Introduction
Hypoxia is a prevalent feature of solid tumors.
Normal tissues have an oxygen partial pressure of 50–60 mmHg,
compared with 10 mmHg or less in most solid tumors (1,2). The
hypoxic environment induces adaptive changes in tumor cell
metabolism, which can distort the local microenvironment. These
changes are clinically important because hypoxia enhances
resistance to chemotherapy and radiation therapy and it is
predictive of metastasis and malignancy (1).
Members of the HIF family of transcription factors
are crucial regulators of adaptive cellular responses to hypoxia.
Overexpression of HIF-1α is a hallmark of diverse tumors and its
constitutive activation is frequently observed in aggressive tumor
phenotypes (3). HIF-1α is degraded
via a ubiquitin-mediated, proteasome-dependent pathway under
normoxic conditions (4). The
oxygen-dependent turnover of HIF-1α is regulated by prolyl HIF
hydroxylase (PHD) enzymes that hydroxylate two conserved proline
residues located in the oxygen-dependent degradation domain of
HIF-1α. During normoxia, hydroxylation of HIF-1α allows it to bind
to the von Hippel-Lindau (VHL) protein, a recognition component of
the E3 ubiquitin ligase complex. The interaction promotes the
ubiquitination of HIF-1α, which is mediated by a complex that
includes VHL, Elongin-B, Elongin-C, Cullin-2 and Rbx1, and leads to
the degradation of HIF-1α (5,6).
Pancreatic cancer has an extremely poor prognosis,
with a 5-year survival rate of <5% (7,8). The
only potential curative treatment for pancreatic cancer is surgery,
but only 10–20% of patients are candidates for surgery at the time
of presentation. Because most patients are diagnosed with malignant
pancreatic cancers of advanced metastatic stages, the therapeutic
options are very limited (9).
Moreover, the efficacy of current treatments, such as monoclonal
antibodies (10,11) or small molecule tyrosine kinase
inhibitors (12), is low. The use
of viral vectors derived from adenovirus, vaccinia virus,
herpesvirus, or H-1 parvovirus has been suggested as a promising
way to expand the current options for pancreatic cancer therapy
(13–15).
The autonomous H-1 parvovirus comprises a small,
non-enveloped icosahedral particle with a single-stranded DNA
genome of ~5 kb (16). H-1
parvovirus has received attention because of its oncotropic and
oncotoxic properties (17). Its
lytic cycle leads to tumor cell death via an apoptotic or a
lysosomal pathway (18). It was
shown that administration of H-1 virus prolongs the survival of
rats with transplanted glioma cells in the brain without having
cytotoxic effects on other tissues (19). Furthermore, clinical phase I/IIa
trials have been performed in patients with progressive primary or
recurrent glioblastoma multiforme, the most malignant type of glial
tumor (20). In pancreatic tumors,
another aggressive cancer model, IFN-γ released from immune cells
accelerates the efficacy of H-1 virus-mediated oncolysis in
pancreatic cancer cells (21). H-1
virus uses cellular SMAD4, a transcription factor that can bind to
the viral P4 promoter, for efficient replication in pancreatic
cancer cells (22).
In this study, we investigated whether H-1
parvovirus could downregulate HIF-1α, a malignant tumor marker, to
trigger apoptosis in pancreatic cancer cells. We found that H-1
parvovirus reduces HIF-1α protein levels independently of VHL and
RACK1. Furthermore, combined treatment with H-1 parvovirus and YC-1
accelerated the apoptosis of pancreatic cancer cells constitutively
expressing HIF-1α, suggesting that H-1 parvovirus could be used
with YC-1 as a therapeutic agent against aggressive pancreatic
tumors.
Materials and methods
Cell culture and virus amplification
Normal rat kidney (NRK) cells and MIA PaCa-2 cells
were cultured in DMEM supplemented with 10% FBS and 1% penicillin
and streptomycin. MIA PaCa-2 cells with stable knockdown of VHL
(Miapaca-shVHL cells) and the corresponding control cells
(Miapaca-shGFP cells) were additionally supplemented with puromycin
(1 μg/ml). H-1 parvovirus was purchased from American Type Culture
Collection (Manassas, VA, USA) and was propagated in NRK cells. The
virus was purified as described elsewhere (23) and the viral titer was determined as
TCID50/ml.
Reagents and antibodies
Cycloheximide and YC-1 were purchased from Sigma
(St. Louis, MO, USA) and MG132 was obtained from Calbiochem (San
Diego, CA, USA). For immunoblotting, anti-β-tubulin and
HRP-conjugated secondary antibodies were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA) and the anti-HIF-1α antibody
was obtained from BD Biosciences (San Jose, CA, USA). The
anti-caspase-8 antibody was purchased from Cell Signaling (Danvers,
MA, USA). Polyclonal anti-H-1 parvovirus antibodies were prepared
after immunization of rabbit 3 times with purified H-1 virus. For
construction of stable pancreatic cancer cells in which VHL protein
expression was stably suppressed, the pRS-shVHL vector was
purchased from OriGene (Rockville, MD, USA).
Western blot assay
Cells were harvested and lysed with lysis buffer
(150 mM NaCl, 1% NP-40, 50 mM Tris-HCl, pH 7.5) containing 0.1 mM
Na2VO3, 1 mM NaF and protease inhibitors
(Sigma). For immunoblotting, proteins from whole cell lysates were
resolved by 10 or 12% SDS-PAGE and then transferred to
nitrocellulose membranes. Primary antibodies were used at 1:1,000
or 1:2,000 dilutions and secondary antibodies conjugated with
horseradish peroxidase were used at a dilution of 1:2,000 in 5%
non-fat dry milk. After the final washing steps, nitrocellulose
membranes were exposed to enhanced chemiluminescence reagent and
imaged using LAS 4000-mini (Fuji, Tokyo, Japan).
RT-PCR analysis
Total RNA was extracted from cells using an RNeasy
Protect Cell Mini kit (Qiagen, Valencia, CA, USA) in accordance
with the manufacturer’s instructions. Three micrograms of total RNA
was converted to cDNA using Superscript II Reverse Transcriptase
(Invitrogen, Carlsbad, CA, USA) and PCR was performed using
specific primers described elsewhere (24). The cDNA in each sample was diluted
and PCR was run for the optimized number of cycles. β-actin mRNA
was measured as an internal standard. After amplification, the
products were subjected to electrophoresis on 1.5% agarose and
detected with ethidium bromide staining.
Statistical analysis
Data are presented as the mean ± standard error of
the mean (SEM). Student’s t-test was used for statistical analysis,
with P values <0.05 defined as statistically significant.
Results
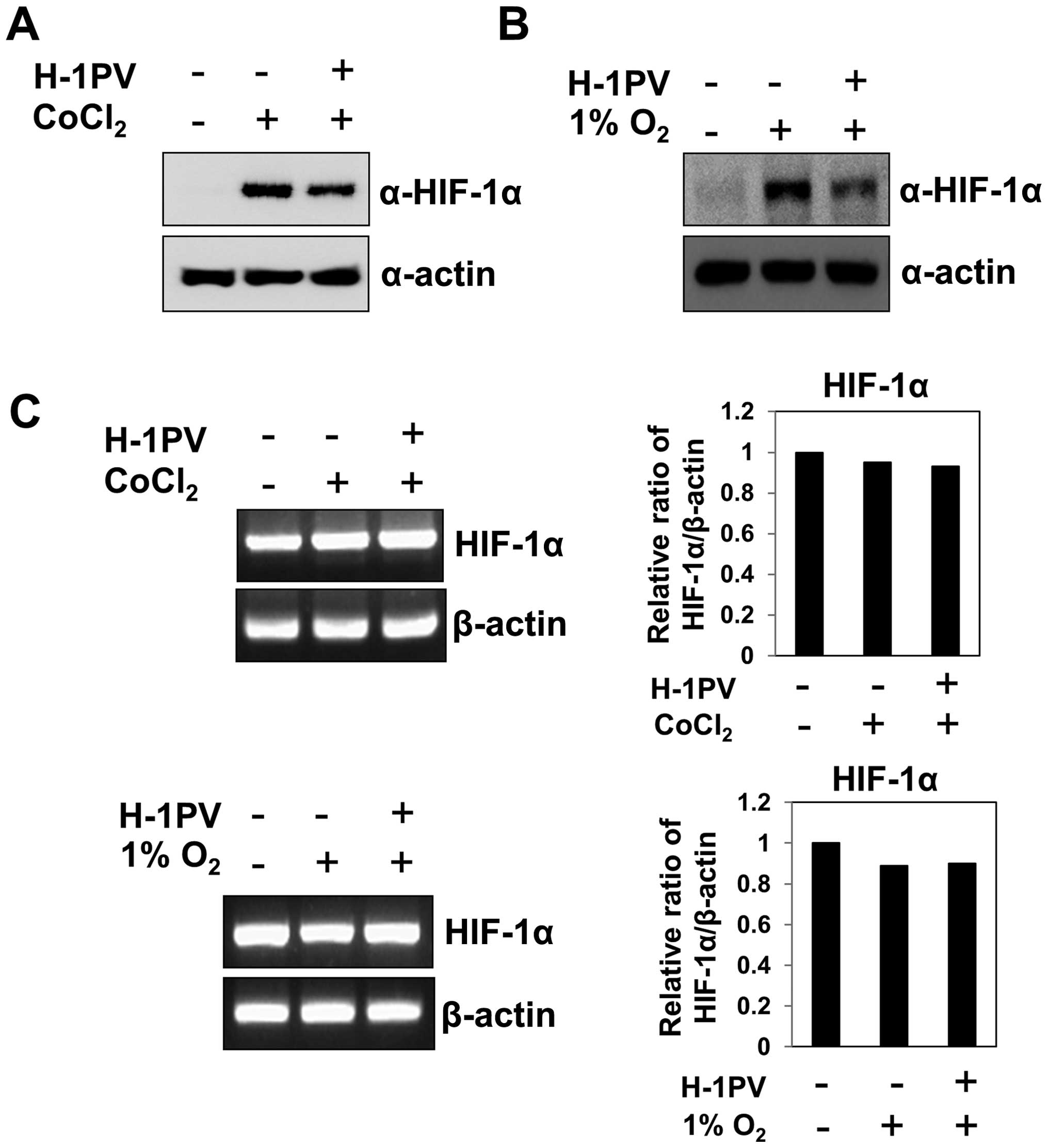
H-1 parvovirus infection induces rapid
degradation of the HIF-1α
Because many solid tumors display features of
hypoxia (1,2), we wondered how tumor cells growing
under hypoxic conditions would respond to infection with the
oncotropic H-1 virus. To test this, we grew MIA PaCa-2 pancreatic
cancer cells under CoCl2 (which mimics hypoxic
conditions), infected them with H-1 virus and measured the levels
of HIF-1α protein at 12 h post-infection. We found that H-1 virus
infection significantly reduced the levels of HIF-1α protein, which
had been increased by CoCl2 (Fig. 1A). Furthermore, when cells under
hypoxia (1% O2) were treated with H-1 virus, HIF-1α
levels were again markedly reduced at 12 h post-infection (Fig. 1B). Thus, H-1 viral infection
significantly reduces the abundance of HIF-1α protein, which is
otherwise increased by hypoxia. To test whether the H-1
virus-induced decrease in HIF-1α abundance occurred at the
transcriptional level, we analyzed HIF-1α mRNA levels in cells
infected with H-1 virus during hypoxia. We detected no significant
alteration in the abundance of HIF-1α transcript after H-1 virus
treatment under hypoxia (Fig. 1C),
even though HIF-1α protein levels changed dramatically (Fig. 1A and B). These results suggest that
H-1 virus induces a rapid decrease in HIF-1α protein abundance at
the post-transcriptional level.
The H-1 virus-induced decrease in HIF-1α
is independent of VHL but requires proteasomes
HIF-1α is degraded via its binding to VHL protein,
which functions as a recognition component of the E3 ubiquitin
ligase complex under normoxia (5,6).
Therefore, we asked whether VHL protein was involved in the H-1
virus-induced decrease in HIF-1α abundance. To answer this
question, we suppressed VHL protein levels using siRNA, which
resulted in stable expression of HIF-1α in MIA PaCa-2 cells even
under normoxic conditions. When the VHL-suppressed MIA PaCa-2 cells
were treated with H-1 virus, HIF-1α protein levels decreased at 12
h post-infection (Fig. 2A). In
addition, we used a vector encoding ubiquitin carrier protein
(UCP), an E2-EPF ubiquitin-conjugating enzyme, which promotes the
ubiquitination and degradation of VHL (25). Introduction of UCP stabilized the
expression of HIF-1α in MIA PaCa-2 cells even under normoxic
conditions. When the MIA PaCa-2 cells with enforced expression of
UCP were treated with H-1 virus, HIF-1α protein levels decreased at
12 h post-infection (Fig. 2B).
These results suggest that H-1 virus-induced HIF-1α degradation can
proceed independent of VHL E3 ubiquitin ligase activity. Next, we
investigated whether H-1 infection induced HIF-1α protein
degradation at the post-translational level without the involvement
of VHL. MIA PaCa-2 cells with VHL knockdown or ectopic expression
of UCP were treated with H-1 virus alone or H-1 virus plus MG132, a
proteasome inhibitor. MG132 blocked H-1 virus-induced HIF-1α
degradation, such that the degree of HIF-1α recovery was greater
than in MIA PaCa-2 cells with VHL knockdown or ectopic expression
of UCP alone (Fig. 2C). These
results suggest that HIF-1α degradation induced by H-1 infection
occurs via proteasomes regardless of VHL protein levels. We
wondered whether H-1 virus-induced HIF-1α degradation required a
new protein synthesis in the infected cells. To test this, MIA
PaCa-2 cells with VHL knockdown or ectopic expression of UCP were
infected with H-1 virus in the presence of cycloheximide. As shown
in Fig. 2D, the inhibition of
protein synthesis with cycloheximide further enhanced the
degradation of HIF-1α induced by H-1 virus in MIA PaCa-2 cells
under these conditions. This result implies that HIF-1α degradation
induced by H-1 virus does not require a new protein synthesis,
although the mechanism underlying the cycloheximide-induced
acceleration of H-1 virus-induced HIF-1α degradation remains
unclear.
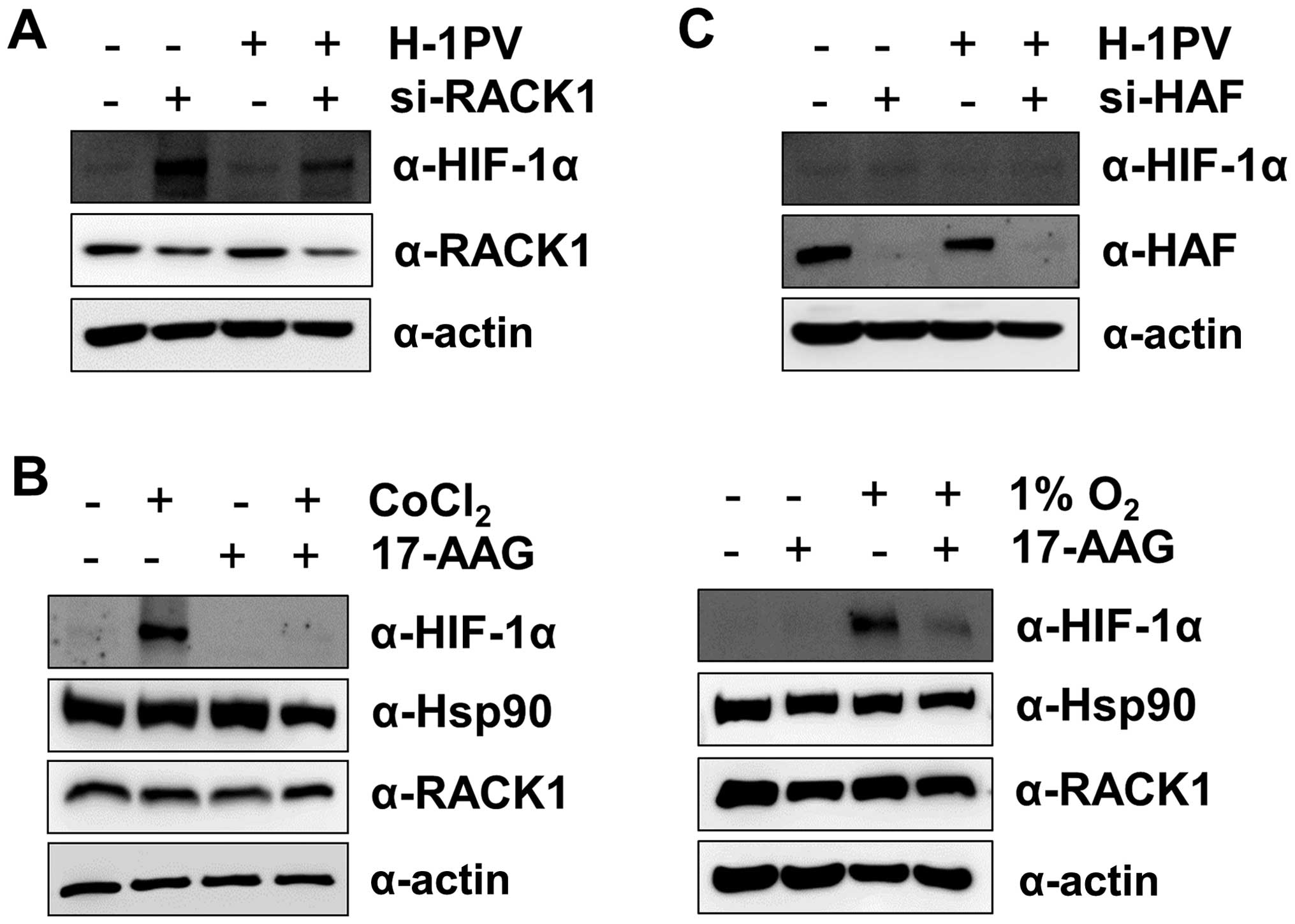
The H-1 virus-induced degradation in
HIF-1α is independent of RACK1 and HAF
Because the receptor of activated protein kinase C
(RACK)1 interacts with the PAS-A domain of HIF-1α and promotes
HIF-1α degradation by recruiting Elongin-C/B ubiquitin ligase
(26) in an
O2/PHD/VHL-independent manner, we next asked whether
RACK1 was involved in the H-1 virus-induced degradation of HIF-1α.
We suppressed RACK1 levels with siRNA, which resulted in stable
expression of HIF-1α even under normoxic conditions. When
RACK1-suppressed MIA PaCa-2 cells were treated with H-1 virus,
HIF-1α protein levels decreased at 12 h post-infection (Fig. 3A). This result indicates that H-1
virus-induced HIF-1α degradation is independent of RACK1.
Furthermore, we treated MIA PaCa-2 cells with 17-AAG, an HSP90
inhibitor, under CoCl2 or hypoxia because RACK1 competes
with HSP90 for binding to the PAS-A domain of HIF-1α (26). Treatment with 17-AAG enhanced the
degradation of HIF-1α protein (Fig.
3B), as seen in a previous study (26). In addition, because
hypoxia-associated factor (HAF) promotes HIF-1α degradation in an
O2/VHL-independent manner (27), we wondered whether HAF played a
role in the H-1 virus-induced degradation of HIF-1α. We repressed
HAF levels with siRNA, but did not observe a significant increase
in HIF-1α protein levels under normoxic conditions, in contrast to
the effects of VHL or RACK1 knockdown (Fig. 3C). Nonetheless, H-1 viral infection
reduced HIF-1α protein levels in HAF-suppressed MIA PaCa-2 cells
(Fig. 3C). This result also
implies that H-1 virus-induced HIF-1α degradation is independent of
HAF.
Constitutive expression of HIF-1α limits
H-1 virus-induced apoptosis
To investigate the biological consequence of the
constitutive expression of HIF-1α in cells infected with H-1 virus,
we first established MIA PaCa-2 cells in which VHL was stably
suppressed (Miapaca-shVHL cells) and prepared corresponding control
(Miapaca-shGFP) cells. We then evaluated H-1 virus-mediated
cytotoxicity in Miapaca-shVHL cells and Miapaca-shGFP cells.
Interestingly, Miapaca-shGFP cells were more sensitive than
Miapaca-shVHL cells to H-1 virus-induced cell death (Fig. 4A). When live cells lines were
counted at 48 h post-infection using the trypan blue exclusion
assay, the number of live Miapaca-shVHL cells was ~2-fold higher
than the number of live Miapaca-shGFP cells (Fig. 4B). Next, we investigated what
caused the greater sensitivity of Miapaca-shGFP cells to cell death
during H-1 virus infection. We examined H-1 capsid protein levels
in whole cell lysates from H-1 virus-infected Miapaca-shVHL and
Miapaca-shGFP cells. We found that the levels of H-1 capsid protein
VP2 were higher in Miapaca-shGFP cells than in Miapaca-shVHL cells
(Fig. 4C). The result suggests
that greater viral replication is associated with the sensitization
of Miapaca-shGFP cells to cell death. Consistent with this result,
Miapaca-shGFP cells exhibited higher levels of cleaved PARP and
active caspase-8 than did Miapaca-shVHL cells (Fig. 4C). Taken together, our results
suggest that the constitutive expression of HIF-1α in Miapaca-shVHL
cells limits replication of the H-1 virus and inhibits cellular
apoptosis to some extent, although we cannot presently rule out the
possibility that the suppression of VHL protein also modulates
apoptotic responses through other pathways.
YC-1, a drug targeting HIF-1α promotes
H-1 virus-induced apoptosis
Because the constitutive expression of HIF-1α
appears to restrict H-1 viral replication and subsequent cell death
(Fig. 4), we attempted to overcome
this hurdle using YC-1, a compound that targets HIF-1α. First, we
identified the minimal concentration of YC-1 necessary to degrade
HIF-1α in Miapaca-shVHL cells in order to avoid the cytotoxic
effects of YC-1. Although YC-1 at 50 μM efficiently reduced HIF-1α
levels (Fig. 5A), the
concentration induced apoptosis in ~70% of the cells at 12 h
post-treatment (data not shown). We therefore used YC-1 at 30 μM
during treatment with H-1 virus and confirmed that degradation of
HIF-1α was greater after co-administration YC-1 and H-1 virus than
after treatment with H-1 virus or YC-1 alone. We then examined the
efficiency of the co-administration killing pancreatic carcinoma
Miapaca-shVHL cells. As shown in Fig.
5B, YC-1 treatment alone did not induce cell death during the
treatment period. Pancreatic carcinoma Miapaca-shVHL cells showed
~30% cell death at 24 h after H-1 viral infection alone and 50%
cell death at 48 h post-infection (Fig. 5B). On the other hand, combined
treatment with YC-1 and H-1 virus induced more rapid cell death:
~60% at 24 h and 85% at 48 h post-treatment (Fig. 5B). We confirmed that Miapaca-shVHL
cell death induced by H-1 virus alone or by combined treatment
occurred through apoptosis, with activation of caspase 8 leading to
PARP cleavage (Fig. 5C). Thus,
because of its ability to overcome the inhibitory effects of
HIF-1α, co-administration of YC-1 and H-1 virus might be an
efficient strategy for the treatment of solid tumors.
 | Figure 5Combined treatment with H-1 virus and
YC-1 efficiently induces apoptotic cell death in MIA PaCa-2 cells
constitutively expressing HIF-1α. (A) Miapaca-shVHL cells were
treated with YC-1 at 1, 5, 10, 30 and 50 μM for 12 h and harvested
for immunoblotting with an anti-HIF-1α antibody. (B and C)
Miapaca-shVHL cells were treated with YC-1 (30 μM), H-1 (MOI = 10),
or YC-1 (30 μM) and H-1 virus (MOI = 10) for 48 h. The cells were
observed under a light microscope at 12, 24 and 48 h post-treatment
to assess cell viability and the viable cell number was determined
using the trypan blue exclusion assay. Open circle, YC-1; closed
rectangle, H-1; closed diamond, H-1 virus plus YC-1;
*P<0.05 for co-treatment with YC-1 and H-1 virus vs
treatment with H-1 virus alone The cell lysates were separated on
10% SDS-PAGE gels and the expression of HIF-1α, caspase 8 and PARP
was examined by immunoblotting with the corresponding
antibodies. |
Discussion
HIF-1α is activated not only by hypoxia but also by
a number of stimulants, including cytokines, LPS and certain
bacterial infections (28). In
addition, HIF-1α is upregulated upon infection with hepatitis B
virus through its interaction with hepatitis B virus X protein
(29). LMP-1, a primary
oncoprotein of Epstein-Barr virus, has been reported to induce
HIF-1α synthesis (30). Recent
studies have shown that infection with respiratory syncytial virus
(RSV) stabilizes HIF-1α protein via the release of nitric oxide
(31) and that stabilization of
HIF-1α by RSV infection does not require hypoxia (32). In contrast to studies showing that
some viruses can activate HIF-1α, we show here that HIF-1α, which
is often elevated in malignant tumors under hypoxic or stress
conditions, is degraded following oncotropic H-1 viral infection,
with down-regulation achieved at the post-transcriptional level, as
seen in reoviral infection (24).
We found that the downregulation of HIF-1α induced by H-1 viral
infection was independent of oxygen levels and VHL. Because recent
studies have reported that both RACK1 and HAF are able to trigger
HIF-1α degradation in a manner that is independent of oxygen, PHD
and VHL (26,27), we suppressed the expression of
these proteins with siRNA. We found that H-1 viral infection still
reduced HIF-1α protein levels under these conditions. However, the
mechanism underlying H-1 virus-mediated HIF-1α degradation is still
under investigation.
Many lines of evidence have shown that the
constitutive expression of HIF-1α functions as a hurdle to the
treatment of chemotherapy- or radiotherapy-resistant cancer cells.
Reovirus infection induces the degradation of HIF-1α but the
constitutive expression of HIF-1α restricts reovirus replication to
some extent (24). Other studies
have shown that the renal carcinoma 786-O cell line with
constitutive expression of HIF-1α inhibits the replication of
vesicular stomatitis virus through the upregulation of proteins
involved in the immune/defense response, such as IFN-β, OAS and
interferon-stimulated genes (ISGs) (33). Therefore, we examined IFN-β and OAS
transcripts in our study. Surprisingly, we found no significant
difference in the levels of IFN-β or OAS transcripts in
Miapaca-shVHL and Miapaca-shGFP cells (data not shown). This might
be attributable to the different backgrounds of the cell lines and
viruses.
Strategies to target HIF-1α, such as screening small
molecules from chemical libraries, have been developed in an effort
to improve the treatment of solid tumors. Several small molecule
inhibitors of HIF-1α activity have been shown to have antitumor and
anti-angiogenic activity in addition to other known roles. These
include the microtubule depolarizing agent 2-methoxyestiradiol
(34); inhibitors of the redox
protein thioredoxin-1 (35); YC-1,
an agent developed for circulatory disorders (36); and the HSP90 inhibitor geldanamycin
(37). Herein, we have described
the potential utility of YC-1. Some lines of evidence indicate that
the introduction of Mdm2, a HIF-1α binding partner, reverses
YC-1-mediated HIF-1α decrease, suggesting Mdm2 has an effect
opposite that of YC-1 in the regulation of HIF-1α (38). Recent studies have also shown that
the Akt/NF-κB signaling pathway contributes to YC-1-mediated HIF-1α
downregulation (39). Although we
have yet to describe the detailed mechanism of the synergic effect
of combined YC-1 and H-1 virus treatment in MIA PaCa-2 pancreatic
cancer cells constitutively expressing HIF-1α, we suggest that
combined treatment with these two agents offers a novel strategy
for the treatment of solid tumors constitutively expressing
HIF-1α.
Acknowledgements
This study was supported by a National Research
Foundation (NRF) grant funded by the Korean government (NRF-2012
R1A1A2038385).
References
|
1
|
Hockel M and Vaupel P: Tumor hypoxia:
definitions and current clinical, biologic, and molecular aspects.
J Natl Cancer Inst. 93:266–276. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brown JM and Wilson WR: Exploiting tumour
hypoxia in cancer treatment. Nat Rev. 4:437–447. 2004. View Article : Google Scholar
|
|
3
|
Bardos JI and Ashcroft M:
Hypoxia-inducible factor-1 and oncogenic signalling. Bioessays.
26:262–269. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jaakkola P, Mole DR, Tian YM, et al:
Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation
complex by O2-regulated prolyl hydroxylation. Science.
292:468–472. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maxwell PH, Wiesener MS, Chang GW, et al:
The tumour suppressor protein VHL targets hypoxia-inducible factors
for oxygen-dependent proteolysis. Nature. 399:271–275. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tanimoto K, Makino Y, Pereira T and
Poellinger L: Mechanism of regulation of the hypoxia-inducible
factor-1 alpha by the von Hippel-Lindau tumor suppressor protein.
EMBO J. 19:4298–4309. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bardeesy N and DePinho RA: Pancreatic
cancer biology and genetics. Nat Rev Cancer. 2:897–909. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maitra A and Hruban RH: Pancreatic cancer.
Annu Rev Pathol. 3:157–188. 2008. View Article : Google Scholar
|
|
9
|
Hawes RH, Xiong Q, Waxman I, Chang KJ,
Evans DB and Abbruzzese JL: A multispecialty approach to the
diagnosis and management of pancreatic cancer. Am J Gastroenterol.
95:17–31. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Safran H, Iannitti D, Ramanathan R, et al:
Herceptin and gemcitabine for metastatic pancreatic cancers that
overexpress HER-2/neu. Cancer Invest. 22:706–712. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xiong HQ, Rosenberg A, LoBuglio A, et al:
Cetuximab, a monoclonal antibody targeting the epidermal growth
factor receptor, in combination with gemcitabine for advanced
pancreatic cancer: a multicenter phase II trial. J Clin Oncol.
22:2610–2616. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Van Cutsem E, van de Velde H, Karasek P,
et al: Phase III trial of gemcitabine plus tipifarnib compared with
gemcitabine plus placebo in advanced pancreatic cancer. J Clin
Oncol. 22:1430–1438. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kasuya H, Takeda S, Nomoto S and Nakao A:
The potential of oncolytic virus therapy for pancreatic cancer.
Cancer Gene Ther. 12:725–736. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kuhlmann KF, Gouma DJ and Wesseling JG:
Adenoviral gene therapy for pancreatic cancer: where do we stand?
Dig Surg. 25:278–292. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cornelis JJ, Lang SI, Stroh-Dege AY,
Balboni G, Dinsart C and Rommelaere J: Cancer gene therapy through
autonomous parvovirus-mediated gene transfer. Curr Gene Ther.
4:249–261. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Paradiso PR, Williams KR and Costantino
RL: Mapping of the amino terminus of the H-1 parvovirus major
capsid protein. J Virol. 52:77–81. 1984.PubMed/NCBI
|
|
17
|
Chen AY and Qiu J: Parvovirus
infection-induced cell death and cell cycle arrest. Future Virol.
5:731–743. 2010. View Article : Google Scholar
|
|
18
|
Di Piazza M, Mader C, Geletneky K, et al:
Cytosolic activation of cathepsins mediates parvovirus H-1-induced
killing of cisplatin and TRAIL-resistant glioma cells. J Virol.
81:4186–4198. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kiprianova I, Thomas N, Ayache A, et al:
Regression of glioma in rat models by intranasal application of
parvovirus h-1. Clin Cancer Res. 17:5333–5342. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Geletneky K, Huesing J, Rommelaere J, et
al: Phase I/IIa study of intratumoral/intracerebral or
intravenous/intracerebral administration of Parvovirus H-1
(ParvOryx) in patients with progressive primary or recurrent
glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer.
12:992012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Grekova SP, Aprahamian M, Daeffler L, et
al: Interferon gamma improves the vaccination potential of
oncolytic parvovirus H-1PV for the treatment of peritoneal
carcinomatosis in pancreatic cancer. Cancer Biol Ther. 12:888–895.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dempe S, Stroh-Dege AY, Schwarz E,
Rommelaere J and Dinsart C: SMAD4: a predictive marker of PDAC cell
permissiveness for oncolytic infection with parvovirus H-1PV. Int J
Cancer. 126:2914–2927. 2010.
|
|
23
|
Halder S, Nam HJ, Govindasamy L, et al:
Production, purification, crystallization and structure
determination of H-1 Parvovirus. Acta Crystallogr Sect F Struct
Biol Cryst Commun. 68:1571–1576. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cho IR, Koh SS, Min HJ, et al:
Down-regulation of HIF-1alpha by oncolytic reovirus infection
independently of VHL and p53. Cancer Gene Ther. 17:365–372. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jung CR, Hwang KS, Yoo J, et al: E2-EPF
UCP targets pVHL for degradation and associates with tumor growth
and metastasis. Nat Med. 12:809–816. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu YV, Baek JH, Zhang H, Diez R, Cole RN
and Semenza GL: RACK1 competes with HSP90 for binding to HIF-1alpha
and is required for O(2)-independent and HSP90 inhibitor-induced
degradation of HIF-1alpha. Mol Cell. 25:207–217. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Koh MY, Darnay BG and Powis G:
Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and
ubiquitinates hypoxia-inducible factor 1alpha, leading to its
oxygen-independent degradation. Mol Cell Biol. 28:7081–7095. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim HY, Kim YH, Nam BH, et al: HIF-1alpha
expression in response to lipopolysaccaride mediates induction of
hepatic inflammatory cytokine TNFalpha. Exp Cell Res.
313:1866–1876. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yoo YG, Oh SH, Park ES, et al: Hepatitis B
virus X protein enhances transcriptional activity of
hypoxia-inducible factor-1alpha through activation of
mitogen-activated protein kinase pathway. J Biol Chem.
278:39076–39084. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wakisaka N, Kondo S, Yoshizaki T, Murono
S, Furukawa M and Pagano JS: Epstein-Barr virus latent membrane
protein 1 induces synthesis of hypoxia-inducible factor 1 alpha.
Mol Cell Biol. 24:5223–5234. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kilani MM, Mohammed KA, Nasreen N, Tepper
RS and Antony VB: RSV causes HIF-1alpha stabilization via NO
release in primary bronchial epithelial cells. Inflammation.
28:245–251. 2004. View Article : Google Scholar
|
|
32
|
Haeberle HA, Durrstein C, Rosenberger P,
et al: Oxygen-independent stabilization of hypoxia inducible factor
(HIF)-1 during RSV infection. PLoS One. 3:e33522008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hwang II, Watson IR, Der SD and Ohh M:
Loss of VHL confers hypoxia-inducible factor (HIF)-dependent
resistance to vesicular stomatitis virus: role of HIF in antiviral
response. J Virol. 80:10712–10723. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Escuin D, Kline ER and Giannakakou P: Both
microtubule-stabilizing and microtubule-destabilizing drugs inhibit
hypoxia-inducible factor-1alpha accumulation and activity by
disrupting microtubule function. Cancer Res. 65:9021–9028. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Welsh SJ, Williams RR, Birmingham A,
Newman DJ, Kirkpatrick DL and Powis G: The thioredoxin redox
inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin
inhibit hypoxia-induced factor 1alpha and vascular endothelial
growth factor formation. Mol Cancer Ther. 2:235–243.
2003.PubMed/NCBI
|
|
36
|
Yeo EJ, Chun YS, Cho YS, et al: YC-1: a
potential anticancer drug targeting hypoxia-inducible factor 1. J
Natl Cancer Inst. 95:516–525. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mabjeesh NJ, Post DE, Willard MT, et al:
Geldanamycin induces degradation of hypoxia-inducible factor 1alpha
protein via the proteosome pathway in prostate cancer cells. Cancer
Res. 62:2478–2482. 2002.PubMed/NCBI
|
|
38
|
Lau CK, Yang ZF, Lam CT, Tam KH, Poon RT
and Fan ST: Suppression of hypoxia inducible factor-1alpha
(HIF-1alpha) by YC-1 is dependent on murine double minute 2 (Mdm2).
Biochem Biophys Res Commun. 348:1443–1448. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun HL, Liu YN, Huang YT, et al: YC-1
inhibits HIF-1 expression in prostate cancer cells: contribution of
Akt/NF-kappaB signaling to HIF-1alpha accumulation during hypoxia.
Oncogene. 26:3941–3951. 2007. View Article : Google Scholar : PubMed/NCBI
|