Introduction
According to GLOBOCAN 2012, an estimated 14.1
million new cancer cases and 8.2 million cancer-related deaths
occurred in 2012 compared with 12.7 million and 7.6 million,
respectively, in 2008. Gastric cancer (GC) is the fifth most common
cancer (0.9 million, 6.8%) in the world after lung, breast,
colorectal, and prostate cancers and the second leading cause of
cancer death worldwide (0.7 million, 8.8%) after lung cancer.
Historically, GC is one of the major cancers in Asian countries,
such as Korea and Japan. Although the mortality and incidence of GC
have decreased worldwide, 31,269 new GC cases were reported in
2012, accounting for 14.2% of all cancer occurrences (219,520
case/year; data available at http://www.cancerresearchuk.org/cancer-info/cancerstats/world/incidence/).
The most effective treatment for localized GC is surgery, but, even
after curative resection, recurrence is noted in more than half of
the cases with advanced-stage disease. Moreover, although the
effective treatments that are available have increased the survival
rates of GC patients during the last three decades, the prognoses
of these patients remain relatively poor, with a 5-year survival
rate of 63.1% during 2004–2008 (http://www.cancer.go.kr/mbs/cancer/subview.jsp?id=cancer_040101000000).
Due to the high recurrence rate after resection surgery, patients
with GC are treated with adjuvant chemotherapy (including
cisplatin, 5-fluorouracil, capecitabine, oxaliplatin, mitomycin C,
and etoposide) and/or radiation therapy. However, the efficacies of
these adjuvant therapies are limited because of the side effects,
such as nausea, vomiting, the high risk of infection, and diarrhea
(1). Thus, the development of
novel anticancer agents for patients with GC and effective
treatment strategies are urgently required to increase their
efficacy.
Autophagy is a process that involves the cellular
degradation of unnecessary or dysfunctional cellular components and
the maintenance of cellular homeostasis. During autophagy, parts of
the cytoplasm and cellular organelles are engulfed within
double-membraned autophagosomes, and the autophagosomes fuse with
lysosomes to form autolysosomes. This results in the degradation of
the sequestered cargo by lysosomal hydrolytic enzymes. The
degradation is followed by the generation of cellular building
blocks for biosynthesis and energy production. Because autophagy
reuses cellular components when dealing with cellular stress by
clearing damaged proteins, organelles, or pathogens and provides
the resources for biosynthesis (e.g., adenosine triphosphate or
anabolic building blocks) during starvation, this process was
initially recognized as a mechanism that was cytoprotective against
environmental stress. Conversely, increasing amounts of recent
evidence have revealed that autophagy can also culminate in cell
death, which is designated as type-2 programmed cell death or
autophagic cell death (2).
The essential role of autophagy in cancer is not
clearly understood; however, its role in cell death is conflicting
depending on the type of tumor, the stage of tumorigenesis, and the
nature and extent of the stimuli (3–5). It
is generally believed that these two self-destructive processes,
autophagy and apoptosis, often occur in the same cell and that
autophagy mostly precedes apoptosis (6,7).
Furthermore, some chemo-therapeutics that are known to trigger
apoptosis also induce autophagy (8). In addition, emerging evidence
indicates that the inhibition of autophagy appears to enhance the
sensitivity of cancer cells towards anticancer drugs (9). Based on the aforementioned findings,
it would be helpful to develop a new anticancer agent that
simultaneously induces both autophagy and apoptosis. Thus, the
elucidation of the role of autophagy and the determination of
whether it is induced by agents in the response of tumor cells to
that agent will provide therapeutic advantages by manipulating the
autophagic process.
The anticancer effects of the synthetic hydroxamic
acid derivative MHY218
[N1-hydroxy-N8-(4-phenoxyphenol)
octanedianide], was first reported by Jeon and colleagues (10). It was found to cause apoptosis in
SKOV-3 ovarian cancer cells. This histone deacetylase (HDAC)
inhibitor exhibits growth inhibitory effects in several tumor cell
lines, including ovarian, breast, and colon cancer cells (10–12).
Two studies in animals have shown that MHY218 has antineoplastic
activities against tamoxifen-resistant breast cancer and ovarian
cancer (10,11). The anticancer mechanisms of MHY218
are still under investigation, and few studies have reported its
effects on cell death. It has been shown to induce the
pro-apoptotic Bax protein, inhibit the anti-apoptotic Bcl-2 protein
(10–12), and suppress the nuclear
translocation of nuclear factor-κB (NF-κB) (12). Thus, modulations of the
NF-κB-regulated genes such as cyclooxygenase-2, matrix
metalloproteinase-9, 5-lipoxygenase (12), and HDACs (10,11)
have been implicated in MHY218-induced apoptotic cell death. In
addition, this synthetic hydroxamic acid derivative induces G2/M
phase arrest in cells, and this is most likely mediated through the
regulation of cyclin B1 and its activating partners Cdc25C and
Cdc2, and a G2/M phase inhibitor p21WAF1/CIP1
(10–12). MHY218 has been shown to
significantly inhibit the growth of tumors in mice bearing human
ovarian cancer (10). A recent
study has indicated that MHY218 can suppress tamoxifen-resistant
breast cancer growth in vivo through the downregulation of
the proliferating cell nuclear antigen (PCNA), the induction of
apoptotic cell death, and the upregulation of light-chain 3 (LC3),
which is a biomarker for autophagy (10,11).
Notably, MHY218 possesses more potent anticancer effects compared
to those of suberoylanilide hydroxamic acid (SAHA, which is also
known as vorinostat) against both ovarian and breast cancer in
vivo animal models (10,11).
The aim of the present study was to determine
whether MHY218 can induce apoptotic cell death and autophagy in AGS
cells, and finally to access the inhibition of autophagy can
potentiate the proapoptotic effect of MHY218 in AGS cells.
Materials and methods
Chemicals
The structure of MHY218
[N1-hydroxy-N8-(4-phenoxyphenol)octanedianide]
that was used in this study is shown in Fig. 1. This compound was kindly provided
by Professor Hyung Ryong Moon (Pusan National University, Busan,
Korea), dissolved in dimethyl sulfoxide (DMSO), and stored at −20°C
before the experiments, and dilutions were made in culture medium.
The maximum concentration of DMSO did not exceed 0.1% (v/v) in the
treatment range in which there was no influence on cell growth. The
DMSO and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium
bromide (MTT) were obtained from AMRESCO LLC (Solon, OH, USA).
Antibodies that were specific for procaspase-3, -8, and -9,
poly(ADP-ribose) polymerase (PARP), Bax, Bcl-2, Beclin-1, and
Z-VAD-FMK were obtained from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). The polyclonal antibody against LC3B was
obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Propidium iodide (PI), acridine orange, 3-methyladenine,
chloroquine (CQ), bafilomycin A1 (Baf-A1), cycloheximide (CHX), and
a monoclonal antibody against β-actin were purchased from
Sigma-Aldrich Co. LLC (St. Louis, MO, USA).
Cell culture and cell viability
assay
The human GC AGS cell line was cultured in RPMI-1640
(GE Healthcare Life Sciences, Logan, UT, USA) that was supplemented
with 10% fetal bovine serum (GE Healthcare Life Sciences), 100 U/ml
of penicillin and 100 μg/ml of streptomycin (GE Healthcare Life
Sciences) at 37°C in a humidified 5% CO2. Cell viability
was determined with an MTT assay. For the MTT assay, AGS cells were
seeded in a 24-well culture plate, cultured for 24 or 48 h in the
growth media, and then treated with or without various reagents for
the indicated concentrations. The cells were incubated in the dark
with 0.5 mg/ml MTT at 37°C for 2 h. The formazan granules that were
generated by the live cells were dissolved in DMSO, and the
absorbance at 540 nm was monitored with a multiwell reader (Thermo
Fisher Scientific Inc., Vantaa, Finland).
Nuclear staining with Hoechst 33342
Cells were stained with 4 μg/ml of Hoechst 33342
(Life Technologies Corp., Grand Island, NY, USA) at 37°C for 10
min. The cells were then examined under a fluorescence
microscope.
Measurement of apoptotic cell death by
flow cytometry
Apoptotic cell death was determined with Annexin
V/PI staining and sub-G1 phase analyses. For the Annexin V/PI
staining, the cells were treated with the appropriate conditions
for 24 h, subsequently harvested, trypsinized, washed once with
cold phosphate-buffered saline (PBS), and then suspended in 1×
binding buffer (BD Biosciences, San Jose, CA, USA). The cells were
stained in PI and Annexin V-fluorescein isothiocyanate (FITC)
solution (BD Pharmingen FITC Annexin V Apoptosis Detection kit) at
room temperature for 15 min in the dark. The stained cells were
analyzed by flow cytometry within 1 h. For the sub-G1 phase
analysis, the cells were trypsinized, washed once with cold PBS,
and then fixed in 70% ethanol at −20°C overnight. The fixed cells
were stained with cold PI solution (50 μg/ml in PBS) at 37°C for 30
min in the dark. The flow cytometry analysis was performed on
Accuri C6 (BD Biosciences).
DNA fragmentation assay
The cells were lysed in buffer containing 5 mM
Tris-HCl (pH 7.5), 5 mM EDTA, and 0.5% Triton X-100 for 30 min on
ice. The lysates were vortexed and cleared by centrifugation at
27,000 × g for 20 min. Fragmented DNA in the supernatant was
treated with RNase, which was followed by proteinase K digestion,
phenol/chloroform/ isoamyl alcohol mixture (25:24:1, v/v/v)
extraction, and isopropanol precipitation. The DNA was separated in
a 1.6% agarose gel, stained with 0.1 μg/ml ethidium bromide, and
visualized with an ultraviolet source.
Western blot analysis
The total cells were lysed in lysis buffer [25 mM
Tris (pH 7.5), 250 mM NaCl, 5 mM EDTA, 1% Nonidet P-40, 100 μg/ml
phenymethylsulfonyl fluoride, and protease inhibitor cocktail
(Sigma-Aldrich Co. LLC)]. Equal amounts of the protein extracts
were denatured by boiling at 100°C for 5 min in sample buffer
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The total proteins
were subjected to 6–15% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and transferred to polyvinylidene fluoride
membranes. The membranes were probed with the desired primary
antibodies overnight, incubated with horseradish
peroxidase-conjugated secondary antibodies (Santa Cruz
Biotechnology, Inc.), and then visualized with the enhanced
chemiluminescence (ECL) detection system (GE Healthcare,
Piscataway, NJ, USA).
Caspase activity
The cells were harvested and washed with cold PBS.
The total cells were incubated with the lysis buffer (R&D
Systems, Inc., Minneapolis, MN, USA) on ice for 10 min. The lysed
cells were centrifuged at 10,000 × g for 1 min, and 100 μg of
protein was incubated with 2X reaction buffer and substrates of
colorimetric tetrapeptides, including Z-DEVD for caspase-3, Z-IETD
for caspase-8, and Ac-LEHD for caspase-9, respectively. The
reaction mixture was incubated at 37°C for 2 h, and the
enzyme-catalyzed release of p-nitroaniline was quantified at 405 nm
with a multiwell reader (Thermo Fisher Scientific Inc.).
Detection of acidic vesicular organelles
(AVOs)
The formation of AVOs is a well-known feature of
autophagy. Cells were treated under the appropriate conditions for
24 h, stained with acridine orange (1 μg/ml) for 15 min,
trypsinized, and then washed with PBS. The stained cells were then
analyzed with an Accuri C6 flow cytometer (BD Biosciences).
GFP-LC3 assay
Cells were transfected with the LC3-GFP plasmid with
the Lipofectamine 2000 reagent (Life Technologies Corp.) according
to the manufacturer’s protocol. The cells were seeded in Lab-Tek II
chamber slides (Thermo Fisher Scientific Inc.). After transfection
for 24 h, the cells were treated with 5 μM of MHY218 for 24 h,
washed with PBS twice, and then fixed with 4% paraformaldehyde for
20 min. The formation of punctate LC3-positive structures was
examined with confocal microscopy. Confocal images were obtained
with a FV10i FluoView Confocal Microscope (Olympus Corp., Tokyo,
Japan).
Statistical analysis
The results are expressed as the mean ± standard
deviation of three separate experiments, and they were analyzed by
Student’s t-tests and ANOVA. The means were considered
significantly different at *p<0.05,
**p<0.01 or ***p<0.001.
Results
MHY218 exhibited a potent cytotoxic
effect on AGS cells
In order to determine whether MHY218 inhibited the
growth and proliferation of AGS cells in vitro, we treated
the cells for 24 or 48 h with increasing concentrations of MHY218
that ranged from 0 to 10 μM. An MTT assay was conducted to measure
the MHY218-mediated growth inhibition and estimate the
concentration of the compound at which cell growth was cut in half
(IC50). As shown in Fig.
2, MHY218 effectively suppressed the cell growth of AGS cells
in a concentration-dependent manner. Moreover, the IC50
value of MHY218 was ~5 and 3 μM at 24 and 48 h, respectively. The
results presented in Fig. 2
indicate that MHY218 suppressed the growth of AGS cells in a
concentration- and time-dependent manner.
MHY218 triggers apoptotic cell death in
AGS cells
In order to address the question of whether the
growth-suppressive effects of MHY218 were associated with apoptotic
cell death, the morphological changes of the cellular structures
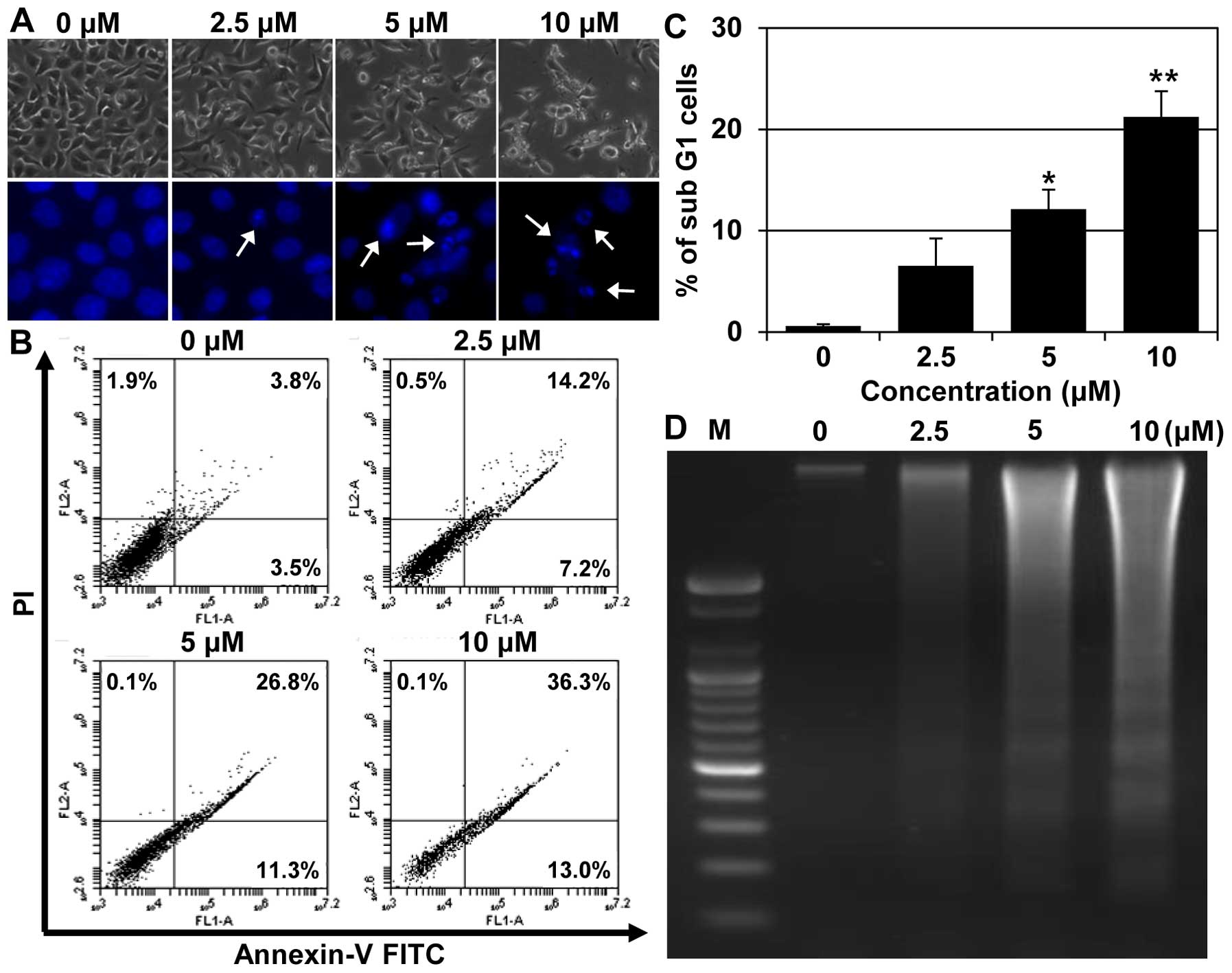
were assessed with Hoechst 33342 staining. Fig. 3A shows the morphological and
nuclear changes in AGS cells after 24 h of MHY218 treatment. MHY218
treatment caused nuclear condensation and cell death, whereas
untreated control cells displayed intact nuclear structures
(Fig. 3A). The number of cells
with highly condensed nuclei (representing programmed cell death)
significantly increased in cells treated with 10 μM of MHY218
(Fig. 3A). MHY218-induced
apoptotic cell death in AGS cells was further verified with Annexin
V and PI double staining. As shown in Fig. 3B, there was a prominent increase in
the percentage of late apoptotic cells (36.3%, with higher Annexin
V and PI-positive signals) in the cells after they were treated
with 10 μM of MHY218 for 24 h compared to the untreated controls (0
μM, 3.8%). Cells with higher Annexin V but lower PI signals were
indicative of early apoptotic cell death. The percentages of total
apoptotic cells (early and late apoptotic) significantly increased
in AGS cells in a concentration-dependent manner (Fig. 3B). In addition, we examined
MHY218-induced cell death in AGS cells with a cell cycle analysis.
As shown in Fig. 3C, a cell cycle
assay showed that MHY218 significantly increased AGS cells in the
sub-G1 population in a concentration-dependent manner (Fig. 3C).
Finally, we examined the effects of MHY218 on DNA
fragmentation. AGS cells were treated with various concentrations
of MHY218, and inter-nucleosomal DNA fragmentation was evaluated
with a DNA ladder on agarose gel electrophoresis. The genomic DNA
of the cells treated with MHY218 for 24 h displayed the
characteristic ladder pattern of discontinuous DNA fragments, while
untreated cells did not show any signs of fragmentation, as shown
in Fig. 3D.
MHY218 modulates the expression of
caspases, Bax, and Bcl-2 in AGS cells
The molecular events underlying the MHY218-induced
apoptosis in AGS cells were investigated. The activations of
several caspases are important in apoptosis that is induced by
various apoptotic stimuli (13).
In order to elucidate the mechanisms of MHY218-induced apoptotic
cell death in AGS cells, caspase involvement was investigated with
a western blot analysis. As shown in Fig. 4A, the exposure of AGS cells to
MHY218 markedly decreased the protein levels of pro-caspase-8, -9
and -3 in a concentration-dependent manner. Thus, after treatment
with MHY218, the full-length form of the PARP protein (116 kDa),
which is a selective substrate for caspase-3, was degraded to the
cleaved form (85 kDa) (Fig.
4A).
We next examined the expression of
apoptosis-associated proteins after treatment with MHY218. With the
balance of anti- and pro-apoptotic proteins arbitrating
life-or-death decisions, the Bcl-2 family proteins may regulate
mitochondria-dependent apoptosis (14). The expression of pro-apoptotic Bax
was increased, while anti-apoptotic Bcl-2 expression was reduced in
MHY218-treated cells in a concentration-dependent manner (Fig. 4B). These results suggested that
MHY218 promoted cell death in AGS cells through apoptosis with both
the intrinsic and extrinsic pathways.
MHY218 induces apoptosis through caspase
activation
Because the activation of caspases plays a critical
role in apoptosis (15), we
examined the activation of caspases by MHY218 in AGS cells.
Therefore, the activities of caspases in AGS cells that were
treated with various concentration of MHY218 were investigated.
After 24 h of treatment with MHY218, the activities of caspase-8,
-9, and -3 were increased in a concentration-dependent manner
(Fig. 5A). MHY218 markedly
stimulated caspase-3 to a >2-fold increase in activity in a
concentration-dependent manner, while it produced less of an effect
on the caspase-8 and -9 activities.
In order to demonstrate the contributions of
caspases to MHY218-induced apoptosis, we next examined the effects
of the caspase inhibitor Z-VAD-FMK on apoptosis. As shown in
Fig. 5B, Z-VAD-FMK, which is a
broad-spectrum caspase inhibitor, markedly decreased the proportion
of apoptotic cells from 26.7 to 16%, which was the percentage
observed in untreated cells. In addition, the effect of the caspase
inhibitor on the MHY218-induced cleavage of PARP was examined with
a western blot analysis. As shown in Fig. 5C, pretreatment with Z-VAD-FMK
inhibited the cleavage of PARP compared to the cells that were
treated with MHY218 alone, but not completely. It is interesting to
note that MHY218-induced apoptosis was only partially suppressed by
Z-VAD-FMK. Taken together, these results suggested that the
apoptogenic effect of MHY218 in AGC cells, at least in part, was
mediated by activating the caspase cascade.
MHY218 induces autophagy in AGS
cells
In recent years, many modes of cell death other than
apoptosis have been found to exist, and these include autophagy,
necroptosis, and PARP1-mediated cell death (16). In the present study, we examined
whether MHY218 also induced autophagy in AGS cells. Cells were
treated with the indicated concentrations of MHY218 for 24 h, and
the formation of autophagic vacuoles was examined with phase
contrast microscopy. As shown in Fig.
6A, MHY218 induced the formation of autophagic vacuoles in AGS
cells in a concentration-dependent manner.
We further conducted a series of experiments to
confirm the effects of MHY218 on the autophagy process. Because the
formation of cytosolic AVOs is one of the typical features of
autophagic cell death, a flow cytometry analysis was performed
after staining the cells with acridine orange for the
quantification of the AVOs. The number of AVOs in MHY218-treated
cells clearly increased in a concentration-dependent manner
(Fig. 6B).
Beclin-1 levels and the conversion of
microtubule-associated protein 1 LC3 are selective autophagic
markers. Next, in order to further confirm that autophagy was
induced by MHY218, we examined the LC3 distribution in
MHY218-treated AGS cells with fluorescence microscopy. As shown in
Fig. 6C, green fluorescent protein
(GFP)-tagged-LC3 (GFP-LC3) formed cytoplasmic puncta in cells that
were treated with MHY218 but not in untreated control cells. In
addition, a western blot analysis showed that LC3 underwent a
conversion from LC3-I (the soluble form) to LC3-II (the lipidized
form) in MHY218-treated cells, thus indicating the induction of
autophagy (Fig. 6D). Consistent
with the findings of LC3 conversion, the upregulation of Beclin-1
by MHY218 was observed in AGS cells (Fig. 6D).
The suppression of autophagy modulates
MHY218-induced apoptotic cell death in AGS cells
Accumulating data have suggested that the inhibition
of autophagy may enhance chemosensitization in human cancer cells
(17). Thus, we investigated the
role of autophagy in MHY218-induced cell death by examining the
effects of pharmacological autophagy inhibitors. Thus, we first
treated cells with various autophagy inhibitors for 1 h, incubated
them with MHY218 for another 24 h, and then performed Annexin V/PI
staining to evaluate cell death. Apoptotic cell death was induced
at a level of 27.2% by MHY218, at a level of 12% by LY294002, which
is an autophagy inhibitor that acts by blocking the class-III
phosphoinositide 3-kinases (PI3Ks) that are critical during the
late stage of vesicle expansion (autophagosome formation), and at a
level of 67.2% by the combination of the two agents (Fig. 7A). However, cells that were treated
with 3-methylad-enine (3-MA), which is an autophagy inhibitor that
acts by blocking the class-III PI3Ks, which was followed by the
addition of MHY218, exhibited an additive effect on the induction
of apoptosis. Similar results were obtained with the use of two
autophagy flux blockers, CQ and Baf-A1, on MHY218-induced
apoptosis. CQ, which is a lysosomotropic agent, prevents lysosome
acidification and thereby the degradation of the products of
autophagy, which results in autophagolysosome accumulation, whereas
Baf-A1, which is a vacuolar-type H+-ATP inhibitor,
prevents the maturation of autophagic vacuoles by inhibiting the
fusion of autophagosomes and lysosomes. We also employed CHX, which
is an inhibitor of protein biosynthesis and which may inhibit
autophagosome formation, to evaluate the effects of MHY218-induced
cell death. As shown in Fig. 7A,
CHX failed to potentiate the apoptosis-inducing effects of MHY218
in AGS cells, indicating that the effects of the autophagy
inhibitor CHX with MHY218 on cell death seemed to be additive.
To verify the observation that the inhibition of
autophagy affected the apoptotic cell death that was induced by
MHY218, we performed a western blot analysis to measure PARP
cleavage, which is an executioner of apoptosis. MHY218 induced PARP
cleavage compared to untreated cells (Fig. 7B). In accordance with the apoptotic
cell data (Fig. 7A), cells
cotreated with MHY218 and 3-MA, CQ, Baf-A1, or CHX exhibited a
measurable increase in PARP cleavage; whereas the combination of
the autophagy inhibitor LY294002 and MHY218 resulted in the most
profound amounts of PARP cleavage of all of the tested autophagy
inhibitors (Fig. 7B). Altogether,
these results indicated that the inhibition of autophagy by
autophagy inhibitors, such as LY294002, enhanced the MHY218-induced
cell death in AGS cells. However, the potency of autophagy
inhibition on apoptotic cell death may vary depending on the
selectivity of the autophagy inhibitors.
Discussion
With the development of new therapeutics and the
advances in diagnostic technology, the incidence of GC has
decreased during the past three decades. However, GC is still the
fifth most lethal neoplasm on a worldwide basis, and, thus, Korea
continues to have one of the highest rates of GC (18). While surgery is the most common
therapy used for patients with stomach cancer, advanced GC (AGC)
needs to be treated with multimodal treatments, including
chemotherapy and radio-therapy. Thus, the modest efficacies in GC
and considerable toxicities that are associated with the current
chemotherapy modalities have prompted the pursuit of novel
treatment strategies. In this study, we investigated the mechanisms
of MHY218, which is a hydroxamic acid derivative, in the induction
of autophagy and the role of autophagy in tumor cell survival in
the presence of MHY218. Our results indicated that MHY218 triggered
both apoptosis and autophagy in AGS cells. Furthermore, we found
that autophagy inhibition resulted in higher levels of apoptotic
cell death in response to MHY218 treatment. Collectively, our
findings suggest that targeting autophagy during cancer treatment
with MHY218 may augment the therapeutic effects.
One of the common features of human tumors is the
deregulation of HDAC isoenzymes, but no conclusive data about the
patterns of HDAC expression in human cancers are available
(19–21). Because studies of GC tissues have
shown an increase in class-I HDAC expression, HDAC is becoming a
prominent therapeutic target for GC treatment (22–24).
HDAC inhibitors (HDACi) have been recognized as a new class of
therapeutic agents with promising outcomes during the treatment of
a broad range of cancer types (25,26).
Therefore, HDACi have so far been approved by the US Food and Drug
Administration for the treatment of cutaneous T cell lymphoma, and
preclinical and clinical trials have provided evidence that HDACi
can be used in the treatment of solid tumors in combination with
other anticancer drugs or radiation (26–28).
MHY218, which is a synthetic hydroxamic acid, was primarily
designed to target HDAC, and it has been shown to suppress HDAC
activity and HDAC expression in cancer cells from two solid tumors
(10,11). These HDAC expression profiles and
previous reports on MHY218 provide the rationale for examining the
anticancer potential of MHY218 in GC cells.
Mechanistically, HDACi have been reported to promote
apoptosis through alterations in the expression of genes that are
involved in apoptotic cell death and activation of the caspase
cascade (29,30). We found that MHY218 inhibited cell
growth and induced apoptosis in AGS cells. The observed
growth-inhibitory and apoptosis-inducing properties of MHY218 were
in agreement with those observed by others in ovarian, breast, and
colon cancer cells (10-12). When we examined the mechanism by
which MHY218 mediated apoptotic cell death in AGS cells, we found
that MHY218 activated caspases in AGS cells. In agreement with
these observations, previous studies have shown that MHY218 induces
caspase activation (10,12). Our observations further revealed
that caspase activation in response to MHY218 may be involved in
part of the MHY218-induced apoptosis. It is estimated that about
half of the induced apoptosis can be inhibited by the pan-caspase
inhibitor Z-VAD-FMK, and this is therefore mediated by caspases,
whereas the other half is executed in a caspase-independent manner.
Likewise, caspase-dependent and -independent apoptosis that is
induced by SAHA, which is a widely used HDACi, has also been
reported in several cell lines (31–34).
Overall, these observations suggest that some other mechanism is
likely to be involved in MHY218-induced apoptosis.
We found that MHY218 mediated anticancer activity by
modulating the expression of genes that are involved in apoptosis.
This hydroxamic acid-derivative treatment resulted in the
upregulation of Bax and the downregulation of Bcl-2 expression
levels, which was in agreement with the findings of previous
reports (10–12). Other HDACi, such as SAHA (35,36),
trichostatin A (37), and valproic
acid (38), have also been
reported to exert apoptosis-inducing effects through the induction
of Bax as well as the inhibition of Bcl-2 expression in cancer
cells.
As with many other anticancer therapies, HDACi
induces autophagy, and this HDACi-mediated autophagy appears to act
as a cytoprotective mechanism to counteract the cytotoxic
activities of HDACi (33,39–41).
We observed the upregulation of Beclin-1 and an increase in LC3-II
conversion in MHY218-treated cells. These observations were
supported by a previous report that showed that MHY218 induced
autophagy in tamoxifen-resistant MCF-7 cells (11). However, Park et al (11) did not rule out the relationship
between apoptosis and autophagy that were induced by MHY218 and
that cooperate or counteract each other.
Although MHY218 enhanced autophagy, we found that
the inhibition of autophagolysosome formation was ineffective on
the synergistic induction of apoptosis by MHY218. The inhibition of
MHY218-stimulated autophagy by CQ turned out to be ineffective on
the synergistic induction of apoptosis by MHY218-CQ cotreatment.
This conclusion was further supported by our data that showed that
the lysosomal H+-ATPase inhibitor Baf-A1, which
similarly interrupted autophagolysosome formation, failed to
produce a synergistic effect on MHY218-inducing apoptosis (Fig. 7A). These results indicated that the
disruption of autophagolysosome formation per se was not
effective on the sensitization of cells for MHY218-mediated
apoptosis.
A group of PI3K inhibitors, including 3-MA,
wortmannin, and LY294002, have been well established and
extensively used as autophagy inhibitors, while the inhibition of
the PI3K-Akt pathway has previously been reported in certain
cancers to trigger autophagy as a survival mechanism (42–44).
The effects of PI3K inhibitors as autophagy inhibitors on
MHY218-induced apoptosis are intriguing. The combination of MHY218
and the autophagy inhibitor 3-MA enhanced cell death in an additive
manner. LY294002, in comparison, dramatically promoted
MHY218-induced apoptosis. The cells that were treated with MHY218
exhibited different apoptotic percentages in response to
PI3K-targeting autophagy inhibitors. This discrepancy may be
explained by the different properties of 3-MA compared to other
PI3K inhibitors. A recent report by Wu and collogues showed a dual
role of 3-MA in modulating autophagy (45). They found that, surprisingly, 3-MA
promoted autophagy under full (nutrient rich) medium for a
prolonged period (≤9 h), whereas it was still capable of inhibiting
starvation-induced autophagy. However, the PI3K inhibitor
wortmannin was able to suppress autophagy regardless of the
nutrient status. An increase in LC3-II levels was observed after 24
h of treatment with 3-MA (data not shown), even though Wu et
al (45) did not determine the
LC3 levels with the same experimental period, the possibility that
3-MA activated autophagy as a survival mechanism rather than
suppressed MHY218-induced cytoprotective autophagy was proposed. In
addition, the effectiveness of the PI3K inhibitors (i.e., 3-MA,
wortmannin, and LY294002) was varied in order to induce the
autophagic marker LC3-II in different cell types (46), thus further supporting the
discrepancy in the results with 3-MA and LY294002 in our present
study. Future studies are required to investigate the molecular
basis and therapeutic significance of autophagy in the
MHY218-derived anticancer effects.
In patients with advanced GC, the efficacy of
systemic chemotherapy is still limited, and the toxicity of
combination chemotherapy is substantial. Hydroxamate-based HDACi,
such as vorinostat (SAHA), belinostat (PXD101), and panobinostat
(LBH-589), have been shown to exhibit a good toxicity profile, and
they are now under phase-I and -II clinical trials in solid tumors
with promising results in selected neoplasms, such as
hepatocarcinoma (27). A recent
phase-I trial of the safety, tolerability, pharmacokinetic, and
pharmacodynamic characteristics of hydroxychloroquine (HCQ) in
combination with the HDAC inhibitor vorinostat in patients with
advanced solid tumors established that 600 mg of HCQ and 400 mg of
vorinostat was the maximum tolerated dose and recommended phase-II
regimen (47). It is worth noting
that the anticancer potency of MHY218 (10 mg/kg, twice a week for
21 days) is greater than that of SAHA (25 mg/kg, twice a week for
21 days) in tumor-bearing nude mice (10,11).
These findings and the promising results from clinical trials
encourage further consideration of MHY’s ability to be used as a
therapeutic target in gastric cancer. However, evaluations in
non-human primates of the pharmacokinetics, pharmacodynamics,
safety, and efficacy and studies that use clinically relevant
animal models to explore more action mechanisms are needed to fully
realize the potential of this fascinating molecule in the treatment
of gastric cancer.
Altogether, our data suggested that the novel
synthetic hydroxamate MHY218 possessed anticancer activity by
triggering and autophagy induction in GC cells. The results from
the present study provide evidence of MHY218-induced autophagy, and
autophagy inhibition by LY294002 dramatically augmented apoptotic
cell death that was induced by MHY218. Our findings suggest that
MHY218-induced autophagy acts as a prosurvival mechanism against
MHY218-induced apoptosis.
Acknowledgements
This study was supported by the National Research
Foundation of Korea (NRF) grant funded by the Korea government
(MSIP) (no. 2009-0083538). We thank the Aging Tissue Bank for
providing research information.
References
|
1
|
Wöhrer SS, Raderer M and Hejna M:
Palliative chemotherapy for advanced gastric cancer. Ann Oncol.
15:1585–1595. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lum JJ, Bauer DE, Kong M, Harris MH, Li C,
Lindsten T and Thompson CB: Growth factor regulation of autophagy
and cell survival in the absence of apoptosis. Cell. 120:237–248.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Debnath J, Baehrecke EH and Kroemer G:
Does autophagy contribute to cell death? Autophagy. 1:66–74. 2005.
View Article : Google Scholar
|
|
4
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
González-Polo RA, Boya P, Pauleau AL,
Jalil A, Larochette N, Souquère S, Eskelinen EL, Pierron G, Saftig
P and Kroemer G: The apoptosis/autophagy paradox: Autophagic
vacuolization before apoptotic death. J Cell Sci. 118:3091–3102.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li X and Fan Z: The epidermal growth
factor receptor antibody cetuximab induces autophagy in cancer
cells by downregulating HIF-1alpha and Bcl-2 and activating the
beclin 1/hVps34 complex. Cancer Res. 70:5942–5952. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Amaravadi RK, Yu D, Lum JJ, Bui T,
Christophorou MA, Evan GI, Thomas-Tikhonenko A and Thompson CB:
Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest. 117:326–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jeon HS, Ahn MY, Park JH, Kim TH, Chun P,
Kim WH, Kim J, Moon HR, Jung JH and Kim HS: Anticancer effects of
the MHY218 novel hydroxamic acid-derived histone deacetylase
inhibitor in human ovarian cancer cells. Int J Oncol. 37:419–428.
2010.PubMed/NCBI
|
|
11
|
Park JH, Ahn MY, Kim TH, Yoon S, Kang KW,
Lee J, Moon HR, Jung JH, Chung HY and Kim HS: A new synthetic HDAC
inhibitor, MHY218, induces apoptosis or autophagy-related cell
death in tamoxifen-resistant MCF-7 breast cancer cells. Invest New
Drugs. 30:1887–1898. 2012. View Article : Google Scholar
|
|
12
|
Kim MK, Kang YJ, Kim DH, Hossain MA, Jang
JY, Lee SH, Yoon JH, Chun P, Moon HR, Kim HS, et al: A novel
hydroxamic acid derivative, MHY218, induces apoptosis and cell
cycle arrest through downregulation of NF-κB in HCT116 human colon
cancer cells. Int J Oncol. 44:256–264. 2014.
|
|
13
|
Kumar S: Caspase function in programmed
cell death. Cell Death Differ. 14:32–43. 2007. View Article : Google Scholar
|
|
14
|
Tsujimoto Y: Role of Bcl-2 family proteins
in apoptosis: Apoptosomes or mitochondria? Genes Cells. 3:697–707.
1998. View Article : Google Scholar
|
|
15
|
Thornberry NA and Lazebnik Y: Caspases:
Enemies within. Science. 281:1312–1316. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Degterev A and Yuan J: Expansion and
evolution of cell death programmes. Nat Rev Mol Cell Biol.
9:378–390. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Carew JS, Nawrocki ST and Cleveland JL:
Modulating autophagy for therapeutic benefit. Autophagy. 3:464–467.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jung KW, Won YJ, Kong HJ, Oh CM, Lee DH
and Lee JS: Cancer statistics in Korea: Incidence, mortality,
survival, and prevalence in 2011. Cancer Res Treat. 46:109–123.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ropero S and Esteller M: The role of
histone deacetylases (HDACs) in human cancer. Mol Oncol. 1:19–25.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakagawa M, Oda Y, Eguchi T, Aishima S,
Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M, et al:
Expression profile of class I histone deacetylases in human cancer
tissues. Oncol Rep. 18:769–774. 2007.PubMed/NCBI
|
|
21
|
Yoo CB and Jones PA: Epigenetic therapy of
cancer: Past, present and future. Nat Rev Drug Discov. 5:37–50.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Choi JH, Kwon HJ, Yoon BI, Kim JH, Han SU,
Joo HJ and Kim DY: Expression profile of histone deacetylase 1 in
gastric cancer tissues. Jpn J Cancer Res. 92:1300–1304. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Song J, Noh JH, Lee JH, Eun JW, Ahn YM,
Kim SY, Lee SH, Park WS, Yoo NJ, Lee JY, et al: Increased
expression of histone deacetylase 2 is found in human gastric
cancer. APMIS. 113:264–268. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Weichert W, Röske A, Gekeler V, Beckers T,
Ebert MP, Pross M, Dietel M, Denkert C and Röcken C: Association of
patterns of class I histone deacetylase expression with patient
prognosis in gastric cancer: A retrospective analysis. Lancet
Oncol. 9:139–148. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Khan O and La Thangue NB: HDAC inhibitors
in cancer biology: Emerging mechanisms and clinical applications.
Immunol Cell Biol. 90:85–94. 2012. View Article : Google Scholar
|
|
26
|
Slingerland M, Guchelaar HJ and Gelderblom
H: Histone deacetylase inhibitors: An overview of the clinical
studies in solid tumors. Anticancer Drugs. 25:140–149. 2014.
View Article : Google Scholar
|
|
27
|
Grassadonia A, Cioffi P, Simiele F, Iezzi
L, Zilli M and Natoli C: Role of hydroxamate-based histone
deacetylase inhibitors (Hb-HDACIs) in the treatment of solid
malignancies. Cancers (Basel). 5:919–942. 2013. View Article : Google Scholar
|
|
28
|
Nolan L, Johnson PW, Ganesan A, Packham G
and Crabb SJ: Will histone deacetylase inhibitors require
combination with other agents to fulfil their therapeutic
potential? Br J Cancer. 99:689–694. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Emanuele S, Lauricella M and Tesoriere G:
Histone deacetylase inhibitors: Apoptotic effects and clinical
implications (Review). Int J Oncol. 33:637–646. 2008.PubMed/NCBI
|
|
30
|
Ellis L and Pili R: Histone deacetylase
inhibitors: Advancing therapeutic strategies in hematological and
solid malignancies. Pharmaceuticals (Basel). 3:2411–2469. 2010.
View Article : Google Scholar
|
|
31
|
Dong G, Wang L, Wang CY, Yang T, Kumar MV
and Dong Z: Induction of apoptosis in renal tubular cells by
histone deacetylase inhibitors, a family of anticancer agents. J
Pharmacol Exp Ther. 325:978–984. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu W, Ngo L, Perez G, Dokmanovic M and
Marks PA: Intrinsic apoptotic and thioredoxin pathways in human
prostate cancer cell response to histone deacetylase inhibitor.
Proc Natl Acad Sci USA. 103:15540–15545. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shao Y, Gao Z, Marks PA and Jiang X:
Apoptotic and autophagic cell death induced by histone deacetylase
inhibitors. Proc Natl Acad Sci USA. 101:18030–18035. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hrzenjak A, Kremser ML, Strohmeier B,
Moinfar F, Zatloukal K and Denk H: SAHA induces
caspase-independent, autophagic cell death of endometrial stromal
sarcoma cells by influencing the mTOR pathway. J Pathol.
216:495–504. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang YC, Kong WZ, Xing LH and Yang X:
Effects and mechanism of suberoylanilide hydroxamic acid on the
proliferation and apoptosis of human hepatoma cell line Bel-7402. J
BUON. 19:698–704. 2014.PubMed/NCBI
|
|
36
|
Zhang XD, Gillespie SK, Borrow JM and
Hersey P: The histone deacetylase inhibitor suberic bishydroxamate
regulates the expression of multiple apoptotic mediators and
induces mitochondria-dependent apoptosis of melanoma cells. Mol
Cancer Ther. 3:425–435. 2004.PubMed/NCBI
|
|
37
|
Park H, Lee YJ, Kim TH, Lee J, Yoon S,
Choi WS, Myung CS and Kim HS: Effects of trichostatin A, a histone
deacetylase inhibitor, on the regulation of apoptosis in
H-ras-transformed breast epithelial cells. Int J Mol Med.
22:605–611. 2008.PubMed/NCBI
|
|
38
|
Shen WT, Wong TS, Chung WY, et al:
Valproic acid inhibits growth, induces apoptosis, and modulates
apoptosis-regulatory and differentiation gene expression in human
thyroid cancer cells. Surgery. 138:979–985. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Choi EJ, Cho BJ, Lee DJ, Hwang YH, Chun
SH, Kim HH and Kim IA: Enhanced cytotoxic effect of radiation and
temozolomide in malignant glioma cells: Targeting PI3K-AKT-mTOR
signaling, HSP90 and histone deacetylases. BMC Cancer. 14:172014.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
El-Khoury V, Pierson S, Szwarcbart E,
Brons NH, Roland O, Cherrier-De Wilde S, Plawny L, Van Dyck E and
Berchem G: Disruption of autophagy by the histone deacetylase
inhibitor MGCD0103 and its therapeutic implication in B-cell
chronic lymphocytic leukemia. Leukemia. 28:1636–1646. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Watanabe M, Adachi S, Matsubara H, Imai T,
Yui Y, Mizushima Y, Hiraumi Y, Watanabe K, Kamitsuji Y, Toyokuni
SY, et al: Induction of autophagy in malignant rhabdoid tumor cells
by the histone deacetylase inhibitor FK228 through AIF
translocation. Int J Cancer. 124:55–67. 2009. View Article : Google Scholar
|
|
42
|
Degtyarev M, De Mazière A, Orr C, Lin J,
Lee BB, Tien JY, Prior WW, van Dijk S, Wu H, Gray DC, et al: Akt
inhibition promotes autophagy and sensitizes PTEN-null tumors to
lysosomotropic agents. J Cell Biol. 183:101–116. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mirzoeva OK, Hann B, Hom YK, Debnath J,
Aftab D, Shokat K and Korn WM: Autophagy suppression promotes
apoptotic cell death in response to inhibition of the PI3K-mTOR
pathway in pancreatic adenocarcinoma. J Mol Med Berl. 89:877–889.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fan QW, Cheng C, Hackett C, Feldman M,
Houseman BT, Nicolaides T, Haas-Kogan D, James CD, Oakes SA,
Debnath J, et al: Akt and autophagy cooperate to promote survival
of drug-resistant glioma. Sci Signal. 3:ra812010.PubMed/NCBI
|
|
45
|
Wu YT, Tan HL, Shui G, Bauvy C, Huang Q,
Wenk MR, Ong CN, Codogno P and Shen HM: Dual role of
3-methylad-enine in modulation of autophagy via different temporal
patterns of inhibition on class I and III phosphoinositide
3-kinase. J Biol Chem. 285:10850–10861. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cheng J, Ohsaki Y, Tauchi-Sato K, Fujita A
and Fujimoto T: Cholesterol depletion induces autophagy. Biochem
Biophys Res Commun. 351:246–252. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mahalingam D, Mita M, Sarantopoulos J,
Wood L, Amaravadi RK, Davis LE, Mita AC, Curiel TJ, Espitia CM,
Nawrocki ST, et al: Combined autophagy and HDAC inhibition: A phase
I safety, tolerability, pharmacokinetic, and pharmacodynamic
analysis of hydroxychloroquine in combination with the HDAC
inhibitor vorinostat in patients with advanced solid tumors.
Autophagy. 10:1403–1414. 2014. View Article : Google Scholar : PubMed/NCBI
|