Introduction
Biochemical alterations leading to the
trans-differentiation of immobile polarized epithelial cells to
mesenchymal cells, which are characterized by increased motility
and invasiveness, are summarized as epithelial-to-mesenchymal
transition (EMT) (1–5). Many aggressive tumors seem to undergo
EMT, and this process has been described to facilitate tumor cell
invasion, progression and metastasis (1–5).
However, EMT seems to be transient as tumor cells in distant
metastases of many cancers resemble an epithelial rather than a
mesenchymal phenotype. Thus, a process named
mesenchymal-to-epithelial transition (MET) describes the reverse
phenotypic transition of disseminated mesenchymal tumor cells after
recruitment and settlement in target organs (2,6–8).
Nevertheless, it is still controversially discussed how both EMT
and MET are involved in cancer progression and metastasis (4).
For glioblastomas, highly malignant brain tumors
with a median survival of <15 months despite multimodal therapy
(9), EMT seems to be also a
relevant process, although pathophysiological properties and
progression modalities are diverging from epithelial tumors
(10–14). As one example, the majority of
glioblastomas do not show intrinsic E-cadherin expression (15), the classical ‘E-cadherin to
N-cadherin switch’ is unlikely to correlate with mesenchymal
transition in glioblastomas (16).
Therefore, Mahabir et al proposed the term ‘glial to
mesenchymal transition (GMT)’ based on the aspect that glial cells
are developmentally derived from the neuroepithelial lineage
(11).
Essential molecules described in glioma EMT
processes are e.g., ZEB-1 (δ-crystalline enhancer binding factor-1)
and ZEB-2, which are critical regulators of TGF-β (transforming
growth factor-β) signaling and are responsible for disrupting
cell-cell contact inhibition (10,13).
In this context, TGF-β is not only one of the most potent EMT
inducers, but also released by glioma cells in large quantities
boostering glioma progression by promoting tumor-angiogenesis,
invasion, migration and inhibiting immune responses (17,18).
Additionally, TGF-β can confer chemoresistance in human
glioblastomas e.g., by induction of the neural adhesion molecule
L1CAM expression (19). Further
EMT-promoting factors are the transcription factors Snail1,
Snail2/Slug, and Twist with WNT/β-catenin, all having shown to
favor glioblastoma cell motility and invasiveness (10–13,20–22).
A decrease in the expression of desmoplakin, an obligate component
of functional desmosomes that anchors intermediate filaments of
desmosal plaques, and desmosome components, has been described in
EMT (17,23). Moreover, recently a multi-cancer
mesenchymal transition gene expression signature has been
identified (24) which contains
numerous EMT markers, additional to the above named ones also
fibronectin, vimentin and biglycan. Low levels of this signature
were shown to be associated with prolonged time until recurrence of
the glioblastomas (25,26).
Taken together, all these investigations pointed to
a pivotal role of EMT in glioma progression. In fact, Mahabir and
colleagues recently showed by qPCR, that in paired probes of
malignant gliomas of 7 donors the expression of collagen, matrix
metalloproteinase-9, smooth muscle α-actin (α-SMA), CD44,
fibronectin, and YKL-40 were elevated in the recurrent glioma
samples. Additionally, they demonstrated by immunohistochemistry
that in 22 cases of clinically recurrent gliomas the expression of
vimentin, α-SMA, and CD44 was increased in relation to primary
glioma samples (11).
Interestingly, they identified Snail1 as a master regulator of
irradiation-induced GMT resulting in enhanced migration and
invasion of glioma cells. These processes were possibly triggered
through phosphorylation of GSK-3β and ERK1/2 activation in a
TGF-β-dependent manner (11).
However, the group investigated only a small cohort of 7 matched
primary versus recurrent glioblastoma samples and took not into
account that transition patterns might be a patient’s individual
more than a general feature. Additionally, the aspect of the
reverse transition MET/MGT has not been addressed properly for
matched glioblastoma samples, nor the distribution of epithelial,
glial and mesenchymal markers within different cell types of solid
human glioblastoma tissues. Thus, we investigated the expression of
Twist1, Snail1, Snail2/Slug, desmoplakin, biglycan, β-catenin,
L1CAM, fibronectin, vimentin, and TGF-β1 with its receptors TGF-βR1
and TGF-βR2 in 17 matched probes of solid primary and recurrent
human glioblastomas by real-time RT-PCR and
double-immunofluorescence staining to identify the individual EMT
molecule-expressing cell types. Additionally, to determine the
influence of temozomolide, the main chemotherapeutic drug in
glioblastoma treatment, on EMT molecules, we stimulated glioma
cells with temozolomide and analyzed the expression profile of the
above named EMT markers.
Materials and methods
Tumor specimens
Glioblastoma samples were surgical dissected tissues
from the Department of Neurosurgery (Kiel, Germany) and were
obtained in accordance with the Helsinki Declaration of 1975 with
approval of the ethics committee of the University of Kiel, Germany
after written informed consent of donors. Tumors were classified
according to the WHO criteria, and the diagnosis was established by
a pathologist. In this study, a total number of 34 glioblastomas
(17 primary and 17 recurrent tumors, paired samples for each donor)
was included. In case of enough material available, matched probes
of individual tumor samples were used for different
investigations.
Reverse transcription and Real-time PCR
(qRT-PCR)
RNA was isolated with the TRIzol reagent
(Invitrogen, Carlsbad, CA, USA), digested by DNase (Promega,
Madison, WI, USA), cDNA was synthesized using RevertAid™ H Minus
Reverse Transcriptase (Thermo Scientific, Schwerte, Germany) and
quantitative reverse transcription real-time PCR (qRT-PCR) was
performed as described before (27,28)
using TaqMan primer probes (Applied Biosystems, Foster City, CA,
USA): β-catenin (Hs-00172016_m1), biglycan (BGN) (Hs-00156076_m1),
desmoplakin (DSP) (Hs-00950591_m1), fibronectin 1 (Hs-00277509_m1),
glial acidic fibrillary protein (GFAP) (Hs-00157674_m1),
glycerinaldehyde-3-phosphate-dehydrogenase (GAPDH) (Hs99999905_m1),
ionized calcium-binding adapter molecule 1 (Iba1) (Hs-00610419_g1),
L1CAM (Hs-00240928_m1), platelet endothelial cell adhesion molecule
(Pecam) (Hs-00169777_m1), Snail1 (Hs-00195591_m1), Sail2/Slug
(Hs-00950344_m1), TGF-β (Hs-00171257_m1), TGF-βR1 (Hs-00610319_m1),
TGF-βR2 (Hs-00234253_m1), Twist1 (Hs-01675818_s1), vimentin
(Hs-00185584_m1). All 34 different primary and recurrent
glioblastoma samples as well as temozolomide treated T98G glioma
cells were analyzed by qRT-PCR. The reaction was carried out with
the MyiQ™ Single Color Real-time PCR Detection system (Bio-Rad,
München, Germany) and fluorescent data were converted into cycle
threshold (CT) measurements. ΔCT values of each sample
were calculated as CTgene of interest − CT
GAPDH. Relative gene expression was calculated with
2(normalized CT non-stimulated − normalized CT
stimulated) = n-fold of control. A ΔCT value of
3.33 corresponds to one magnitude lower gene expression compared to
GAPDH. For each gene, logarithmic linear dependence of CT-values
from the number of copies was verified by using different amounts
of cDNA.
To visualize possible similarities according to EMT
expression markers, relative gene expression data of individual
primary-recurrent glioblastoma pairs were assigned to grey shades.
A relative gene expression value of 1 (= equal expression in
primary and recurrent glioblastomas) was assigned as 30% grey,
lower n-fold expression values (lower expression in recurrent
compared to primary) were displayed with increasing lighter shading
with 0 corresponding to white. Relative expression values >1
(higher expression in recurrence compared to primary) were assigned
with increasing darker grey shades until 3-fold induction which was
assigned as maximum (black). Afterwards, a ‘heatmap-like’
arrangement of individual primary-recurrent glioblastoma pairs was
performed orientating particularly to n-fold values of GFAP,
desmoplakin, Snail1, Snail2, Twist1 and vimentin.
Cell culture
The human glioblastoma cell line T98G was obtained
from the DKFZ, Heidelberg, Germany, and cultured as reported in
Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) plus 10%
fetal calf serum (FCS; Invitrogen) as described (29,30).
T98G cells were checked for purity by immunostaining with cell
type-specific markers and for the absence of Mycoplasma
contamination by staining with bisbenzimide as described (29,30).
For stimulation experiments T98G cells were treated
with/without temozolomide (Sigma-Aldrich, Taufkirchen, Germany) at
400 μg/ml (adjusted to stock concentrations of 20 mg/ml in
dimethylsulfoxide (DMSO; Sigma-Aldrich) or equal volumes DMSO alone
(solvent control) for 24 and 48 h in DMEM plus 0.5% FCS,
respectively, and afterwards RNA isolation and qPCR were performed
as described above.
Immunofluorescence
Cryostat sections of different primary and recurrent
glioblastoma tissues were fixed in acetone/methanol (1:1; 10 min)
at −20°C, washed with Tris-buffered saline plus 0.1% Tween-20 (3X
TBS-T, room temperature), incubated in 20%, then 70% ethanol (each
2 min), blocked with Sudan black (1% in 70% ethanol) for 10 min,
rinsed with 70% ethanol until dye free, then for 2 min with 20%
ethanol, washed with TBS-T (3X), blocked with 0.1% bovine serum
albumin (PAA Laboratories GmbH, Cölbe, Germany) and 0.2% glycine in
TBS (1 h), then without washing incubated with primary antibodies
in TBS-T at 4°C overnight (with exception of bovine serum albumin
all reagents were purchased from Roth Karlsruhe, Germany). Primary
antibodies were omitted for negative controls. After a washing step
(3X TBS-T, 10 min) the first secondary antibody was incubated for 1
h at 37°C in the dark. The sections were washed with TBS-T (3×10
min). Since all primary antibodies were obtained from mouse, an
additional blocking step with donkey anti-mouse
F(ab)2-fragments (1:1,000 1 h, room temperature, from
Dianova, Hamburg, Germany) was performed. The second primary
antibody was applied after another washing step (3X TBS-T) and
incubated overnight at 4°C. Second primary antibodies were omitted
for negative controls. The slides were washed again (3X TBS-T) and
incubated with the second secondary antibody for 1 h at 37°C. After
washing with TBS-T (1×10 min), TBS (2×10 min), nuclei were stained
with 4′, 6-diamidino-2-phenylindole (DAPI; Invitrogen; 1:30,000, 30
min, room temperature), washed with TBS (3X) and finally distilled
water. After embedding in Immu-Mount (Shandon, Pittsburgh, PA, USA)
digital photography was performed using a Zeiss microscope and
Zeiss camera (Zeiss, Oberkochem, Germany).
In combination with anti-GFAP (1:500, MAB3402, mouse
monoclonal, Millipore, Darmstadt, Germany), anti-von Willebrand
factor protein (vWF) (1:1,000, sc-53465, mouse monoclonal, Santa
Cruz Biotechnology, CA, USA), and anti-CD11b (1:100, sc-1186, mouse
monoclonal, Santa Cruz), the specific EMT antibodies anti-Twist1
(1:500, H00007291-M03, mouse monoclonal, Abnova, Heidelberg,
Germany); β-catenin (1:150, 610153, mouse monoclonal; BD
Transduction Laboratories, Franklin Lakes, NJ, USA); DSP
(undiluted, 651109, mouse monoclonal, Progen Biotechnology GmbH,
Heidelberg, Germany), vimentin (1:50, sc-6260, mouse monoclonal,
Santa Cruz), and L1CAM [1:100, L1-11A (subclone UJ127.11);
(31) mouse monoclonal], were
always stained first with Alexa Fluor 488 coupled secondary
antibodies (green, 1:1,000, donkey anti-mouse IgG, Invitrogen), the
second secondary antibody detecting the cellular markers was donkey
anti-mouse IgG Alexa Fluor 555 (red, 1:1,000, Invitrogen).
Statistical analysis
For statistical analyses the two-tailed Student’s
t-test with dependent samples was used. Significance levels ranged
between *p<0.05, **p<0.01 and
***p<0.001.
Results
mRNA expression of EMT markers in matched
human primary and recurrent glioblastomas
To evaluate mRNA expression levels of different EMT
markers in matched probes of human primary and recurrent
glioblastomas qRT-PCR analysis was performed. Results are shown in
Fig. 1, single ΔCT
values for primary glioblastoma samples are demonstrated in white
circles and ΔCT values for recurrent ones in grey
triangles. It should be kept in mind that a ΔCT value of
3.33 corresponds to one magnitude lower gene expression. Black
filled symbols identify tumor samples which were also used for
double-immunofluorescence staining.
 | Figure 1(1) Expression of EMT markers
(Twist1, Snail1, Snail2/Slug, desmoplakin (DSP), biglycan (BGN),
β-catenin, L1CAM, fibronectin, vimentin, TGF-β1, TGF-βR1, TGF-βR2)
and cellular markers [glial fibrillary acidic protein (GFAP), Iba1
and Pecam] in 17 paired primary (prim; white circles) and recurrent
(rec; grey triangles) glioblastoma samples was evaluated by
real-time RT-PCR (logarithmic scale, ΔCT=3.33 corresponds to a
10-fold difference; black filled symbols identify samples which
were used for immunofluorescence staining); and (2) ‘heatmap-like’
analysis of n-fold expression differences [calculated with
2(normalized CT non-stimulated − normalized CT
stimulated) = n-fold of control) between 15 primary-recurrent
glioblastoma pairs [analysis orientated particularly to n-fold
values of GFAP, desmoplakin, Snail1, Snail2, Twist1 and vimentin;
equal n-fold expression in primary and recurrent pairs was assigned
as 30% grey, lower n-fold expression values with increasing lighter
shading with 0 corresponding to white, and relative n-fold
expression values >1 with increasing darker grey shades until
3-fold induction which was assigned as maximum (black)]. (1)
Twist1, Snail1, Snail2/Slug and BGN mRNA expression in recurrent
glioblastoma was significantly lower than for primary one
(*p<0.05; **p<0.01), whereas L1CAM was
found in higher amounts in recurrent samples
(*p<0.05). (2) In total five different glioblastoma
groups could be distiguished, the general de-differentiation group
[1], the mesenchymal-epithelial/glial transition group [2], the
mesenchymal-epithelial transition group [3], the
epithelial-/glial-mesenchymal transition group [4], and the
mesenchymal trend group [5]. |
Irrespective of the tumor source (primary versus
recurrent), EMT markers could be detected at considerable levels in
glioblastoma tissues. Highest mRNA expression was found for
vimentin, fibronectin and TGF-β1, respectively, and β-catenin,
TGF-βR2 and BGN showed also prominent mRNA expression levels. In
contrast, TGF-βR1, Sail2/Slug and DSP, respectively, were found at
lower expression levels, and Twist1, Snail1 and L1CAM,
respectively, were measurable at lowest levels, although a clear
detection of all these markers was possible. The normalized
averaged ΔCT values for investigated primary/recurrent
glioblastoma samples were: −0.70/−0.22 (vimentin), 2.68/2.59
(fibronectin), 3.64/3.94 (TGF-β1), 5.49/6.23 (β-catenin), 4.98/5.45
(TGF-βR2), 5.82/6.64 (BGN), 9.01/8.98 (TGF-βR1), 8.11, 9.22
(Snail2/Slug), 8.96/9.20 (DSP), 9.97/11.63 (Twist1), 10.66/11.62
(Snail1) and 10.35/8.57 (L1CAM), respectively (Fig. 1).
Moreover, although a wide range between single
samples was observed and in some samples no detection of certain
EMT markers was possible, significant expression differences of
some EMT markers were determined in primary and recurrent
glioblastomas. Twist1, Snail1, Snail2/Slug and BGN mRNA expression
was significantly lower in recurrent glioblastomas than in primary
tumors (p<0.05 for Snail1, Snail2/Slug and BGN, p<0.01 for
Twist1). In contrast, mRNA of L1CAM was found in higher amounts in
recurrent glioblastoma samples (p<0.05). For all other
investigated EMT markers no statistically significant different
mRNA expression level between matched pairs of primary and
recurrent tumor samples in the presented glioblastoma collective
was measurable.
To test whether the cellular composition of the
tumors significantly differs between investigated matched pairs of
glioblastoma samples accounting for the above mentioned findings,
in addition mRNA expression levels of GFAP (identifying glial
cells), Iba1 (identifying microglia/macrophages) and Pecam
(identifying endothelial cells) were also determined. With
exception of GFAP, which showed a slightly reduced expression in
recurrent glioblastoma samples, no clear expression differences
between primary and recurrent glioblastomas concerning these
cellular markers were observed.
Since not all of the investigated EMT markers
differed between individual primary and recurrent glioblastoma,
while for others a wide range of expression amounts were
detectable, qRT-PCR results of the individual patients were
analyzed in more detail. Therefore, we arranged the n-fold
expression changes of 15 individual primary-recurrent glioblastoma
pairs in a manner of a heatmap (two pairs which showed constantly
no mRNA expression of different EMT markers were omitted for this
analysis): equal n-fold expression in primary and recurrent
glioblastomas was assigned as 30% grey, lower n-fold expression
values were displayed with increasing lighter shading with 0
corresponding to white, and relative n-fold expression values >1
were assigned with increasing darker grey shades until 3-fold
induction which was assigned as maximum (black). To identify
possible groups of primary-recurrent glioblastoma pairs according
to EMT expression patterns in the values of GFAP, desmoplakin,
Snail1, Snail2, Twist1 and vimentin were inspected.
Five different glioblastoma groups in our collective
were identified: the first group, containing five primary-recurrent
glioblastoma pairs, was characterized by a general loss of mRNA
expression of all investigated markers, with the exception of
L1CAM, which showed higher expression in recurrent glioblastomas.
In this group not only GFAP downregulation (designed as our ‘glial’
marker within a hypothetical glial-mesenchymal transition) but also
loss of desmoplakin as classical known epithelial marker was found.
Since the mesenchymal markers Snail1, Snail2, Twist1 and vimentin
were additionally found in lower amounts in recurrent samples, this
group was defined as ‘general de-differentiation’ group. The second
group, containing four primary-recurrent glioblastoma pairs, was
characterized in addition to L1CAM induction by clearly increased
GFAP mRNA expression amounts in recurrent samples, and, moreover,
in most cases by higher expression amounts of DSP and the TGF-β
members, including TGF-β1, TGF-βR1, TGF-βR2, and biglycan. In
contrast, expression of other mesenchymal markers was mostly
reduced in recurrent glioblastomas. Thus, these cases were
summarized as ‘mesenchymal-epithelial/-glial transition’ group. The
third to fifth group included two glioblastoma pairs, respectively.
Whereas the third one was mainly characterized by an increased DSP
expression combined with loss of GFAP and inconsistent mesenchymal
expression profile in recurrent samples, the
‘mesenchymal-epithelial transition’ group, the fourth group showed
a combination of inconsistent loss of GFAP and DSP with clearly
higher mRNA expression amounts of mesenchymal markers in recurrent
samples, the ‘epithelial-/glial-mesenchymal transition’ group. The
fifth group was characterized by a clear consistent loss of GFAP
and DSP in combination with inconsistent higher expression amounts
of mesenchymal markers, the ‘mesenchymal trend’ group.
Summarized, our results indicate that a broad range
of expression profiles of different epithelial, mesenchymal and
glial (exemplified for glial acidic fibrillary protein) markers
exists in our collective of paired primary and recurrent
glioblastoma samples. Various combinations of loss or gain of EMT
marker expression are apparently detectable, and that besides
screening a whole collective in total, each individual pair has to
be analyzed in detail allowing a more precise evaluation of this
process underlying the progression from primary to recurrent
glioblastoma in individual patients.
Cellular assignment of EMT markers in
human primary and recurrent glioblastomas
In order to determine in more detail which cells
might account for expression of the EMT markers Twist1, DSP,
β-catenin, L1CAM and vimentin in primary and recurrent
glioblastomas, co-stainings with anti-glial fibrillary acidic
protein (GFAP), anti-von Willebrand factor protein (vWF), and
anti-CD11b was performed and analyzed by fluorescence microscopy.
The filament protein GFAP identified glial cells, and vWF is a
multimeric glyco-protein that is widely used for the detection of
endothelial cells in tumor tissues. CD11b, also designated
complement component receptor α3 chain or integrin αM, is a cell
adhesion molecule that acts as a receptor for cell surface ligands
such as intracellular adhesion molecules and is an established
marker for microglial/macrophages/monocytes in glioblastoma
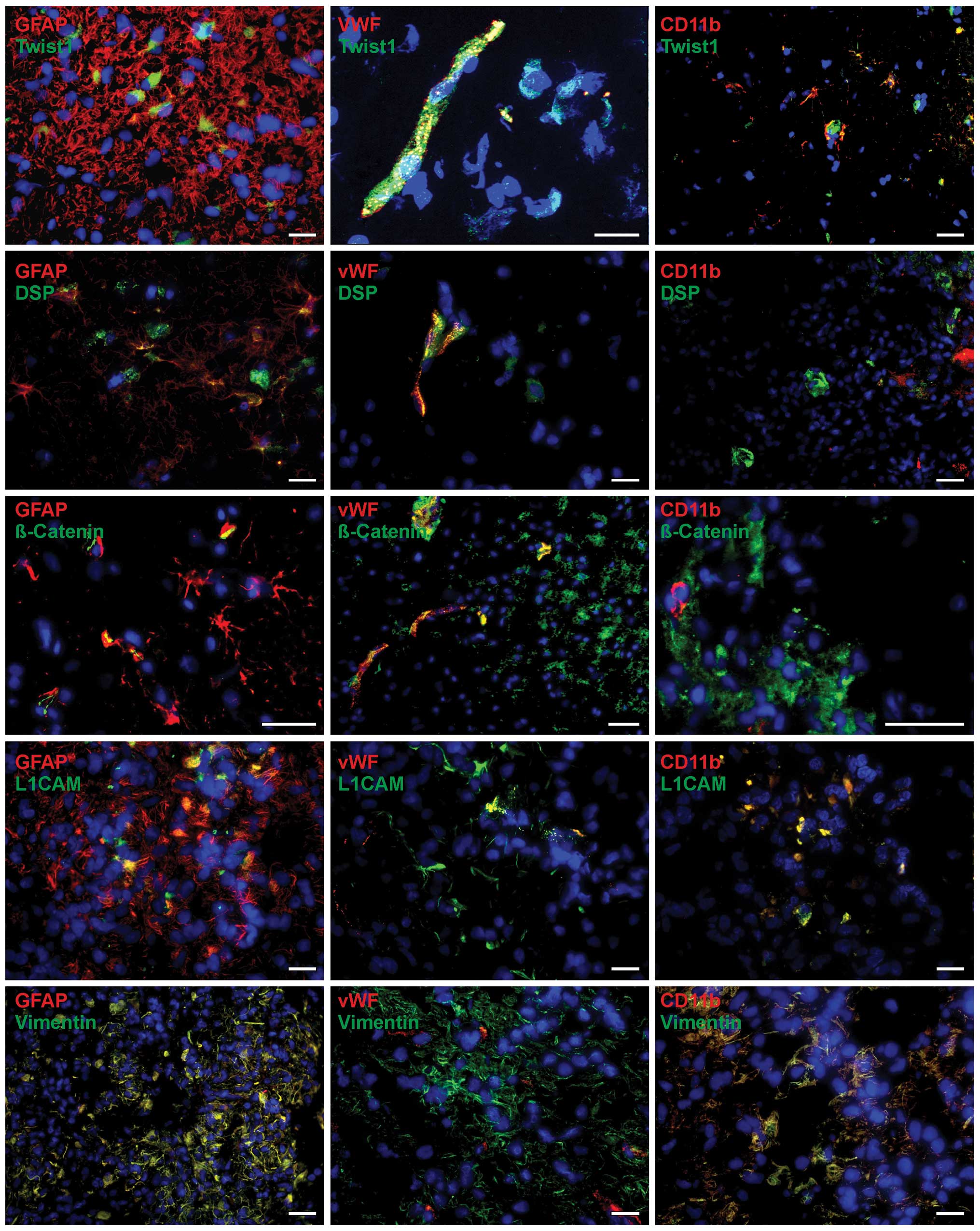
tissues. Results for primary glioblastomas are shown in Fig. 2, for recurrent ones in Fig. 3.
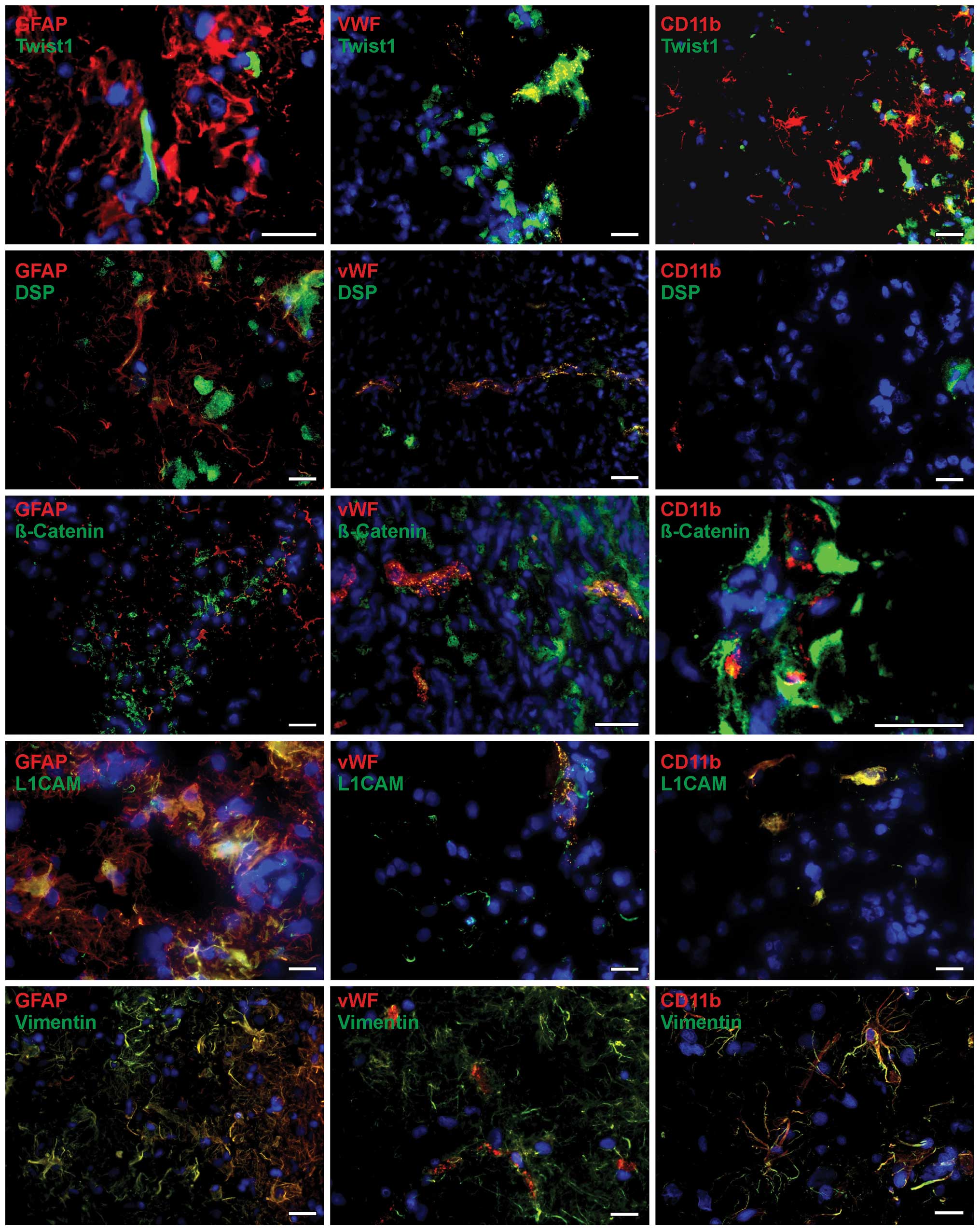
 | Figure 2Representative co-staining of EMT
markers [Twist1, desmoplakin (DSP), β-catenin, L1CAM and vimentin]
with cellular markers [glial fibrillary acidic protein (GFAP), von
Willebrand factor protein (vWF) and CD11b] in primary glioblastoma
as determined by immunofluorescence microscopy. EMT markers (green)
are found in different combinations with all cellular markers (red)
in a complex pattern. Since different markers are not all localized
within the same structures in the cells, signals did not merge
(yellow) in all cases, but were found in the same regions. Besides
double-positive cells also single-positive, either for stained EMT
or cellular marker, and double-negative ones were detectable.
Magnification, ×400; bar, 20 μm. |
Since this method does not allow a valid
quantification of both staining intensities and amounts of
positively stained cells, this analysis exclusively focused on the
determination of the cell-specific expression and localisation of
EMT markers.
The basic helix-loop-helix transcription factor
Twist1 was found to be expressed in GFAP-positive glial cells
within both primary and recurrent glioblastoma samples (Figs. 2 and 3, top). Since different markers were not
localized within the same structures in the cells, signals did not
merge in all cases, but were found in the same regions. Moreover,
cells which were positive for either GFAP or Twist1 (or negative
for both) existed within the tumor sections. Interestingly, besides
a cytoplasmic staining pattern, a nuclear expression of Twist was
also observed. Moreover, Twist1 was clearly expressed in
vWF-positive endothelial cells and in CD11b-positive
microglia/macrophages within primary and recurrent glioblastoma
samples. Twist1 expression in endothelial cells was clearly more
obvious in relation to microglia/macrophages, which showed in some
cases only slight Twist1 expression (Figs. 2 and 3, top).
In comparison to Twist1, staining for desmoplakin,
an obligate component of functional desmosomes that anchors
intermediate filaments of desmosomal plaques, was detectable in
lower amounts in primary and recurrent glioblastoma tissues.
Nevertheless, expression of desmoplakin could be related to
GFAP-positive cell regions, where it was found in clusters as well
as in single distributed positive cells within the tissue sections
(Figs. 2 and 3; second from top). Since different
markers were not localized within the same structures in the cells,
signals rarely merged, but were found in the same regions. Whereas
vWF-positive cells were also desmoplakin-positive, CD11b-positive
cells were almost desmoplakin-negative. As described for Twist1,
cells which were solely positive for GFAP, vWF or desmoplakin (or
negative for both) existed within the tumor sections. β-catenin is
a dual function protein, regulating the coordination of cell-cell
adhesion and gene transcription, and induces within the
WNT/β-catenin pathway cell migration which is necessary for pattern
formation and differentiation during embryonic development and
tumor progression. Staining of β-catenin in primary and recurrent
glioblastomas revealed β-catenin-positive cells in GFAP-positive
regions, but also, often as clusters of positive cells, near the
vWF and CD11b-positive cells (Figs.
2 and 3; third of top).
Whereas for endothelial cells a co-staining of vWF and β-catenin
was detectable, CD11b-positive cells seem to be β-catenin-negative
or only slightly positive. In case of GFAP, single glial cells were
positive for both GFAP and β-catenin, whereas several others showed
only β-catenin expression. However, GFAP is a filament protein and
β-catenin has mostly cytoplasmic localization in glioblastoma
samples, so these markers have not necessarily to co-localized.
Nevertheless, β-catenin and GFAP were found in the same
regions.
The neural adhesion molecule L1CAM (CD171) and the
class-III intermediate filament protein vimentin have been
associated with EMT along with increased invasion and metastasis in
various carcinomas. Indeed, both proteins were expressed in
investigated glioblastoma samples revealing robust staining
intensities (Figs. 2 and 3). Thus, nearly all GFAP-positive cells
were also stained for vimentin, while L1CAM could be localized in
GFAP-positive cells but either GFAP-positive or L1CAM-positive
cells were detectable, as well. Additionally, it seems that
vWF-positive endothelial cells were found to be also positive for
either L1CAM or vimentin, those costaining L1CAM with vWF seemed to
be more prominent. CD11b-positive cells showed L1CAM and
vimentin-positive staining, respectively. As described before, in
all tissues the cells solely positive for CD11b GFAP, vWF, L1CAM or
vimentin (or negative for both) existed also within the tumor
sections.
Summarized, the EMT markers Twist1, DSP, β-catenin,
L1CAM and vimentin were co-stained with GFAP-, vWF- and
CD11b-positive cells in a complex pattern identifying not only
tumor cells but also stromal cells (endothelial cells, macrophages)
as possible sources of EMT marker expression. Additionally, no
clear expression differences concerning various cell types within
solid glioblastoma tissues were detectable between primary and
recurrent glioblastoma samples.
Temozolomide treatment induces mRNA
expression of different EMT markers
To analyze a possible influence of temozolomide, the
most common chemotherapeutic drug in glioblastoma treatment, on the
mRNA expression levels of different EMT markers, we stimulated the
glioma cell line T98G with temozolomide for 24 and 48 h,
respectively. Since this chemotherapeutic drug is dissolved in DMSO
equal amounts of this solvent were applied as controls that were
used to determine the n-fold induction of mRNA expression as base
line values (n-fold expression for DMSO = 1). Additionally, for
determining basal expression levels of all EMT markers,
unstimulated T98G cells were investigated. Results are shown in
Fig. 4.
 | Figure 4Expression of EMT markers [Twist1,
Snail1, Snail2/Slug, desmoplakin (DSP), biglycan (BGN), β-catenin,
L1CAM, fibronectin, vimentin, TGF-β1, TGF-βR1, TGF-βR2] in
untreated (top) and temozolomide-treated (middle, 24 h; bottom, 48
h) T98G glioma cells evaluated by real-time RT-PCR (logarithmic
scale, ΔCT=3.3 corresponds to a 10-fold difference; relative gene
expression was calculated with 2(normalized CT
non-stimulated − normalized CT stimulated) = n-fold of
control; control = DMSO = 1). With exception of DSP and BGN all
investigated EMT markers were expressed in considerable amounts in
T98G cells. Temozolomide treatment for up to 48 h was able to
induce mRNA expression of nearly all EMT markers
(*p<0.05; **p<0.01;
***p<0.01). |
The basal expression of all EMT markers was moderate
to high, except for DSP and BGN (Fig.
4, top). High mRNA expression was detectable for vimentin,
TGF-β1 and fibronectin (normalized averaged ΔCT values
were 1.75±0.99, 3.88±0.86 and 4.11±1, respectively), moderate
expression for Snail2, L1CAM, Twist1 and TGF-βR2 (normalized
averaged ΔCT values were 6.15±0.70, 6.68±1.07, 8.56±1.93
and 7.89±0.78, respectively), and lower expression for Snail1,
β-catenin and TGF-βR1 (normalized averaged ΔCT values
were 10.31±0.73, 9.19±1.32 and 10.29±1.70, respectively).
Especially for the highly expressed markers (fibronectin, vimentin
and TGF-β1), these results were in accordance with those found in
primary and recurrent glioblastoma samples (Fig. 1). Moreover, as described for solid
glioblastomas, T98G cells showed a higher expression of TGF-βR2 in
relation to TGF-βR1, and Snail2 was more abundantly expressed than
Snail1. Only L1CAM and β-catenin expression was divergent in T98G
cells and tumor tissues (L1CAM moderate in T98G versus low in solid
samples and vice versa for β-catenin). Nevertheless, T98G cells can
be regarded as a suitable model system to study the effect of
temozolomide on EMT marker expression in glioblastoma cells.
For all investigated EMT markers (with exception of
BGN which was below detection limit; not shown) an induction of
mRNA expression levels after temozolomide treatment was detectable
(Fig. 4, middle and bottom).
Especially, after 48-h incubation time statistical significant
induction of Twist1, DSP, vimentin (p<0.001), Snail1, β-catenin,
TGF-βR1 (p<0.01) and L1CAM (p<0.05) mRNA was observed. In
detail, n-fold induction values were: 5.98±3.52 (Twist1), 5.46±1.99
(DSP), 3.47±1.59 (vimentin), 4.67±2.26 (Snail1), 2.93±1.67
(β-catenin), 2.10±0.95 (TGF-βR1) and 2.93±2.39 (L1CAM). Expression
of Snail2, fibronectin, TGF-β1 and TGF-βR2 was also induced but
without any statistical significance.
Taken together, we were able to show that in paired
samples of primary and recurrent glioblastomas a broad range of
different EMT markers are expressed in various combinations, and
that these markers are expressed in different cell types within
solid glioblastoma samples in a complex pattern. Moreover,
temozolomide was able to induce expression of nearly all EMT
markers, giving a first hint that a chemotherapeutic treatment not
only reduces tumor growth by induction of apoptosis, but may also
influence the tumor progression in other ways.
Discussion
EMT is an evolutionary conserved developmental
process (32) comprising
co-ordinated molecular and cellular changes that result in
reduction of cell-cell adhesion, apical-basolateral polarity and
epithelial marker expression along with an acquisition of a
spindle-cell shape, increased expression of mesenchymal markers and
enhanced motility (4).
Additionally, EMT is well known to support cancer progression,
although its importance is still controversially discussed in the
process of cancer metastasis regarding the fact that metastases
appear histopathologically similar to the primary tumors from which
they are derived (4). Thus, a
reversion of this process, called MET, has been postulated which
could explain this phenomenon (3).
However, experimental data supporting MET in cancer metastasis are
still rare mainly due to the limited availability of metastatic
tissues (4). Additionally, many
invasive and metastatic carcinomas have not undergone complete EMT
or invade adjacent connective tissue as individual mesenchymal-like
cells or even as epithelial cell clusters (4,33).
Thus, up to now it is not completely understood which functional
role both EMT and MET play in cancer progression and metastasis,
particularly applying for glioblastomas.
Malignant gliomas including also their most
malignant form, the glioblastomas, frequently occur in the adult
brain and represent one of the most aggressive neoplasms in humans
(9,11). Despite drastic therapeutic options
such as surgical resection, fractionated radiotherapy and adjuvant
chemotherapy with alkylating drugs like temozolomide, the incidence
of recurrence, regrowth and dissemination of gliomas is still
obvious. Thus, several groups are focused on the investigation of
EMT processes in gliomas in order to understand in which way this
process is involved in glioma progression (10). It was shown that ZEB1 and ZEB2 are
responsible for disrupting cell-cell contact inhibition, Twist and
Snail family members augment glioblastoma cell motility and
invasiveness, and β-catenin and miR-21 enhance the extracellular
matrix-cleavage, suggesting that EMT is a major molecular event in
glial tumors (10). However, since
the majority of glioblastomas do not show intrinsic E-cadherin
expression (15), the classical
‘E-cadherin to N-cadherin switch’ is unlikely to correlate with
mesenchymal transition in glioblastomas (16), and therefore the term glial to
mesenchymal transition (GMT) was coined (11). Up to now only one group has
investigated the EMT process in pairs of primary and recurrent
glioblastomas (11). They found by
PCR analysis that the expression of collagen, matrix
metalloproteinase (MMP)-9, smooth muscle α-actin (α-SMA), CD44,
fibronectin, and YKL-40 were elevated in the recurrent glioma
samples, and demonstrated by immunohistochemistry that in 22 cases
of clinically recurrent gliomas the expression of vimentin, α-SMA,
and CD44 was increased (11).
Nevertheless, the group investigated only a small cohort of 7
matched primary and recurrent glioblastoma samples, and did not
distinguish in detail which cell types within solid human
glioblastomas mainly expressed the EMT markers. Additionally, they
did not address the relevance of other processes such as MET or
GMT. Thus, we investigated the expression of Twist1, Snail1,
Snail2/Slug, desmoplakin, biglycan, β-catenin, L1CAM, fibronectin,
vimentin, and TGF-β1 with its receptors TGF-βR1 and TGF-βR2 in 17
matched probes of solid primary and recurrent human glioblastomas
by real-time RT-PCR as well as by double-immunofluorescence
staining to precisely identify EMT molecule-expressing cell
types.
We showed that Twist1, Snail1, Snail2/Slug and BGN
mRNA expression in recurrent glioblastomas was significantly lower
than in primary tumors, whereas mRNA expression of L1CAM was found
in higher amounts in recurrent glioblastoma samples. For the other
investigated EMT markers no expression differences were detectable.
Nevertheless, when investigating each individual pair in detail, a
‘heatmap-like’ analysis revealed five different groups within our
investigated glioblastoma collective. According to the regulation
of mRNA expression of mainly GFAP, desmoplakin, Snail1, Snail2,
Twist1 and vimentin, we defined a mesenchymal-epithelial/glial
transition group, the ‘general-de-differentiation’ group, the
mesenchymal-epithelial transition group, the
epithelial-/glial-mesenchymal transition group, and the mesenchymal
trend group. Additionally, double-immunofluorescence staining
revealed that the EMT markers Twist1, DSP, β-catenin, L1CAM and
vimentin could be co-stained with GFAP-, vWF- and CD11b-positive
cells in a complex pattern identifying all of these three different
cellular types as possible sources for EMT marker expression. Thus,
our results demonstrate that the EMT, MET and also GMT can all be
involved in glioma progression but in a specialized, individual way
for each patient’s primary-recurrent pair. Since the cellular
architecture of glioblastomas is very heterogeneous this result is
not really surprising, in particular when considering the aspect
that the different cell types within glioblastomas, the tumor cells
themselves, endothelial cells and microglia/macrophages, are all
possible sources of EMT markers. Nevertheless, our results support
the view of the existence of distinct ‘EMT-phenotypes’ in glioma
progression which are characterized by congruent expression
profiles for certain EMT markers. These results are in accordance
with recent investigations of Zarkoob et al (14) showing that the mesenchymal and
neural subtypes of glioblastomas have the strongest correlation
with a CD133 genetic signature, identifying an aggressive
subpopulation of glioblastomas. While the mesenchymal subtype of
glioblastomas displays similarity to the signature of both EMT and
CD133, it also differs from each of these signatures, possibly due
to the fact that signatures of EMT and CD133 are inversely related
to each other (14). Additionally,
an important role of Snail1 (11,34),
Snail2/Slug (25), BGN (25), and Twist1 (12,35)
in glioma EMT was previously demonstrated supporting our results,
which demonstrated expression differences for these EMT markers
between primary and recurrent glioblastomas. Surprisingly, L1CAM,
which is well known in glioma progression (19), but was up to now not identified as
a classical EMT marker in this tumor entity also showed robust
expression differences between paired primary-recurrent samples.
The observation that L1CAM expression in gliomas was correlated
with high expression of TGF-β (36), and that binding of Snail2/Slug has
been shown to be essential for TGF-β1-induced L1CAM expression in
human pancreatic ductal epithelial and pancreatic adenocarcinoma
cells (37), point to a similar
mechanism underlying L1CAM expression in glioblastomas as well as
to its role in EMT in these tumors. In order to substantiate these
findings and to clarify the role of EMT and its reversion in
glioblastoma progression in more detail, further investigations
implying the analysis of a larger cohort of patient-derived samples
are certainly required.
Since temozolomide is the main chemotherapeutic drug
of glioma therapy, we speculated whether EMT associated processes
can be promoted by temozolomide application. To test this, EMT
marker expression was analyzed in T89G glioma cells after
temozolomide treatment. Indeed, temozolomide was able to induce
expression of nearly all EMT markers to considerable amounts in
T98G glioma cells. These results are in accordance with the
investigations of Mahabir et al (11) showing that especially Snail1 was
found in higher amounts in glioma cells 21 days after irradiation.
Moreover, these cells exhibited an increased migration, invasion
and MMP-2 production all these features being abrogated after
Snail1 knockdown (11). Altogether
these results support the view that different therapeutic options
of glioma therapy, including irradiation and chemotherapeutic
treatment, are not only beneficial for reducing tumor burden, but
may concomitantly favor tumor progression in different ways, e.g.,
by promoting EMT.
Summarized, EMT/GMT and also MET seemed to be
involved in glioma progression in a complex way, the complete
understanding requires an individualized analysis. Moreover, these
processes seemed to be modulated by certain therapeutic options of
glioma therapy potentially antagonizing the expected antitumor
effects and providing an explanation for the still high recurrence
rate of this tumor entity.
Acknowledgements
We thank Brigitte Rehmke, Fereshteh Ebrahim and Jörg
Krause for expert technical assistance. This study was supported by
the University of Kiel and by the popgen 2.0 network [(P2N;
supported by a grant from the German Ministry for Education and
Research (01EY1103)].
Abbreviations:
|
BGN
|
biglycan
|
|
CT
|
cycle of threshold
|
|
DMEM
|
Dulbecco’s modified Eagle’s medium
|
|
DSP
|
desmoplakin
|
|
EMT
|
epithelial-to-mesenchymal
transition
|
|
FCS
|
fetal calf serum
|
|
GFAP
|
GAPDH,
glycerinaldehyde-3-phosphate-dehydrogenase; glial fibrillary acidic
protein
|
|
GMT
|
glial to mesenchymal transition
|
|
Iba1
|
ionized calcium-binding adapter
molecule 1
|
|
MET
|
mesenchymal-to-epithelial
transition
|
|
Pecam
|
platelet endothelial cell adhesion
molecule
|
|
qRT-PCR
|
quantitative reverse transcription
polymerase chain reaction
|
|
TGF-β (R)
|
transforming growth factor-β
(receptor)
|
|
vWF
|
von Willebrand factor protein
|
|
WHO
|
World Health Organization
|
References
|
1
|
Gao D, Vahdat LT, Wong S, Chang JC and
Mittal V: Micro-environmental regulation of epithelial-mesenchymal
transitions in cancer. Cancer Res. 72:4883–4889. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Scheel C and Weinberg RA: Cancer stem
cells and epithelial-mesenchymal transition: Concepts and molecular
links. Semin Cancer Biol. 22:396–403. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tsuji T, Ibaragi S and Hu GF:
Epithelial-mesenchymal transition and cell cooperativity in
metastasis. Cancer Res. 69:7135–7139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zavadil J, Haley J, Kalluri R, Muthuswamy
SK and Thompson E: Epithelial-mesenchymal transition. Cancer Res.
68:9574–9577. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tarin D, Thompson EW and Newgreen DF: The
fallacy of epithelial mesenchymal transition in neoplasia. Cancer
Res. 65:5996–6001. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thompson EW, Newgreen DF and Tarin D:
Carcinoma invasion and metastasis: A role for
epithelial-mesenchymal transition? Cancer Res. 65:5991–5995. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yates C: Prostate tumor cell plasticity: A
consequence of the microenvironment. Adv Exp Med Biol. 720:81–90.
2011.PubMed/NCBI
|
|
9
|
Ohgaki H and Kleihues P: Epidemiology and
etiology of gliomas. Acta Neuropathol. 109:93–108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kahlert UD, Nikkhah G and Maciaczyk J:
Epithelial-to-mesenchymal(-like) transition as a relevant molecular
event in malignant gliomas. Cancer Lett. 331:131–138. 2013.
View Article : Google Scholar
|
|
11
|
Mahabir R, Tanino M, Elmansuri A, Wang L,
Kimura T, Itoh T, Ohba Y, Nishihara H, Shirato H, Tsuda M and
Tanaka S: Sustained elevation of Snail promotes glial-mesenchymal
transition after irradiation in malignant glioma. Neuro-oncol.
16:671–685. 2014. View Article : Google Scholar :
|
|
12
|
Mikheeva SA, Mikheev AM, Petit A, Beyer R,
Oxford RG, Khorasani L, Maxwell J-P, Glackin CA, Wakimoto H,
González-Herrero I, et al: TWIST1 promotes invasion through
mesenchymal change in human glioblastoma. Mol Cancer. 9:194–211.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pala A, Karpel-Massler G, Kast RE, Wirtz
CR and Halatsch M-E: Epidermal to mesenchymal transition and
failure of EGFR-targeted therapy in glioblastoma. Cancers (Basel).
4:523–530. 2012. View Article : Google Scholar
|
|
14
|
Zarkoob H, Taube JH, Singh SK, Mani SA and
Kohandel M: Investigating the link between molecular subtypes of
glioblastoma, epithelial-mesenchymal transition, and CD133 cell
surface protein. PLoS One. 8:e641692013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kahlert UD, Maciaczyk D, Doostkam S, Orr
BA, Simons B, Bogiel T, Reithmeier T, Prinz M, Schubert J,
Niedermann G, et al: Activation of canonical WNT/β-catenin
signaling enhances in vitro motility of glioblastoma cells by
activation of ZEB1 and other activators of
epithelial-to-mesenchymal transition. Cancer Lett. 325:42–53. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brabletz T: To differentiate or not -
routes towards metastasis. Nat Rev Cancer. 12:425–436. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu J, Lamouille S and Derynck R:
TGF-β-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qiao B, Johnson NW and Gao J:
Epithelial-mesenchymal transition in oral squamous cell carcinoma
triggered by transforming growth factor-beta1 is Snail
family-dependent and correlates with matrix metalloproteinase-2 and
-9 expressions. Int J Oncol. 37:663–668. 2010.PubMed/NCBI
|
|
19
|
Held-Feindt J, Schmelz S, Hattermann K,
Mentlein R, Mehdorn HM and Sebens S: The neural adhesion molecule
L1CAM confers chemoresistance in human glioblastomas. Neurochem
Int. 61:1183–1191. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Elias MC, Tozer KR, Silber JR, Mikheeva S,
Deng M, Morrison RS, Manning TC, Silbergeld DL, Glackin CA, Reh TA
and Rostomily RC: TWIST is expressed in human gliomas and promotes
invasion. Neoplasia. 7:824–837. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Han SP, Kim JH, Han ME, Sim HE, Kim KS,
Yoon S, Baek SY, Kim BS and Oh SO: SNAI1 is involved in the
proliferation and migration of glioblastoma cells. Cell Mol
Neurobiol. 31:489–496. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang HW, Menon LG, Black PM, Carroll RS
and Johnson MD: SNAI2/Slug promotes growth and invasion in human
gliomas. BMC Cancer. 10:3012010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
De Wever O, Pauwels P, De Craene B, Sabbah
M, Emami S, Redeuilh G, Gespach C, Bracke M and Berx G: Molecular
and pathological signatures of epithelial-mesenchymal transitions
at the cancer invasion front. Histochem Cell Biol. 130:481–494.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim H, Watkinson J, Varadan V and
Anastassiou D: Multi-cancer computational analysis reveals
invasion-associated variant of desmoplastic reaction involving
INHBA, THBS2 and COL11A1. BMC Med Genomics. 3:512010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cheng WY, Kandel JJ, Yamashiro DJ, Canoll
P and Anastassiou D: A multi-cancer mesenchymal transition gene
expression signature is associated with prolonged time to
recurrence in glioblastoma. PLoS One. 7:e347052012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kaufhold S and Bonavida B: Central role of
Snail1 in the regulation of EMT and resistance in cancer: A target
for therapeutic intervention. J Exp Clin Cancer Res. 33:622014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tong Y, Mentlein R, Buhl R, Hugo H-H,
Krause J, Mehdorn HM and Held-Feindt J: Overexpression of midkine
contributes to anti-apoptotic effects in human meningiomas. J
Neurochem. 100:1097–1107. 2007. View Article : Google Scholar
|
|
28
|
Mentlein R, Forstreuter F, Mehdorn HM and
Held-Feindt J: Functional significance of vascular endothelial
growth factor receptor expression on human glioma cells. J
Neurooncol. 67:9–18. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hattermann K, Held-Feindt J, Lucius R,
Müerköster SS, Penfold MET, Schall TJ and Mentlein R: The chemokine
receptor CXCR7 is highly expressed in human glioma cells and
mediates antiapoptotic effects. Cancer Res. 70:3299–3308. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Held-Feindt J, Hattermann K, Müerköster
SS, Wedderkopp H, Knerlich-Lukoschus F, Ungefroren H, Mehdorn HM
and Mentlein R: CX3CR1 promotes recruitment of human
glioma-infiltrating microglia/macrophages (GIMs). Exp Cell Res.
316:1553–1566. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mechtersheimer S, Gutwein P, Agmon-Levin
N, Stoeck A, Oleszewski M, Riedle S, Postina R, Fahrenholz F, Fogel
M, Lemmon V and Altevogt P: Ectodomain shedding of L1 adhesion
molecule promotes cell migration by autocrine binding to integrins.
J Cell Biol. 155:661–673. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thiery JP: Epithelial-mesenchymal
transitions in development and pathologies. Curr Opin Cell Biol.
15:740–746. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Friedl P and Wolf K: Tumour-cell invasion
and migration: Diversity and escape mechanisms. Nat Rev Cancer.
3:362–374. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Myung JK, Choi SA, Kim S-K, Wang K-C and
Park S-H: Snail plays an oncogenic role in glioblastoma by
promoting epithelial mesenchymal transition. Int J Clin Exp Pathol.
7:1977–1987. 2014.PubMed/NCBI
|
|
35
|
Velpula KK, Dasari VR, Tsung AJ, Dinh DH
and Rao JS: Cord blood stem cells revert glioma stem cell EMT by
down regulating transcriptional activation of Sox2 and Twist1.
Oncotarget. 2:1028–1042. 2011.PubMed/NCBI
|
|
36
|
Tsuzuki T, Izumoto S, Ohnishi T, Hiraga S,
Arita N and Hayakawa T: Neural cell adhesion molecule L1 in
gliomas: Correlation with TGF-beta and p53. J Clin Pathol.
51:13–17. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Geismann C, Arlt A, Bauer I, Pfeifer M,
Schirmer U, Altevogt P, Müerköster SS and Schäfer H: Binding of the
transcription factor Slug to the L1CAM promoter is essential for
transforming growth factor-β71 (TGF-β)-induced L1CAM expression in
human pancreatic ductal adenocarcinoma cells. Int J Oncol.
38:257–266. 2011.
|