sox2 antibody. As shown in
Fig. 3, all the cells were
positive for all the analyzed proteins except for Sox2 protein,
whose antibody did not give any signal in our experimental
condition (data not shown). CD44 was mainly localized at membrane
level (Fig. 3A–F), while
pan-cytokeratine and vimentin were localized at the cytoskeleton
level (Fig. 3A and B) and, as
expected, β-catenin was localized at level of membrane, cytoplasm
and nucleus (Fig. 3C). Snail, Oct4
and Nanog showed a turnover between nucleus and cytoplasm (Fig. 3D–F). Furthermore, Oct4 and Nanog
were mostly expressed at the level of cancer cell spheres and
clones (data not shown). They were also able to grow as a sphere in
a conditioned medium without serum and in low adhesion condition
(Fig. 3G and H)
LiCl incubation of primary colon cancer
cell culture induces MET and differentiation in vitro
Following incubation with the GSK3β inhibitor LiCl,
cells differentiated, as shown in Fig.
4A, B, E and F and β-catenin, like CD44, become mainly
localized at membrane level, as shown by immunofluorescence assay
(Fig. 4C, D, G and H). Twist1,
Snail, COX2 and CD44 expression was downregulated (Fig. 5A, B, E and F) and cells, originally
negative for E-cadherin, started to express this epithelial marker
both at RNA (Fig. 5C and D) and
protein level (Fig. 5E and F),
suggesting a mesenchymal-to-epithelial reverting transition
process. The T88-untreated cells were more mesenchymal-like than
the T93 cells, and did not express E-cadherin. On the other hand,
the T93-untreated cells, that showed more epithelial-like
morphology, had low level of E-cadherin transcription (Fig. 5C and D).
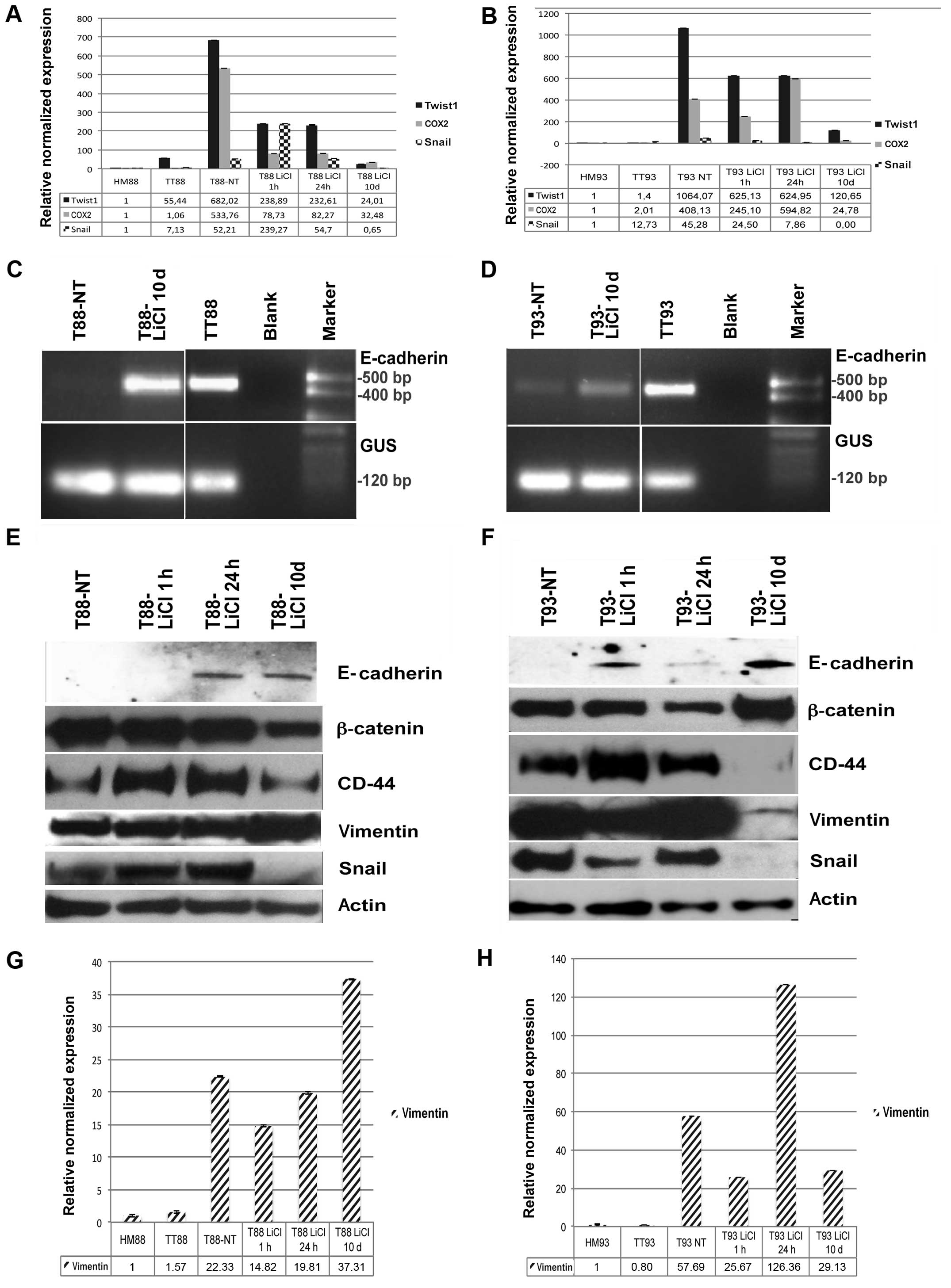 | Figure 5LiCl incubation of primary cancer
cell cultures induces mesenchymal-to-epithelial transition (MET)
in vitro. (A and B) Real-time RT-PCR analysis of Twist1,
Snail and cyclooxygenase-2 (COX2) performed on healthy mucosa (HM),
tumor tissue (TT), untreated tumor cell cultures (T), and tumor
cells after 1 and 24 h as well as 10 days of LiCl incubation from
patient (A) no. 88 and (B) no. 93. (C and D) RT-PCR analysis of
E-cadherin performed on cDNA from HM, TT and untreated T from
patient (C) no. 88 and (D) no. 93. (E and F) Western blot assay of
β-catenin, E-cadherin, CD44, vimentin, Snail and actin performed on
proteins extracted from untreated T, and tumor cells after 1 and 24
h as well as 10 days of LiCl incubation of (E) T88 and (F) T93
cells. (G and H) Real-time RT-PCR analysis of vimentin performed on
HM, TT, untreated T, and tumor cells after 1 and 24 h as well as 10
days of LiCl incubation from patient (G) no. 88 and (H) no. 93. |
When analyzed by western blot assay, Snail and CD44
gave no signal after 10 days of LiCl incubation in T93 cells, while
they strongly decreased in T88 cells (Fig. 5E and F). Interestingly, β-catenin
and vimentin showed the opposite trend in response to GSK3β
inhibition (Fig. 5E and F). After
10 days of LiCl incubation, expression of vimentin mRNA and protein
strongly decreased in T93 cells (Fig.
5F and H) while they increased in T88 cells (Fig. 5E and G). On the other hand, in the
same conditions, β-catenin was upregulated in T93 cells and
downregulated in T88 cells (Fig. 5E
and F).
Discussion
We have set up a protocol to isolate and establish
primary CRC cell cultures highly enriched in mesenchymal cancer
cells derived by an EMT process from epithelial cells. To our
knowledge, this is the first primary CRC cell culture isolated from
CRC patients expressing mesenchymal and epithelial biomarkers
together with high level of EMT transcription factors.
The T88 and T93 cultures were further analysed. T88
cells appeared more mesenchymal-like than T93 cells, that instead
showed a more epithelial-like morphology. The cancer nature of
these cells was confirmed and validated by MSI and kariotype
analysis.
MSI, CpG island methylator phenotype (CIMP) and CIN
play a significant role in CRC (6).
CIN occurs in ~60% of CRC and concerns different
cellular phenomena chacterised by the presence of abnormal
chromosome number or complement (23).
MSI consists in variations in the number of
repetitive units in each microsatellite. It is caused by failure of
the DNA mismatch repair system to repair errors occurring during
replication. These mistakes are usually corrected by the MMR which
include hMLH1, hMSH2 and hMSH6 (24). There are two well-established MSI
phenotypes, namely MSI-H and MSI-low (MSI-L or MSS). We found that
T88 cells showed MIN, whereas T93 cells were carriers of a
46,XX,t(20;22) (q13;q13) translocation in 95% of metaphases. Both
regions on chromosome 20 and 22, involved in this translocation,
are reported to be alterated in sporadic CRC (25–27).
We concluded that both cell cultures were highly enriched with
colon cancer cells.
Furthermore, we showed that these cells expressed
mesenchymal markers, EMT-TF and epithelial markers together,
therefore they have to be considered mesenchymal colon cancer cells
that have undergone EMT from epithelial adenocarcinoma cells. In
agreement with each morphological phenotype, T88 cells, that were
more mesenchymal-like than T93 cells, did not express E-cadherin,
not even at mRNA level and showed a higher increment in Twist1 mRNA
expression in TT compared to T93 cells, that showed more
epithelial-like morphology and had a low level of E-cadherin
transcription (Fig. 5C and D).
According to literature data (28,29),
these cells express some stem cell markers, such as Oct4 and Nanog
and are able to grow as spheres in a conditioned medium without
serum and in low adhesion condition.
We were able to induce a mesenchymal-to-epithelial
reverting transition under incubation with the GSK3β inhibitor
LiCl. LiCl is the most studied between GSK3β inhibitors and exerts
its action through two well-known mechanisms. As a direct
inhibitor, lithium is a competitive inhibitor of the
Mg2+, which results in inhibition of the activity of
this enzyme. On the other end, it also acts in an indirect manner,
causing a large increase in the phosphorylation of Ser9 of GSK3β
(30).
In our experience, after 10 days of LiCl incubation,
cells were able to transcribe E-cadherin; Twist1 and Snail mRNA was
strongly downregulated as well as CD44 and Snail protein, when
investigated by western blot assay. Interestingly, COX2 mRNA was
highly overexpressed in both cell cultures, compared to their
matched colon mucosae and it was also highly downregulated after
LiCl incubation. According to these results, literature data
indicate a strong association between COX2 expression and cancer
progression and metastasis (31).
As stated in the Introduction, GSK3β can act as
pro-apoptotic signal negatively regulating Wnt signaling, by
controlling the degradation of β-catenin. On the other hand, it
positively regulates NF-κB pathway by mediating the degradation of
IκB, a central inhibitor of NF-κB, so inducing an anti-apoptotic
responce (32).
In colon and pancreatic cancer cells, GSK3β
activates a proliferative signal by activation of NF-κB, and
inactivation of GSK3β inhibits NF-κB activity (33,34).
Interestingly, Snail, CD44,
G3BP2, and YAP1 are targets of Wnt5A, a gene
involved in invasion and metastasis of many cancers, that is
regulated by NF-κB signaling pathway (35,36).
E-cadherin and its transcriptional repressor Snail
orchestrate a functional cross-regulation between Wnt and NF-κB
pathways during EMT. Expression of Snail promotes E-cadherin
downregulation destroying epithelial organization and inducing
mesenchymal phenotype of epithelial cells, that takes place
together with upregulation of mesenchymal genes expression.
E-cadherin and other cell adhesion components directly bound both
β-catenin and NF-κB, sequestering them into adherens junctions and
therefore preventing the transcription of target genes (37,38).
As recently described (39), we suggest that GSK3β inhibition
could represent a good strategy targeting the cross-regulation
between these two pathways and may be a promising direction for
future cancer therapy that needs to be better elucidated.
Cell culture models obviously do not mimic the
complex interaction that are found in the intestinal mucosa and a
functional change in the cell cultures cannot tell us whether there
will be the same effect in vitro. Nevertheless cell cultures
are used in an attempt to make complex systems more simple by
isolating certain functions and investigate them in detail. From
this point of view, we speculate that our cell culture could
represent an interesting model to further investigate the molecular
biology of mesenchymal CRC cells, clinical relevance of EMT in
human CRC and the molecular basis of pharmacological resistance and
metastasis.
In conclusion, our preliminary data suggest that EMT
could represent a common mechanism among very different colon
cancer types and LiCl, a GSK3β inhibitor, induces MET in both cell
types despite their genotype, suggesting that LiCl and GSK3β could
represent, respectively, interesting drug and target for CRC
therapy. However, it seems to be more efficient in T93 cells,
characterized by a CIN phenotype, than in T88 cells, that showed
MIN phenotype, even if the first one was a more advanced stage
carcinoma. Obviously, larger population studies and further
investigations will be necessary to confirm our results.
Acknowledgements
This study has been supported by the agreement
2010–2012 between CEINGE and Campania Regional Authority; POR
Campania FSE 2007–2013.
Abbreviations:
|
EMT
|
epithelial-to-mesenchymal
transition
|
|
MET
|
mesenchymal-to-epithelial
transition
|
|
CRC
|
colorectal cancer
|
|
CIN
|
chromosomal instability
|
|
MIN
|
microsatellite instability
|
|
MMR
|
mismatch repair genes
|
|
HM
|
healthy mucosa
|
|
TT
|
tumor tissue
|
|
CIMP
|
CpG island methylator phenotype
|
References
|
1
|
Ewing I, Hurley JJ, Josephides E and
Millar A: The molecular genetics of colorectal cancer. Frontline
Gastroenterol. 5:26–30. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yeung AT, Patel BB, Li XM, Seeholzer SH,
Coudry RA, Cooper HS, Bellacosa A, Boman BM, Zhang T, Litwin S,
Ross EA, Conrad P, Crowell JA, Kopelovich L and Knudson A: One-hit
effects in cancer: altered proteome of morphologically normal colon
crypts in familial adenomatous polyposis. Cancer Res. 68:7579–7586.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Phelps RA, Chidester S, Dehghanizadeh S,
Phelps J, Sandoval IT, Rai K, Broadbent T, Sarkar S, Burt RW and
Jones DA: A two-step model for colon adenoma initiation and
progression caused by APC loss. Cell. 137:623–634. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sideris M and Papagrigoriadis S: Molecular
biomarkers and classification models in the evaluation of the
prognosis of colorectal cancer. Anticancer Res. 34:2061–2068.
2014.PubMed/NCBI
|
|
7
|
Fearon ER: Molecular genetics of
colorectal cancer. Annu Rev Pathol. 6:479–507. 2011. View Article : Google Scholar
|
|
8
|
Moon RT, Kohn AD, De Ferrari GV and Kaykas
A: WNT and beta-catenin signalling: diseases and therapies. Nat Rev
Genet. 5:691–701. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
McCubrey JA, Steelman LS, Bertrand FE,
Davis NM, Sokolosky M, Abrams SL, Montalto G, D’Assoro AB, Libra M,
Nicoletti F, Maestro R, Basecke J, Rakus D, Gizak A, Demidenko ZN,
Cocco L, Martelli AM and Cervello M: GSK-3 as potential target for
therapeutic intervention in cancer. Oncotarget. 5:2881–2911.
2014.PubMed/NCBI
|
|
10
|
Hoesel B and Schmid JA: The complexity of
NF-κB signaling in inflammation and cancer. Mol Cancer. 12:862013.
View Article : Google Scholar
|
|
11
|
Li H, Huang K, Liu X, Liu J, Lu X, Tao K,
Wang G and Wang J: Lithium chloride suppresses colorectal cancer
cell survival and proliferation through ROS/GSK-3β/NF-κB signaling
pathway. Oxid Med Cell Longev. 2014:2418642014. View Article : Google Scholar
|
|
12
|
Mishra R: Glycogen synthase kinase 3 beta:
can it be a target for oral cancer. Mol Cancer. 9:1442010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Loboda A, Nebozhyn MV, Watters JW, Buser
CA, Shaw PM, Huang PS, Van’t Veer L, Tollenaar RA, Jackson DB,
Agrawal D, Dai H and Yeatman TJ: EMT is the dominant program in
human colon cancer. BMC Med Genomics. 4:92011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Batlle E, Sancho E, Francí C, Domínguez D,
Monfar M, Baulida J and García De Herreros A: The transcription
factor snail is a repressor of E-cadherin gene expression in
epithelial tumor cells. Nat Cell Biol. 2:84–89. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kreso A and O’Brien CA: Colon cancer stem
cells. Curr Protoc Stem Cell Biol. Chapter 3(Unit 3):
12008.PubMed/NCBI
|
|
18
|
Liehr T, Weise A, Heller A, Starke H,
Mrasek K, Kuechler A, Weier HU and Claussen U: Multicolor
chromosome banding (MCB) with YAC/BAC-based probes and
region-specific micro-dissection DNA libraries. Cytogenet Genome
Res. 97:43–50. 2002. View Article : Google Scholar
|
|
19
|
Duraturo F, Cavallo A, Liccardo R, Cudia
B, De Rosa M, Diana G and Izzo P: Contribution of large genomic
rearrangements in Italian Lynch syndrome patients: characterization
of a novel alu-mediated deletion. Biomed Res Int. 2013:2198972013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Galatola M, Miele E, Strisciuglio C,
Paparo L, Rega D, Delrio P, Duraturo F, Martinelli M, Rossi GB,
Staiano A, Izzo P and De Rosa M: Synergistic effect of
interleukin-10-receptor variants in a case of early-onset
ulcerative colitis. World J Gastroenterol. 19:8659–8670. 2013.
View Article : Google Scholar :
|
|
21
|
Galatola M, Paparo L, Duraturo F, Turano
M, Rossi GB, Izzo P and De Rosa M: Beta catenin and cytokine
pathway dysregulation in patients with manifestations of the ‘PTEN
hamartoma tumor syndrome’. BMC Med Genet. 13:282012. View Article : Google Scholar
|
|
22
|
Shaffer LG, McGowan-Jordan J and Schmid M:
ISCN 2013: An International System for Human Cytogenetic
Nomenclature. 1st edition. S. Karger AG; Basel: pp. 1402013
|
|
23
|
Alberici P and Fodde R: The role of the
APC tumor suppressor in chromosomal instability. Genome Dyn.
1:149–170. 2006. View Article : Google Scholar
|
|
24
|
Sameer AS, Nissar S and Fatima K: Mismatch
repair pathway: molecules, functions, and role in colorectal
carcinogenesis. Eur J Cancer Prev. 23:246–257. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Therkildsen C, Jönsson G,
Dominguez-Valentin M, Nissen A, Rambech E, Halvarsson B, Bernstein
I, Borg K and Nilbert M: Gain of chromosomal region 20q and loss of
18 discriminates between Lynch syndrome and familial colorectal
cancer. Eur J Cancer. 49:1226–1235. 2013. View Article : Google Scholar
|
|
26
|
Castellví-Bel S, Castells A, Johnstone CN,
Piñol V, Pellisé M, Elizalde JI, Romo N, Rustgi AK and Piqué JM:
Evaluation of PARVG located on 22q13 as a candidate tumor
suppressor gene for colorectal and breast cancer. Cancer Genet
Cytogenet. 144:80–82. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zheng HT, Peng ZH, Zhou CZ, Li DP, Wang
ZW, Qiu GQ and He L: Detailed deletion mapping of loss of
heterozygosity on 22q13 in sporadic colorectal cancer. World J
Gastroenterol. 11:1668–1672. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li X, Pei D and Zheng H: Transitions
between epithelial and mesenchymal states during cell fate
conversions. Protein Cell. 5:580–591. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell
LL, Polyak K, Brisken C, Yang J and Weinberg RA: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jope RS: Lithium and GSK-3: one inhibitor,
two inhibitory actions, multiple outcomes. Trends Pharmacol Sci.
24:441–443. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Elzagheid A, Emaetig F, Alkikhia L,
Buhmeida A, Syrjänen K, El-Faitori O, Latto M, Collan Y and
Pyrhönen S: High cyclooxygenase-2 expression is associated with
advanced stages in colorectal cancer. Anticancer Res. 33:3137–3143.
2013.PubMed/NCBI
|
|
32
|
Beurel E and Jope RS: The paradoxical pro-
and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic
apoptosis signaling pathways. Prog Neurobiol. 79:173–189. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shakoori A, Ougolkov A, Yu ZW, Zhang B,
Modarressi MH, Billadeau DD, Mai M, Takahashi Y and Minamoto T:
Deregulated GSK3beta activity in colorectal cancer: its association
with tumor cell survival and proliferation. Biochem Biophys Res
Commun. 334:1365–1373. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shakoori A, Mai W, Miyashita K, Yasumoto
K, Takahashi Y, Ooi A, Kawakami K and Minamoto T: Inhibition of
GSK-3 beta activity attenuates proliferation of human colon cancer
cells in rodents. Cancer Sci. 98:1388–1393. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Du Q and Geller DA: Cross-regulation
between Wnt and NF-κB signaling pathways. For Immunopathol Dis
Therap. 1:155–181. 2010. View Article : Google Scholar
|
|
36
|
Katoh M and Katoh M: Transcriptional
mechanisms of WNT5A based on NF-κB, Hedgehog, TGFβ, and Notch
signaling cascades. Int J Mol Med. 23:763–769. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Conacci-Sorrell M, Zhurinsky J and
Ben-Ze’ev A: The cadherin-catenin adhesion system in signaling and
cancer. J Clin Invest. 109:987–991. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Solanas G, Porta-de-la-Riva M, Agusti C,
Casagolda D, Sánchez-Aguilera F, Larriba MJ, Pons F, Peiró S,
Escrivà M, Muñoz A, Duñach M, de Herreros AG and Baulida J:
E-cadherin controls beta-catenin and NF-kappaB transcriptional
activity in mesenchymal gene expression. J Cell Sci. 121:2224–2234.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Benoit YD, Guezguez B, Boyd AL and Bhatia
M: Molecular Pathways: Epigenetic modulation of wnt/glycogen
synthase kinase-3 signaling to target human cancer stem cells. Clin
Cancer Res. 20:5372–5378. 2014. View Article : Google Scholar : PubMed/NCBI
|