Introduction
High expression of surface GD2 ganglioside,
MYCN gene amplification and aberrant activation of the PI3K
pathway are major hallmarks of high risk neuroblastoma (1,2). GD2
ganglioside-targeted therapies using monoclonal antibodies are
studied in phase III clinical trials for neuroblastoma patients.
The mAbs can inhibit tumor growth by means of antibody-dependent
cellular cytotoxicity (ADCC), complement-dependent cytotoxicity
(CDC) and generation of the anti-idiotypic network (3,4).
Moreover, the mAb therapies supplemented with additional agents,
like GM-CSF, IL-2 or retinoic acid are now accepted as a standard
treatment of high risk patients (5).
Several studies have evidenced that GD2-specific
antibodies inhibit tumor growth without involvement of the immune
system (6–8). The mechanisms of the
anti-proliferative effects of the antibodies are only partially
elucidated. In small cell lung cancer cell cultures anti-GD2
ganglioside monoclonal antibodies induced apoptosis through
reduction of the phosphorylation levels of focal adhesion kinase
(FAK) and the activation of p38 (6) as well as activation of c-Jun N
terminal kinase (JNK) (7).
Moreover, the anti-GD2 ganglioside mAbs were very efficient in
combination with anticancer drugs to exert enhanced cytotoxicity
against the afore-mentioned cells (7). Our studies showed that the anti-GD2
14G2a mAb decreases survival of IMR-32 human neuroblastoma cells in
a dose-dependent manner through induction of apoptosis and also
exerts a synergistic effect with doxorubicin and topotecan in
killing IMR-32 cells in culture (8). It was also recently shown by us that
the same mAb inhibits IMR-32 and LA-N-1 human neuroblastoma cells
survival in vitro, through significant decrease in
expression of all three Aurora kinases and phosphorylation
(9). Moreover, the Aurora A kinase
binding partners: P53, PHLDA1 and MYCN, were either upregulated as
in case of P53 and PHLDA1, or downregulated (MYCN) in the 14G2a
mAb-treated IMR-32 and CHP-134 human neuroblastoma cells
contributing to the observed decrease in cell viability (9). Finally, Cochonneau et al,
demonstrated that an O-acetyl-GD2 ganglioside specific mAb
treatment inhibited tumor growth of IMR-5 neuroblastoma cells in
vivo in a NOD/SCID mouse model in the absence of operating ADCC
and CDC mechanisms (10).
The phosphoinositide 3-kinase (PI3K), Akt and
mammalian target of rapamycin (mTOR) pathway is the most frequently
altered pathway in human tumors appearing as a central oncogenic
driver, fundamental to all cancer cells (11,12).
The pathway is prominently activated by many growth factors that
signal through receptor tyrosine kinases. PI3Ks are recruited to
receptor kinases and stimulate the conversion of
phosphatidylinositol-4,5-bisphosphate (PIP2) to
phosphatidylinositol-3,4,5-triphosphate (PIP3) that
provides a docking site for Akt, which thereby becomes activated
(13). The serine/threonine kinase
Akt/PKB regulates multiple biological processes including cell
survival, proliferation, growth and glycogen metabolism (13,14).
The serine/threonine kinase mTOR functions as two distinct
complexes in the PI3K signaling network (12,15).
mTOR complex 2 (mTORC2) phosphorylates key residues to activate Akt
and other kinases, thus regulating survival, metabolism and the
cytoskeleton. mTORC1 is a central regulator of cellular metabolism
and biosynthesis, promoting anabolic processes such as ribosome
biogenesis, translation and the synthesis of lipids and nucleotides
while suppressing catabolic processes such as autophagy and
lysosome biogenesis (15).
Inhibition of the PI3K/Akt/mTOR pathway has been the
subject of extensive efforts. Numerous inhibitors have been
developed that target almost every step of the pathway. Most of
these compounds are either in preclinical development, or in
clinical trials (13,16,17).
These include ATP-competitive, dual inhibitors of class I PI3K and
mTORC1/2, ‘pan-PI3K' inhibitors, which inhibit all four isoforms of
class I PI3K (α, β, δ and γ), isoform-specific inhibitors of the
various PI3Ks, allosteric and catalytic inhibitors of Akt and
ATP-competitive inhibitors of mTOR only.
Based on the vast amount of scientific data, it
becomes clear that the clinical application of single inhibitors
targeting only e.g., the PI3K/AKT/mTOR pathway shows limited
activity. Blockage of the pathway usually does not induce cancer
cell death, due to selection of compensatory pathways that maintain
survival and restore tumor growth (12). Therefore, combinations of agents
targeting multiple elements of various signaling networks may be
important for treatment of cancer patients.
The findings prompted us to investigate effects of
the 14G2a mAb on protein members of the PI3K/Akt/mTOR network in
neuroblastoma cells in vitro. There are numerous elements of
the pathway which have been downregulated upon the mAb addition,
including Akt, mTOR, p70 S6 and AMPK kinases. We observed transient
increase in PTEN, a suppressor of the pathway. Additionally, four
different PI3K/Akt/mTOR pathway inhibitors (LY294002, perifosine,
BEZ-235 and SAR245409) were used in combination with the monoclonal
antibody to determine neuroblastoma cell viability. We showed that
BEZ-235 was the most potent of the four drugs tested.
Materials and methods
Cell culture
Three GD2-positive human neuroblastoma cell lines
were used in experiments, i.e., IMR-32 (ATCC, USA, CCL-127), LA-N-1
(ECACC, UK, 06041201) and CHP-134 (ECACC, 06122002). No research
involving human subjects is reported. IMR-32 cells were grown in
EMEM medium with addition of 10% fetal calf serum, 1% non-essential
amino acid solution, 1 mM sodium pyruvate and 50 μg/ml gentamicin.
LA-N-1 cells were grown in EMEM/F-12 (1:1) medium with 10% fetal
calf serum, 1% non-essential amino acid solution, and 50 μg/ml
gentamicin. CHP-134 cells were grown in RPMI-1640 medium with 10%
fetal calf serum and 50 μg/ml gentamicin. All cell lines were
cultured at 37°C in a 5% CO2 atmosphere and were
routinely tested for mycoplasma contamination and were found
negative (Lonza, USA).
Antibody purification
Two mouse monoclonal antibodies, i.e., 14G2a and
PK136 were purified from hybridoma culture supernatants using the
HiTrap Protein G HP column (GE Healthcare Bio-Sciences AB, Sweden)
according to the manufacturer's protocol, dialyzed against PBS, and
assayed for protein content using a BCA method (Sigma-Aldrich,
Poland) (detailed in ref. 9). The
hybridoma cell line producing the 14G2a mAb (IgG2a), binding to
GD2, was provided by Dr R.A. Reisfeld (Scripps Institute, La Jolla,
CA, USA) and the hybridoma cell line clone PK136 (a mouse
isotype-matched control) was purchased from the ATCC, USA. The
PK136 mAb recognizes an antigen expressed on murine NK cells
isolated from some strains of mice (NK1.1) that is not expressed on
non-lymphoid cells.
Antibody and drug treatment
Cells were incubated with the 14G2a mAb or PBS
(control cells) at concentration of 40 μg/ml for 1 h at 4°C and
then seeded on 96-well plates (2×104 cells/100 μl/well
for IMR-32, LA-N-1, and 0.5×104 cells/100 μl/well for
CHP-134, BD Falcon, Belgium) and incubated for 72 h. Concentration
of the 14G2a mAb used in our in vitro model (40 μg/ml), can
be reached in patients sera during immunotherapy. Despite the mAb
side effects, clinical efficacy of the mAb in the treatment of
neuroblastoma patients have been already reported (3). For viability tests, cellular ATP
contents were measured (ATPlite - luminescence ATP detection assay
system, Perkin-Elmer, USA). For protein isolation cells were
cultured in 6-well plates (1×106 cells in 5 ml of
culture medium for IMR-32 and LA-N-1 cells, and 0.25×106
cells in 5 ml of culture medium for CHP-134 cells) and treated for
a given time with the 14G2a mAb or PBS, as described above.
In some experiments the inhibitors perifosine
(KRX-0401, S1037, Selleck, Germany), BEZ-235 (NVP-BEZ235, S1009,
Selleck), SAR245409 (XL765, S1523, Selleck) and LY294002 (#9901,
Cell Signaling, Lab-JOT, Poland) were used on IMR-32, LA-N-1 and
CHP-134 cells. Tested ranges of concentrations of the inhibitors
were perifosine (1–20 μM), BEZ-235 (0.01–2 μM), SAR245409 (1–100
μM) and LY294002 (2.5–80 μM). Inhibitors were added to cell
cultures for 72 h. Control cells were treated with equivalent
volume of DMSO or water (solvents for the inhibitors). In some
experiments, cells were first treated with 14G2a (40 μg/ml) (or
PBS) for 1 h on ice, and then inhibitors or diluents were
added.
RNA isolation and quantitative
RT-PCR
Total RNA samples were isolated from IMR-32 cells
using TRI Reagent® as described in the manufacturer's
protocol (Molecular Research Center, Inc., USA; detailed in ref.
9). Briefly, 1 μg of total RNA was
reverse-transcribed using Oligo(dT)15 Primer (Invitrogen, Poland)
and M-MLV reverse transcriptase (Invitrogen). cDNA was analyzed
using real-time PCR (Rotor-Gene 3000 system, Corbett Life Science,
Australia). A KAPA SYBR FAST qPCR Master Mix (Kapa Biosystems, USA)
was used in reactions. For sample normalization, the amount of
eukaryotic translation elongation factor 2 (eEF2) cDNA was
measured. Following primers were used: P27 (5′-GAAGCGACCTGCAACCGAC
GATT-3′, 5′-CAGGCTTCTTGGGCGTCTGCTC-3′); eEF-2
(5′-GGTGCAGTGCATCATCGAGGAGTC-3′, 5′-TCGCGG TACGAGACGACCGG-3′).
Sample quantification was performed in triplicates using the ΔΔCt
relative quantification method.
Protein extract isolation and proteome
array analysis
The Proteome Profiler Antibody Array from R&D
Systems (Human Phospho-Kinase Array kit, ARY003, UK) was applied to
simultaneously detect relative levels of multiple phosphorylated
proteins in a single sample. Ca 250 μg of proteins was used for
each nitrocellulose membrane. The assay was performed according to
the manufacturer's instructions. Chemiluminescence was detected
with a Tecan Infinite M200 microplate reader and the positive
signals were identified and quantified.
Whole cell extracts were prepared using the TRI
Reagent® method or lysis buffer from the Human
Phospho-Kinase Array kit (see above). Nuclear and cytoplasmic
fractions were isolated with a method described by Suzuki et
al (18).
Immunoblotting
Western blot analyses were performed as previously
described (9). The first group of
antibodies (Ab) was from Cell Signaling Technology (USA), i.e.,
phospho-Akt Pathway Antibody Sampler kit (#9916), mTOR Substrates
Antibody Sampler kit (#9862), and AMPK and ACC Antibody Sampler kit
(#9957). Dilutions of all antibodies were 1:1,000 for:
anti-phospho-Akt (Ser473) Ab, #4060; anti-phospho-Akt (Thr308) Ab,
#2965; anti-Akt (pan) Ab, #4691; anti-phospho-c-Raf (Ser259) Ab,
#9421; anti-phospho-GSK-3β (Ser9) Ab, #9323; anti-phospho-PTEN
(Ser380) Ab, #9551; anti-phospho-PDK1 (Ser241) Ab, #3438;
anti-phospho-mTOR (Ser2448) Ab, #5536; anti-mTOR Ab, #2983;
anti-phospho-p70 S6 kinase (Thr389) Ab, #9234; anti-phospho-p70 S6
kinase (Ser371) Ab, #9208; anti-phospho-4E-BP1 (Thr37/46) Ab,
#2855; anti-phospho-AMPKα (Thr172) Ab, #2535; anti-AMPKα Ab, #2603;
anti-AMPKβ1/2 Ab, #4150; anti-phospho-acetyl-CoA carboxylase
(Ser79) Ab, #3661, anti-TBP Ab, #8515 and acetyl-CoA carboxylase
Ab, #3676. The other antibodies were from Sigma, i.e., anti-GAPDH,
G8795 (1:40,000). The following HRP-conjugated antibodies were
used: anti-rabbit IgG antibodies (Cell Signaling), #7074 (1:2,000)
or anti-mouse IgG antibodies (Sigma), A-9044 (1:40,000).
Immunoreactive bands were visualized by an enhanced
chemiluminescence method (Immobilon Western HRP Substrate,
Millipore, Poland) according to the manufacturer's protocol and
their intensity was quantified by densitometric scanning (Quest
Spot Cutter, Quantity One Analysis software, Bio-Rad).
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), or TATA-box
binding protein (TBP) reference proteins were used for
normalization of protein samples signals. In figures, levels of the
protein expression in control samples were set as 1.
Flow cytometry analysis of the GD2
ganglioside content
FACS analyses were routinely used to measure
contents of GD2 on neuroblastoma cells using the 14G2a mAb. Cells
were incubated for 40 min at 4°C with 1 μg of the 14G2a mAb or the
PK136 mAb in 2% BSA/PBS and then the cells were washed with 2%
FBS/PBS. The binding of antibodies was detected with mouse
Ig-specific FITC-conjugated goat F(ab')2 fragments using
flow cytometry (BD™ LSR II with BD FACSDiva software, BD
Biosciences). Prior to cell collection propidium iodide was added
and populations of alive (propidium iodide-negative) cells were
analyzed for GD2 content, based on the staining with the PK136 mAb
(a negative control) and the 14G2a mAb.
Statistical analyses
Data in the graphs and Table II are presented as means ± SEM (a
standard error of the mean). All experiments were repeated at least
three times. We used one-way ANOVA with repeated measurements to
test for statistically significant differences in means in
experiments with more than two independent groups. IC40
values, i.e., concentrations of inhibitors causing decrease of
cellular ATP contents to 40% of control cells (treated with
appropriate diluents) were obtained from equations of regression
curves applied to mean ATP assay results (of three to four
independent experiments). Uncertainty of each IC40 value
was calculated from the fit parameters of the given regression
curve with application of the law of error propagation. We applied
series of pairwise tests (t-test), comparing, e.g., means of
control and treated cells with following p-values:
*p<0.05, **p<0.01,
***p<0.001. Statistical analyses were performed with
R software (R version 2.15.1 Patched) and Excel (Microsoft,
USA).
 | Table IIIC40 values calculated for
single agent and combined treatment of neuroblastoma cell
lines. |
Table II
IC40 values calculated for
single agent and combined treatment of neuroblastoma cell
lines.
|
IC40a (inhibitor) |
IC40a (inhibitor + 14G2a) | Fold change |
|---|
| LY294002 |
| IMR-32 | 34.72 (±9.57) | 4.12 (±2.66) | 8.4 |
| LA-N-1 | 26.51 (±5.09) | 14.86 (±1.00) | 1.8 |
| CHP-134 | 13.98 (±2.77) | 0.79 (±0.79) | 17.8 |
| Perifosine |
| IMR-32 | 12.53 (±0.01) | 8.248 (±2.09) | 1.5 |
| LA-N-1 | 11.12 (±1.00) | 10.39 (±0.34) | 1.1 |
| CHP-134 | 10.33 (±1.38) | 4.32 (±0.12) | 2.4 |
| BEZ-235 |
| IMR-32 | 1.21 (±0.31) | 0.17 (±0.06) | 7.2 |
| LA-N-1 | 0.18 (±0.26) | 0.16 (±0.03) | 1.2 |
| CHP-134 | 0.16 (±0.03) | 0.07 (±0.01) | 2.4 |
| SAR245409 |
| IMR-32 | 95.75 (±15.30) | 73.34 (±2.41) | 1.3 |
| LA-N-1 | 93.75 (±17.23) | 88.30 (±20.78) | 1.1 |
| CHP-134 | 85.67 (±2.72) | 38.28 (±10.29) | 2.2 |
Results
Key intracellular pathways are affected
in the 14G2a-treated IMR-32 cells
The presence of GD2 ganglioside on the cell lines
used in experiments was routinely tested by flow cytometry and
similar results demonstrating high (ca 95%) GD2 ganglioside content
were obtained (data not shown).
Quantitative proteome profiling performed on
cellular extracts revealed significant temporal changes in
phosphorylation of several intracellular proteins isolated from the
14G2a mAb-treated IMR-32 cells (Table
I). It was shown that the Akt/mTOR pathway is affected by the
14G2a mAb added at 2, 4, 8 and 24 h to the cultured cell line. Some
proteins became hyperphosphorylated early up to 4 h upon the 14G2a
mAb treatment (β-catenin, MEK1/2, ERK1/2, STAT5a/b and AMPKα2),
while others were phosphorylated only at 2 h (MSK1/2, JNKpan,
AMPKα1 and p38α). Moreover, such proteins as Hck and Fyn were
hyperphosphorylated later, with maximum at 8 h.
| Table IChanges in protein phosphorylation in
anti-GD2 ganglioside 14G2a mAb-treated IMR-32 human neuroblastoma
cells. |
Table I
Changes in protein phosphorylation in
anti-GD2 ganglioside 14G2a mAb-treated IMR-32 human neuroblastoma
cells.
| Protein name | Phosphorylation
sitea | 2 h | 4 h | 8 h | 24 h |
|---|
| β-catenin | - | 209 | 153 | 70 | 47 |
| MSK1/2 | S376/S360 | 190 | 92 | 38 | 57 |
| JNKpan | T183/Y185,
T221/Y223 | 187 | 40 | 60 | 95 |
| MEK1/2 | S218/S222,
S222/S226 | 184 | 123 | 31 | 89 |
| STAT5a | Y699 | 177 | 194 | 57 | 70 |
| AMPKα1 | T174 | 176 | 78 | 37 | 72 |
| p38α | T180/Y182 | 174 | 67 | 20 | 9 |
| STAT5b | Y699 | 168 | 157 | 52 | 56 |
| STAT5a/b | Y699 | 161 | 143 | 57 | 49 |
| ERK1/2 | T202/Y204,
T185/Y187 | 159 | 231 | 30 | 29 |
| AMPKα2 | T172 | 154 | 138 | 38 | 74 |
| STAT6 | Y641 | 153 | 101 | 50 | 43 |
| Fgr | Y412 | 141 | 77 | 37 | 29 |
| GSK-3α/β | S21/S9 | 133 | 66 | 32 | 75 |
| STAT3 | Y705 | 130 | 104 | 37 | 45 |
| Akt | S473 | 129 | 102 | 36 | 45 |
| STAT2 | Y689 | 127 | 108 | 47 | 80 |
| HSP27 | S78/S82 | 127 | 72 | 9 | 65 |
| Lck | Y394 | 108 | 81 | 15 | 29 |
| CREB | S133 | 105 | 92 | 37 | 54 |
| FAK | Y397 | 105 | 91 | 35 | 30 |
| Yes | Y426 | 103 | 114 | 37 | 47 |
| Lyn | Y397 | 100 | 101 | 31 | 96 |
| eNOS | S1177 | 98 | 51 | 58 | 76 |
| c-Jun | S63 | 97 | 79 | 75 | 145 |
| STAT1 | Y701 | 97 | 37 | 93 | 54 |
| Chk-2 | T68 | 91 | 89 | 58 | 42 |
| STAT4 | Y693 | 91 | 48 | 70 | 63 |
| RSK1/2 | S221 | 83 | 46 | 69 | 47 |
| mTOR | S2448 | 80 | 105 | 88 | 67 |
| p27 | T157 | 79 | 34 | 37 | 39 |
| Hck | Y411 | 78 | 127 | 260 | 123 |
| RSK1/2/3 | S380 | 73 | 41 | 56 | 40 |
| p70 S6 | T421/S424 | 73 | 39 | 80 | 52 |
| p53 | S46 | 70 | 37 | 51 | 66 |
| p27 | T198 | 69 | 29 | 27 | 23 |
| p70 S6 | T229 | 68 | 47 | 80 | 44 |
| Src | Y419 | 67 | 120 | 106 | 89 |
| p53 | S392 | 67 | 55 | 64 | 60 |
| p53 | S15 | 65 | 39 | 49 | 62 |
| paxillin | Y118 | 64 | 49 | 41 | 50 |
| Akt | T308 | 63 | 38 | 69 | 78 |
| Fyn | Y420 | 61 | 108 | 177 | 105 |
| PLCγ-1 | Y783 | 61 | 36 | 48 | 51 |
| Pyk2 | Y402 | 57 | 47 | 40 | 32 |
| p70 S6 | T389 | 43 | 13 | 35 | 16 |
The PI3K/Akt/mTOR pathway has an important role in
cell metabolism, growth, migration, survival and angiogenesis
(16) and therefore changes of the
network after the 14G2a mAb treatment were analyzed in detail.
Besides this pathway, the other signaling routes were obviously
affected upon targeting of GD2 with the mAb: β-catenin involved in
Wnt signaling as the transcription co-activator (19), was hyperphosphorylated and
therefore destabilized. This leads to inhibition of the canonical
Wnt/β-catenin pathway. The mitogen-activated protein kinases
(MAPKs), have been implicated in a variety of cellular processes
such as proliferation, differentiation, motility, stress response,
apoptosis and survival (20). The
kinases include ERK1/2, JNK1–3 and p38, which became
hyperphosphorylated early upon the mAb treatment (Table I) and next their phosphorylation
was inhibited leading to decrease in their activity. Additionally,
specific MAPK-activated protein kinases, known to amplify the MAPK
signal, such as members of the RSK and MSK family were also
initially hyper- and next hypo-phosphorylated upon the mAb addition
(Table I). c-jun N-terminal kinase
(JNK) is a kinase activated by cellular stress whereby leading to
apoptosis (21). We have shown
that at 2 h of incubation of IMR-32 cells with the 14G2a mAb, the
kinase is activated at key amino acid residues by nearly doubled
phosphorylation (Table I). The
result is similar to other data on JNK activation by cisplatin in
the anti-GD2 ganglioside mAb-treated small cell lung carcinoma
(7). The protein is also activated
by arsenium oxide in acute myelocytic leukemia, leading to cell
death (22). One of
mitogen-activated protein kinases (MAPK), p38α, known to respond to
stress stimuli (23), becomes also
activated in the 14G2a-treated IMR-32 cells at 2 h through
increased phosphorylation exceeding the control >70% (Table I). Similar result was observed in
small cell lung carcinoma treated with an anti-GD2 ganglioside
antibody (6). Increase in p38α
activity was also connected with loss of attachment to collagen
type I in colon cancer cells and their cell death through anoikis
(24).
The signal transducer and activator of transcription
(STAT) family of transcription factors, known from their role in
signaling pathways utilized by a large number of cytokines, growth
factors and hormones (25),
represented by STATs 1–6, was either early (STAT1 and STAT4), or
late (STAT 2, 3, 5 and 6) dephosphorylated and therefore inhibited.
Focal adhesion kinase (FAK), regulates cellular adhesion and
apoptosis, therefore diminished phosphorylation at Tyr397 (Table I) resulting in increased
detachment, decreased cell viability, and increased apoptosis
(26). Based on the experiments we
can conclude that the mAb affects several pivotal signaling routes
that drive or influence the malignant phenotype of the cells.
Akt pathway is inhibited in the
14G2a-treated cells
Our proteomic analysis revealed profound inhibition
of the pro-mitotic mTOR pathway upon the GD2 ganglioside-directed
mAb addition through dephosphorylation of Akt, mTOR, and p70 S6
proteins that leads to decrease in their activities. The pathway
appears to be important in neuroblastoma, therefore we have further
characterized changes in levels and phosphorylation of the
afore-mentioned proteins not only in IMR-32 cells but also in
CHP-134 and LA-N-1 cells. Akt is activated through a dual
phosphorylation mechanism at Thr308 located within its activation
loop by PDK1 and at Ser473 by mTORC2. Activated Akt phosphorylates
a large number of downstream targets such as mTOR and GSK-3 leading
to either promotion of cell growth and G1 cell cycle progression,
or promotion of glycogen metabolism and regulation of Wnt
signaling, respectively (14).
Although Akt protein levels did not change greatly
in the IMR-32 and CHP-134 cells treated with the mAb (Fig. 1A and D), its phosphorylation on key
amino acid residues, Ser473 and Thr308, was statistically
significantly and severely decreased in the IMR-32 cells (Fig. 1B and C) and to smaller extent in
CHP-134, in which p-Thr308 was significantly lower only at 24 h
(Fig. 1E and F). We have shown
that the kinase is dephosphorylated at Thr308 already after 2 h of
incubation of IMR-32 cells with the antibody, and next after 6, 24
and 48 h the effect is more evident reaching ca 20% of the control
at 48 h (Fig. 1C), while in
CHP-134 dephosphorylation is reaching only ca 80% of the control
(Fig. 1F). Interestingly,
phosphorylation of PKD1 itself at Ser241 has not been clearly
changed (as shown in Fig. 3E), in
agreement with our major conclusion on the pathway inhibition under
the 14G2a mAb treatment. Dephosphorylation of Ser473 in Akt takes
place in the IMR-32 cells at 2 h and after 48 h reaching only 20%
of the control (Fig. 1B), while in
the CHP-134 cells the lowest value constitutes ca 65% of the
control (Fig. 1E), this result on
IMR-32 is similar to our proteomic array in which we have also
found significant dephosphorylation of Akt at 8 and 24 h (Table I). Dephosphorylation of Thr308 and
Ser473 by the intracellular phosphatases, protein phosphatase 2
(PP2) and PH domain leucine-rich repeat protein phosphatase
(PHLPP), respectively, terminates Akt signaling (14), and we found similar effect of the
14G2a mAb on the treated cells.
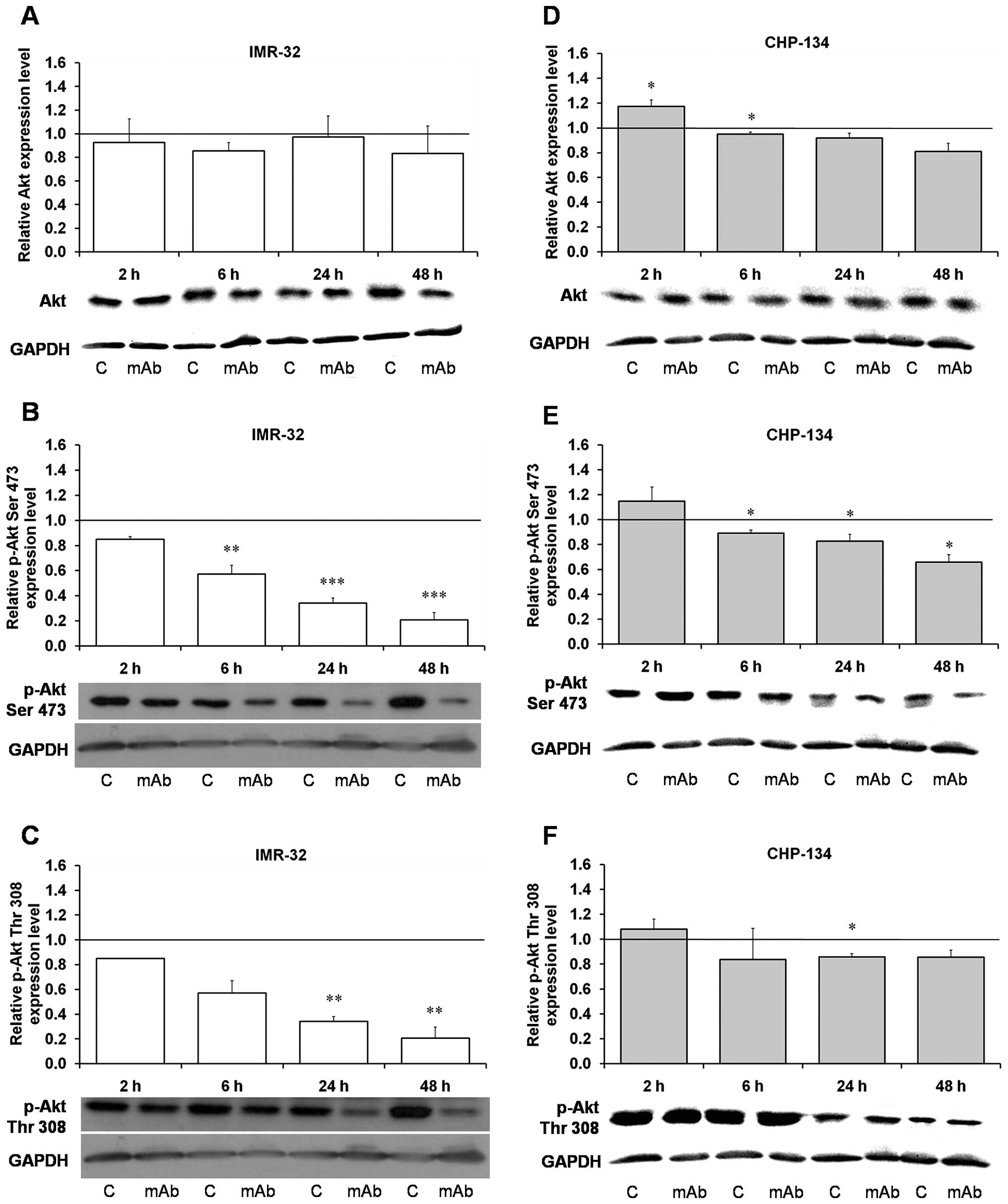 | Figure 1Effect of the 14G2a mAb on Akt
expression and phosphorylation. Akt and phosphorylated Akt (p-Akt,
Ser473 and Thr308) were measured at 2, 6, 24 and 48 h, and
normalized to GAPDH. Mean values of three separate experiments
obtained for the 14G2a mAb-treated IMR-32 (A–C) and CHP-134 (D–F)
cells are presented (± SEM), and calculated versus control values,
set as 1 (black baseline). ANOVA shows statistically significant
changes of protein content in time in IMR-32 cells: p-Akt Ser473
[F(3,8)=61.26, p<0.0001], p-Akt Thr308 [F(3,8)=12.08, p=0.0024]
and in CHP-134 cells: Akt [F(3,9)=12.02, p=0.0017], p-Akt Ser473
[F(3,9)=8.50, p=0.0054]. ANOVA shows no statistically significant
changes of expression in time for the rest of the proteins.
P-values for t-test were as follows: *p<0.05,
**p<0.01, ***p<0.001. Below each chart
representative immunoblottings are presented: C, control cells;
mAb, mAb-treated cells (40 μg/ml). |
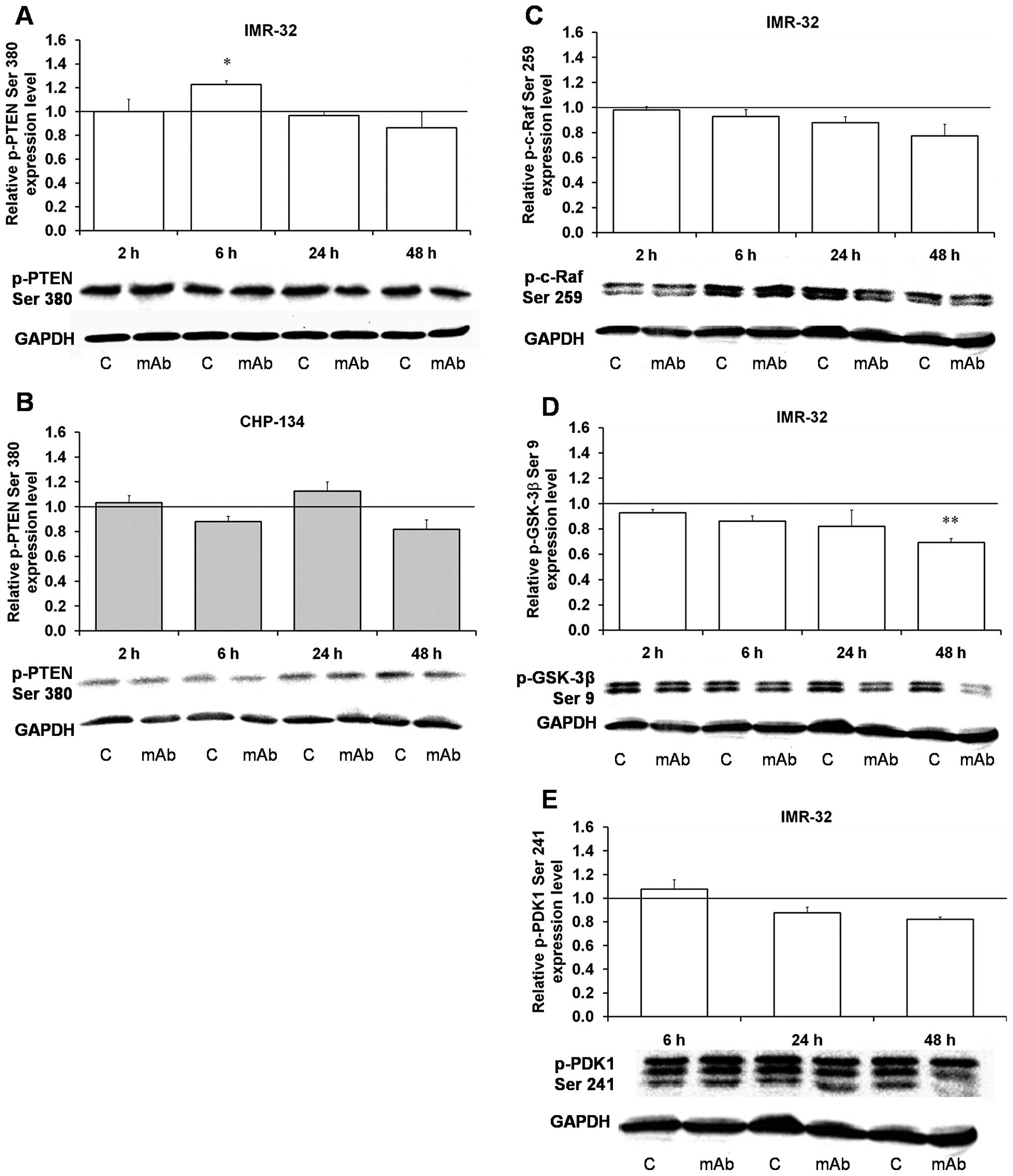 | Figure 3Effect of the 14G2a mAb on PTEN,
c-Raf, Gsk-3β and PDK-1 phosphorylation. Phosphorylated PTEN
(p-PTEN, Ser380), phosphorylated c-Raf (p-c-Raf Ser259) and
phosphorylated GSK-3β (p-GSK-3β Ser9) were measured at 2, 6, 24 and
48 h, and normalized to GAPDH. Phosphorylated PDK1 (p-PDK1 Ser241)
was measured at 6, 24 and 48 h. Mean values of three separate
experiments obtained for the 14G2a mAb-treated IMR-32 (A, C and D)
and CHP-134 (B) are presented, and calculated versus control
values, set as 1 (black baseline). For p-PDK1 mean values of two
separate experiments for the IMR-32 cells are shown (E). ANOVA
shows statistically significant changes of p-PTEN protein content
in time in the IMR-32 cells [F(3,8)=4.57, p=0.0381] and in the
CHP-134 cells [F(3,9)=21.21, p=0.0002]. ANOVA shows no
statistically significant changes of expression in time for the
rest of the proteins. P-values for t-test were as follows:
*p<0.05, **p<0.01,
***p<0.001. Below each chart representative
immunoblottings are presented: C, control cells; mAb, mAb-treated
cells (40 μg/ml). |
P27 is a negative regulator of cell cycle
progression in G1-S phase through inhibition of cyclins E-Cdk2 and
D-Cdk4 and cyclin-dependent kinases (27). Akt kinase is able to phosphorylate
P27 at Thr157 and Thr198 leading to cytoplasmic retention of the
protein (27,28). We found that P27 protein
phosphorylation at both amino acids is inhibited as early as at 2 h
in the 14G2a mAb-treated IMR-32 cells (Table I) and the effect is deepened up to
24 h. Q-RT-PCR revealed an early decrease and next at 48 h a return
to a control level in P27 mRNA content in the IMR-32 cells
treated with the 14G2a mAb for 6–48 h (Fig. 2A). Moreover, the protein total
cellular levels were increased in the IMR-32 and LA-N-1 cells
although statistically significant increase was observed only in
the IMR-32 cells at 24 and 48 h (Fig.
2B and C) probably due to increased transcript stability and/or
efficient translation. Finally, analysis of P27 protein expression
in cytoplasmic fraction of the IMR-32 cells shows decreased levels
at 24 and 48 h (Fig. 2D) with
concomitant increase in nuclear content (Fig. 2E), further supporting our
conclusion of possible participation of P27 in cell cycle
inhibition in the analyzed model.
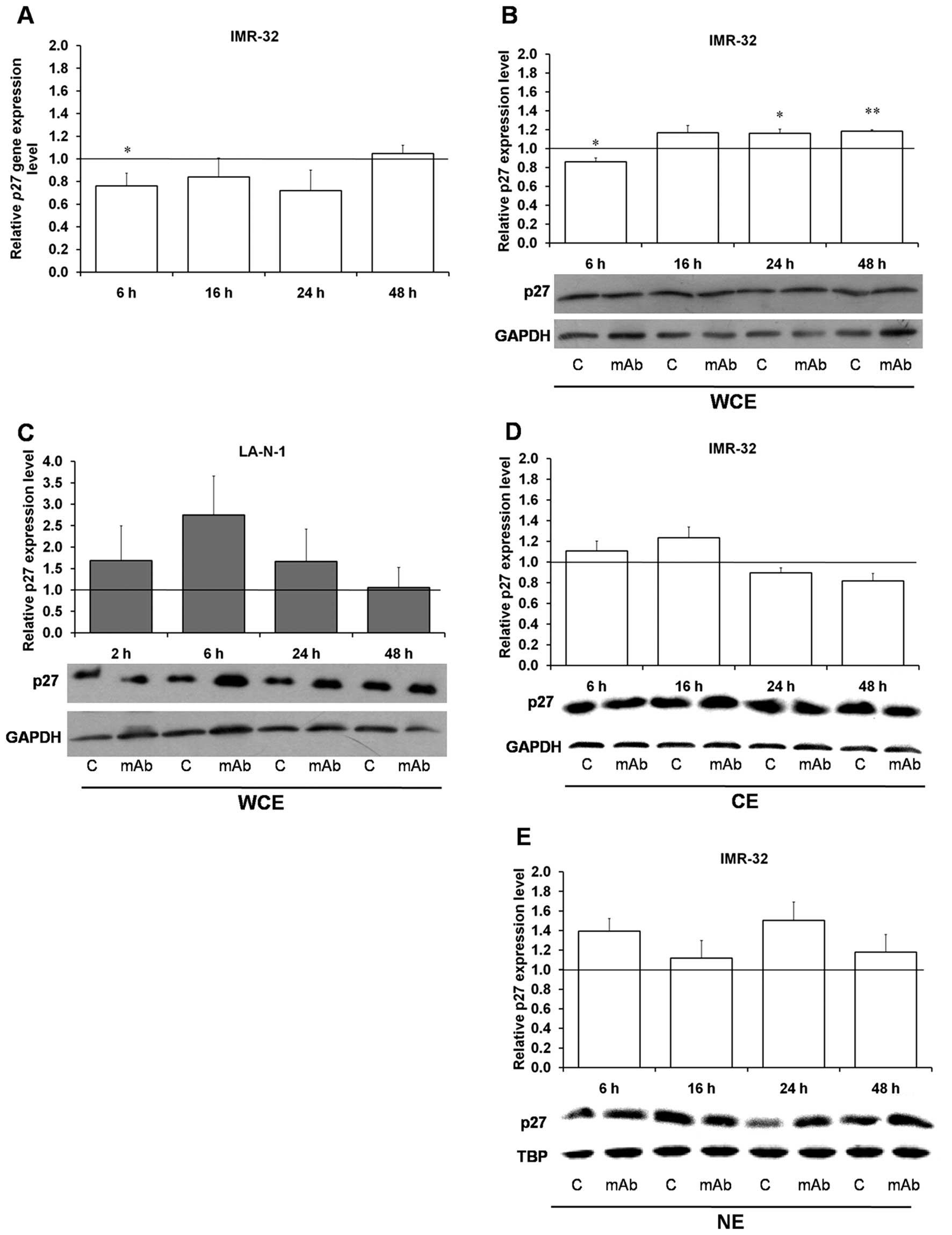 | Figure 2Effect of the 14G2a mAb on P27
gene and P27 protein expression. P27 gene expression level
was measured by qRT-PCR in the IMR-32 cell line treated with the
14G2a mAb (40 μg/ml) for 6, 16, 24 and 48 h (A). EF-2 cDNA was used
as a reference. P27 gene expression in control cells equals
1 (black baseline). Data are presented as means of triplicates from
three independent experiments (± SEM). P27 protein content was
measured in whole cell (WCE) (B), cytoplasmic (CE) (D) and nuclear
(NE) (E) extracts at 6, 16, 24 and 48 h after 14G2a addition (40
μg/ml) into culture media of the IMR-32 cells and normalized to
GAPDH levels (for WCE and CE), or TBP (for NE). P27 protein content
in LA-N-1 (C) was measured in WCE. Mean values of three separate
experiments are presented, and calculated versus control values,
set as 1 (black baseline). ANOVA shows statistically significant
changes of P27 gene expression in time in the IMR-32 cells
[F(3,9)=4.29 p=0.0387]. ANOVA shows also statistically significant
changes of protein content in time in the IMR-32 cells: in WCE
[F(3,8)=10.11, p=0.0043] and in CE [F(3,6)=5.36, p=0.0392]. ANOVA
shows no statistically significant changes of expression in time
for the rest of the proteins. P-values for t-test were as follows:
*p<0.05, **p<0.01,
***p<0.001. Below each chart representative
immunoblottings are presented: C, control cells; mAb, mAb-treated
cells (40 μg/ml). |
Phosphatase and tensin homolog (PTEN), a lipid
phosphatase (mostly for phosphatidylinositol-3,4,5-triphosphate -
PIP3) that catalyzes the dephosphorylation of
PIP3, is a major negative regulator of PI3K and Akt
signaling (11). Thus, the
phosphatase counteracts PI3K by degrading its product,
PIP3. PTEN gene inactivation can be responsible
for aberrant activation of the PI3K signaling in prostate cancer
(29,30). We have shown that neither in
IMR-32, nor in CHP-134 cells, the PTEN phosphatase phosphorylation
at Ser380 is statistically significantly changed upon the 14G2a mAb
treatment except for an increase observed in the IMR-32 cells at 6
h (Fig. 3A and B). PTEN
phosphorylation at Ser380 constitutes a mechanism of PTEN
inactivation in gastric cancer (31), therefore we can conclude that in
the mAb-treated human neuroblastoma cells used in the study, the
phosphatase is still active. Additionally, we have shown that
c-Raf, a member of MAPKKKs (32),
is slightly dephosphorylated at Ser259 (an inhibitory site) in the
IMR-32 cells (Fig. 3C). The
reduction of the phosphorylation state of c-Raf at Ser259 is
following the inhibition of Akt in PC3 cells (a human prostatic
adenocarcinoma cell line) (32).
Akt regulates the storage of glucose in the form of
glycogen by phosphorylating glycogen synthase kinase (GSK)-3β at
Ser9 and -3α at Ser21, whereby blocking its kinase activity.
Inhibitory phosphorylation of GSK-3β and 3α is not only promoting
glycogen metabolism but also cell cycle progression and regulation
of Wnt signaling (14).
Significant inhibition of phosphorylation of GSK-3β at Ser9 found
in the 14G2a mAb-treated IMR-32 cells (Table I and Fig. 3D) follows similar changes as the
phosphorylated Akt and therefore can result in decreased
proliferation of neuroblastoma cells.
mTOR pathway is inhibited in the
14G2a-treated cells
Activation of PI3K and Akt results in mTOR
phosphorylation at Ser2448 (33),
which next stimulates two major pathways leading to increased
protein synthesis. The mTOR phosphorylates eukaryotic translation
initiation factor 4E-binding protein 1 (4E-BP1), responsible for
inhibition of eukaryotic translation initiation factor 4E (eIF-4E),
which leads to release of 4E-BP1 and thus suppression of
inhibition. The other pathway stimulated by mTOR involves
activating phosphorylation of serine-threonine kinase 70S6 (p70
S6). The kinase phosphorylates proteins of ribosomal subunit 40S,
causing recruitment of the subunit to active polysomes (34).
We found that in the 14G2a-treated IMR-32 cells,
mTOR protein expression is decreased to ca 60% of the control,
although in the CHP-134 and the LA-N-1 cells the decrease is
smaller (Fig. 4A, C and E).
However, mTOR phosphorylation at Ser2448, responsible for its
activity, decreases statistically significantly in all three cell
lines, with the most pronounced inhibition in the IMR-32 cells at
48 h (Fig. 4B, D and F). The
latter result was not so pronounced in our proteomic arrays, and
only 33% inhibition of Ser2448 phosphorylation was observed in
IMR-32 cells at 24 h (Table
I).
 | Figure 4Effect of the 14G2a mAb on mTOR
expression and phosphorylation. mTOR and phosphorylated mTOR
(p-mTOR, Ser2448) were measured at 2, 6, 24 and 48 h, and
normalized to GAPDH. Mean values of three or four separate
experiments obtained for the 14G2a mAb-treated IMR-32 (A and B) and
CHP-134 (C and D) and LA-N-1 cells (E and F) are presented, and
calculated versus control values, set as 1 (black baseline). ANOVA
shows statistically significant changes of mTOR content in time in
the IMR-32 cells [F(3,8)=42.15, p<0.0001] and the LA-N-1 cells
[F(3,9)=4.01, p=0.0458] and changes of p-mTOR content in time in
the LA-N-1 cells [F(3,9)=12.62, p=0.0014]. ANOVA shows no
statistically significant changes of expression in time for the
rest of the proteins. P-values for t-test were as follows:
*p<0.05, **p<0.01,
***p<0.001. Below each chart representative
immunoblottings are presented: C, control cells; mAb, mAb-treated
cells (40 μg/ml). |
P70 ribosomal S6 kinase (S6K1), a major substrate of
the mTOR, exists as two protein isoforms, p85 and p70, because of
the use of alternative ATG start codons. There are several
phosphorylation sites in p70 S6 kinase, including Thr389 and
Thr229, vital for its activity (35). Phosphorylation of Thr421 and Ser424
in autoinhibitory domain of the kinase is also contributing to its
increased activity (36). In our
studies, three sites of phosphorylation on kinase p70 S6 at Thr389,
Thr229 and Thr421/Ser424 are characterized by early and significant
decrease, especially at Thr389 in the IMR-32 cells treated with the
mAb (Table I) and similar
spectacular inhibition was observed in the same cell line and
additionally in the LA-N-1 cells at Thr389 for 70 and 85 kDa
isoforms of the enzyme (Fig. 5A, C, E
and G). Phosphorylation at Ser371, indispensable for Thr389
phosphorylation (37), was also
statistically meaningfully downregulated in the IMR-32 cells
(Fig. 5B and D), while in the
LA-N-1 cells only insignificant decrease was observed (Fig. 5F and H).
 | Figure 5Effect of the 14G2a mAb on p70 S6
phosphorylation. Phosphorylated p70 S6 (p-p70 S6, Thr389 and
Ser371) 70 and 85 kDa forms were measured at 2, 6, 24 and 48 h, and
normalized to GAPDH. Mean values of three separate experiments
obtained for the 14G2a mAb-treated IMR-32 (A–D) and LA-N-1 (E–H)
are presented, and calculated versus control values, set as 1
(black baseline). ANOVA shows statistically significant changes of
p-p70 S6 Thr389 content in time in the IMR-32 cells for 70 kDa
[F(11,34), p=0.0021] and 85 kDa isoform [F(3,9)=5.38, p=0.0214].
ANOVA also shows statistically significant changes of p-p70 S6
Ser371 content in time in the LA-N-1 cells for 85 kDa isoform
[F(3,9)=3.93, p=0.0479]. ANOVA shows no statistically significant
changes of expression in time for the rest of the proteins.
P-values for t-test were as follows: *p<0.05,
**p<0.01, ***p<0.001. Below each chart
representative immunoblottings are presented: C, control cells;
mAb, mAb-treated cells (40 μg/ml). |
4E-BP1 phosphorylation at Thr37/46 by mTORC1 leads
to release of its binding protein, eIF-4E, and suppression of
inhibition (38,39). The phosphorylated 4E-BP1 was
statistically significantly decreased at 24 h in the IMR-32 cells
but the decrease was observed already at 6 h (Fig. 6). Dephosphorylation of the protein
may indicate that initiation of translation is inhibited. It is not
exactly clear how the mTORC1/4E-BP1/eIF4E axis contributes to
cancer. It is possible that eIF4E affects cell proliferation and
tumorigenesis by promoting the translation of specific mRNAs coding
for pro-oncogenic proteins regulating cell survival, cell cycle
progression, angiogenesis, energy metabolism and metastasis
(15).
 | Figure 6Effect of the 14G2a mAb on 4E-BP1
phosphorylation. Phosphorylated 4E-BP1 (p-4E-BP1, Thr37/46) was
measured at 2, 6, 24 and 48 h, and normalized to GAPDH. Mean values
of three separate experiments obtained for the 14G2a mAb-treated
IMR-32 cells are presented, and calculated versus control values,
set as 1 (black baseline). ANOVA shows no statistically significant
changes of 4E-BP1 expression in time. P-values for t-test were as
follows: *p<0.05, **p<0.01,
***p<0.001. Below the chart representative
immunoblotting is presented: C, control cells; mAb, mAb-treated
cells (40 μg/ml). |
AMP-activated protein kinase (AMPK) is
inhibited upon the 14G2a mAb treatment
AMPK is one of the principal regulators of mTOR
activation and the main energy-saving intracellular enzyme
activated by the increased AMP/ATP ratio (40). The AMPK consists of catalytic α and
regulatory β and γ subunits (41).
The β1 subunit is partially phosphorylated at three sites,
Ser24/25, Ser182 and Ser108. The α1 subunit Thr172 is a major,
although not exclusive, site of both basal and stimulated
phosphorylation by an upstream AMPK kinase. AMPK phosphorylates a
number of protein substrates including key enzymes involved in the
control of carbohydrate and lipid metabolism, like acetyl-CoA
carboxylase (ACC).
Early (at 2 h upon the 14G2a antibody addition)
hyperphosphorylation of important amino acid residues
(Thr172/Thr174) of the catalytic subunit α of AMPKα1/2 kinase,
followed by severe decrease in phosphorylation were observed in the
IMR-32 cells (Table I), while
decrease in AMPKα isoform protein content was detectable at 2 h (as
shown in Fig. 8). Phosphorylation
of Ser108 of AMPKβ isoform was statistically significantly
decreased in the IMR-32 reaching only 20% of the control level at
48 h (Fig. 7A), while in the
CHP-134 it was not changed (Fig.
7D). Thirty-eight and 34 kDa isoforms of the protein were also
statistically significantly decreased at 48 h in the IMR-32 cells
(Fig. 7B and C), while in CHP-134
the protein level was not changed for the 38 kDa isoform (Fig. 7E) and a significant decrease was
found for the 34 kDa isoform only at 48 h (Fig. 7F).
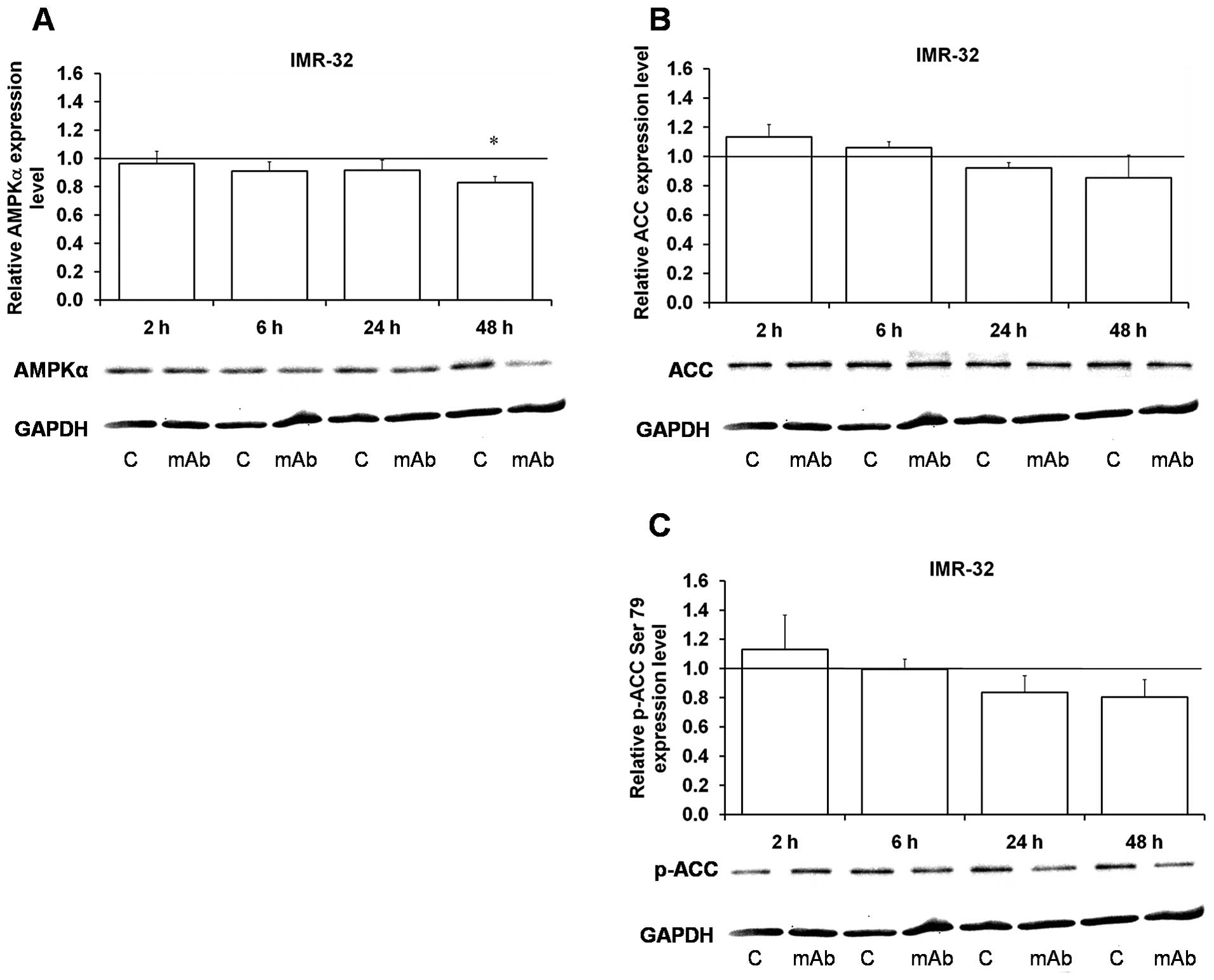 | Figure 8Effect of the 14G2a mAb on AMPKα and
acetyl-CoA carboxylase (ACC) expression and phosphorylation. AMPKα,
ACC and phosphorylated ACC (p-acetyl CoA Ser79) were measured at 2,
6, 24 and 48 h, and normalized to GAPDH. Mean values of three
separate experiments obtained for the 14G2a mAb-treated IMR-32
cells are presented for the AMPKα (A), ACC (B) and p-ACC (C) and
calculated versus control values, set as 1 (black baseline). ANOVA
shows no statistically significant changes of AMPKα, ACC and p-ACC
expression in time. P-values for t-test were as follows:
*p<0.05, **p<0.01,
***p<0.001. Below each chart representative
immunoblottings are presented: C, control cells; mAb, mAb-treated
cells (40 μg/ml). |
 | Figure 7Effect of the 14G2a mAb on AMPKβ
expression and phosphorylation. AMPKβ 38 and 34 kDa forms and
phosphorylated AMPKβ (p-AMPKβ, Ser108) were measured at 2, 6, 24
and 48 h, and normalized to GAPDH. Mean values of three separate
experiments obtained for the 14G2a mAb-treated IMR-32 (A–C) and
CHP-134 (D–F) cells are presented for the AMPKβ 38 kDa (B and E),
AMPKβ 34 kDa (C and F), and p-AMPKβ (A and D), and calculated
versus control values, set as 1 (black baseline). ANOVA shows
statistically significant changes of p-AMPKβ Ser108 protein content
in time in the IMR-32 cells [F(3,8)=32.16, p=0.0001] and AMPKβ1/2
34 kDa in the CHP-134 cells [F(3,9)=5.76, p=0.0176]. ANOVA shows no
statistically significant changes of expression in time for the
rest of the proteins. P-values for t-test were as follows:
*p<0.05, **p<0.01,
***p<0.001. Below each chart representative
immunoblottings are presented: C, control cells; mAb, mAb-treated
cells (40 μg/ml). |
Acetyl coenzyme A carboxylase (ACC) (Fig. 8B) and p-acetyl-ACC (Ser79)
(42) (Fig. 8C) were insignificantly decreased at
24 and 48 h, upon an early (2 h) small increase in their content.
Kinetics of AMPK-dependent inhibitory phosphorylation of ACC, does
not exactly reflect changes in the AMPK itself, which is
significantly decreased by the mAb treatment.
PI3K/Akt/mTOR inhibitors modulate effects
of the anti-GD2 ganglioside mAb
PI3Ks are lipid kinases that regulate diverse
cellular processes including proliferation, adhesion, survival and
motility (43). The PI3K signaling
pathway frequently becomes dysregulated in human cancers and used
by tumor cells for increased proliferation, evasion of apoptosis,
tissue invasion and metastasis (44). Therefore PI3Ks have emerged as
viable targets for novel anticancer therapy leading to design of
potent and selective small molecule inhibitors that have progressed
from preclinical tests to even phase III clinical trials (45).
BEZ-235, a novel imidazo-quinoline derivative, is a
dual ATP-competitive PI3K and mTOR inhibitor with potent antagonist
activity against p110-α, -β, -γ and -δ isoforms and mTOR in
nanomolar concentrations (46).
The inhibitor drives degradation of MYCN in neuroblastoma tumor
cells and decreases angiogenesis (47). Moreover, BEZ235 and the
lysosomotropic agent chloroquine synergize to trigger apoptosis in
neuroblastoma cells via mitochondrial-lysosomal cross-talk
(48). SAR245409 (also known as
XL765), another PI3K/mTOR inhibitor, was shown to inhibit
proliferation and induce apoptosis in various tumor cell lines
(49), in mouse xenograft models
(50) and in patients with
advanced solid tumors (51,52).
Perifosine, a pan-Akt inhibitor of PIP3 binding,
possesses antitumor growth effect alone or in combination with
chemotherapy in neuroblastoma (53). Most importantly, perifosine-induced
inhibition of Akt increases sensitivity of neuroblastoma to
chemotherapy (53). LY294002 is an
ATP-competitive PI3K inhibitor which can also directly inhibit mTOR
due to structural similarity of mTOR and PI3Ks (12).
Based on the above cited literature, we decided to
investigate effects of combinatorial treatment with the 14G2a mAb
and the afore-mentioned inhibitors on ATP levels in cultures the
three GD2-positive, MYCN-amplified neuroblastoma cell lines
used in our studies. We show that in IMR-32, CHP-134 and LA-N-1
cells the viability is decreased by all four inhibitors tested
alone or in combination with the mAb in a dose-dependent manner
after 72 h of treatment (Fig.
9A–L). We calculated and compared IC40 values for
the inhibitors used alone or in combination with the mAb (Table II). This was based on the results
of control experiments with the cell lines treated with the 14G2a
mAb alone for 72 h that showed decrease of the cellular ATP levels
to 0.47 (±0.01) for IMR-32, 0.46 (±0.02) for CHP-134 and 0.64
(±0.02) for LA-N-1 as compared to the control cells (treated with
PBS alone, with p<0.001, t-test). The cell lines tested showed
variability in sensitivity to the inhibitors. BEZ-235 was the most
potent, followed by perifosine, LY294002, and SAR245409 (Table II). The observed effects of the
dual treatment on ATP contents were inhibitor-dependent and cell
line-dependent. The most pronounced changes of IC40
values were calculated for combination of the mAb with the LY294002
inhibitor for the CHP-134 cells (17.8-fold change) and the IMR-32
cells (8.4-fold change) as well as for the BEZ-235 inhibitor for
the IMR-32 cells (7.2-fold change). For other conditions tested the
observed changes in IC40 values were rather small,
ranging from 1.1 to 2.4. However, we observed that the effects of
combinatorial treatment were not always statistically significant
as compared to cells treated with the inhibitors alone, due to
inter-experiment variability that affected calculated values of
means and SEMs. This is especially evident for higher doses of the
used inhibitors (Fig. 9). In such
instances the IC40 values should be only used as a trend
indication.
 | Figure 9Cell viability measurements. The
IMR-32, LA-N-1 and CHP-134 cells were treated with Akt inhibitor
(perifosine), dual mTOR/PI3K inhibitors (BEZ-235 and SAR245409),
and pan-PI3K inhibitor (LY294002) alone or in combination with the
14G2a mAb (40 μg/ml) for 72 h. The cell viability was determined by
measuring ATP content, and compared to respective controls treated
with appropriate diluents (DMSO or water, set as 1). Data are
presented as means (± SEM) from three (B, C, H and J–L) to four (A,
D–G and I) independent experiments (A and B). Logarithmic scales
were used for LY294002 and BEZ-235 and linear scales were used for
perifosine and SAR245409. P-values for t-test were as follows:
*p<0.05, **p<0.01,
***p<0.001. The observed effects were shown by ANOVA
to be statistically significant for all cell lines treated with
either inhibitors alone or combinatorial treatment (data not
shown). |
Discussion
Constitutive activation of the PI3K/Akt/mTOR
signaling network is a frequent event in high-risk neuroblastoma
(2,54) and therefore appears to be an
attractive therapeutic target. Additionally, the malignancy
exhibits increased expression of several growth factor receptors
that transmit their signals through the pathway, such as
insulin-like growth factor I receptor (IGF-IR), epidermal growth
factor receptor (EGFR), tyrosine receptor kinase B (TrkB),
platelet-derived growth factor receptor B (PDGFR B), and c-Kit,
allowing for selective co-targeting based on mechanism-guided
combination that may lead to more effective and durable therapies.
Combinatorial treatment of minimal residual disease with the
anti-GD2 mAb (ch14.18) and 13-cis retinoic acid with
addition of IL-2 and GM-CSF has become a standard treatment in
high-risk neuroblastoma (5). Based
on our previous experimental data showing that the mAb 14G2a causes
decrease in viability of IMR-32, LA-N-1 and CHP-134 cells (among
other cell lines), we aimed to characterize changes induced with
the antibody in the phosphorylation status of several cellular
proteins. Our data allowed us to conclude that there are numerous
alterations of phosphorylation levels of proteins in the
GD2-positive IMR-32 cells treated with the 14G2a mAb. The findings
broaden our knowledge on the mechanisms of the antibody actions on
the cells and were extended with more thorough analyses of chosen
proteins including Akt, PTEN, mTOR, P70 S6, GSK3β, 4E-BP1, AMPK,
and P27. We characterized temporal changes of the protein
expression levels and their phosphorylation and/or subcellular
localization in the mAb-treated cells. Western blot analyses
confirmed statistically meaningful decrease in Akt, and mTOR
phosphorylation in our models 2–48 h after the 14G2a treatment.
The PI3K/Akt/mTOR and RAS/RAF/MEK/ERK pathways
converge to stabilize the expression of the MYC oncoprotein
(55). Additionally, it was
previously reported that the MYCN gene, frequently amplified
in neuroblastoma (54), can confer
resistance to PI3K/Akt/mTOR inhibitors independently of the RAS
pathway (56,12). Also, two PI3K inhibitors, LY294002
and wortmannin, were shown to be able to destabilize MYCN (57). The above literature underlines the
significance of inter-connections between the proteins. In this
context, our previous studies have shown that MYCN is decreased in
the 14G2a mAb-treated, MYCN-amplified IMR-32 and CHP-134
neuroblastoma cells (9). Data
reported here allow us to state that the treatment with the 14G2a
downregulates not only MYCN, but also mTOR as well as decreases
both Akt and mTOR phosphorylation, stressing the ability of the GD2
specific antibody to hit crucial neuroblastoma targets.
It is known that one of the other proteins linked to
the PI3K/Akt/mTOR route i.e., GSK-3β, phosphorylates and stabilizes
the MYCN protein (58), therefore
our result documenting significant decrease in GSK-3β
phosphorylation upon the 14G2a mAb addition, is in agreement with
our earlier finding on MYCN levels (9). Aurora A kinase is another protein
known to stabilize MYCN in human neuroblastoma (59). We have shown before that the
14G2a-induced inhibition of Aurora A kinase accompanied by
significant decrease in the MYCN content (9). Other studies have revealed cross-talk
of Aurora A kinase with the PI3K pathway at Akt activation
(60). We can support this with
our finding showing that Aurora A kinase inhibition with the
specific inhibitor MK5108, in the IMR-32 cells resulted in
suppression of Akt activation (data not shown).
We wish to highlight that observed inhibition of ERK
by the mAb might be important in light of recent findings
concerning independent promotion of the accumulation of MYC
oncoproteins by ERK and mTOR (54). We have found a decrease in ERK
activity in the 14G2a mAb-treated neuroblastoma cells (Table I). Therefore, the kinase may be yet
another factor responsible for the observed downregulation of MYCN
levels in our model.
We show here that Akt is inhibited in the 14G2a
mAb-treated neuroblastoma cell lines, therefore Akt, cannot
efficiently phosphorylate and activate MDM2, a negative regulator
of P53 and thus cannot inhibit apoptosis. As the P53 and Akt
signaling pathways are inter-connected in an opposite manner,
therefore inhibition of Akt correlates with upregulation and
nuclear accumulation of P53 as observed in our earlier studies
(9). Activation of P53 can lead to
either cell cycle arrest and DNA repair, or apoptosis (61). We have found decreased
phosphorylation of the P53 protein at Ser46, Ser392 and Ser15 in
IMR-32 cells (Table I).
Phosphorylation of P53 at Ser46 is strongly associated with its
proapoptotic activity, therefore decrease in phosphorylation at the
amino acid residue could prevent apoptosis. This finding is in
agreement with our earlier results, showing that the mAb-induced
cell death is only partially caused by apoptosis (8). P53 can be also phosphorylated at
Ser15 by different kinases and thus impairs the ability of MDM2 to
bind P53, promoting both the accumulation and activation of P53 in
response to DNA damage (62). Our
results allow us to hypothesize that P53 transcription function can
be decreased. On the other hand, dephosphorylation of P53 at Ser392
found in the mAb-treated IMR32 cells, may indicate that P53 is
stabilized as phosphorylation of Ser392 is coupled with the rapid
turnover of P53 (63).
P27 is a major target of the growth-promoting
activity exerted by tyrosine kinase receptors. Moreover, reduced
expression of P27 correlates with poor prognosis of patients
affected by various types of cancer (29). Akt is responsible for
phosphorylation of P27 at Thr157 and Thr198, leading to its
cytoplasmic retention and decrease in inhibitory activity against
cyclins and Cdks in the nucleus that promotes carcinogenesis
(27,28). We observed dephosphorylation of
Thr157 and Thr198 of P27 which can be linked with decreased
cytoplasmic and increased nuclear accumulation of the cell cycle
inhibitor in the IMR-32 cells treated with the 14G2a mAb. We can
also conclude that the changes parallel Akt deactivation in our
studied model.
The PI3K/Akt/mTOR pathway is dysregulated in cancer
and therefore several inhibitors of the route are currently tested
in preclinical and clinical settings. The present study shows for
the first time, to the best of our knowledge, results of
combinatorial treatment of three MYCN-amplified
neuroblastoma cell lines with the GD2 specific antibody and
inhibitors of PI3K/Akt/mTOR pathway. This adds to our previous data
on effects of the 14G2a mAb in combination with the Aurora A
inhibitor MK-5108 and 13-cis-retinoic acid (9). Based on the results, we reckon that
PI3K/Akt/mTOR signaling network is inhibited in the 14G2a
mAb-treated cells and the antibody can sensitize the cells to the
pathway inhibitors to further enhance their antitumor activity,
albeit the effects vary depending on the cell line, its sensitivity
to the mAb and the small molecule drug used. Among the three PI3K
and mTOR inhibitors tested, BEZ-235 was shown to be most potent as
the IC40 values were lower from 29- to 147-fold as
compared to LY294002, from 10- to 65-fold as compared to
perifosine, and from 79- to 535-fold when compared to SAR245409
(depending on the cell line treated with each inhibitor). The
heterogeneous responses observed advocate for further more detailed
studies that may lead to better characterization of the observed
effects. This should include more cell lines as well as in
vivo studies on applicable GD2-mouse neuroblastoma models.
Our understanding of the mechanisms by which
GD2-specific antibodies decrease neuroblastoma cell survival is
still far from complete. We provide here an explanation of cellular
response to the antibody at the level of the PI3K/Akt/mTOR
signaling network and other PI3K inter-connected wires. There are
numerous elements of the intracellular pathways which are altered
upon the mAb addition, including Akt, mTOR and p70 S6 kinases as
well as AMPK and PTEN, suppressors of the pathway. Additionally,
four different PI3K/Akt/mTOR pathway inhibitors were used in
combination with the monoclonal antibody to determine neuroblastoma
cell viability. We showed that BEZ-235 was the most potent of the
four drugs tested. Effects of combination of the inhibitors with
immunotherapy depends on the cell line susceptibility to the given
drug and the 14G2a mAb. To conclude, our findings indicate that the
14G2a mAb influences numerous, both disparate and overlapping
signal routes. Further studies are warranted in a quest for the
best possible combinations of GD2-specific antibodies and other
drugs to achieve the greatest anti-proliferative effects on cancer
cells.
Acknowledgements
This study was supported by grant no. N301 158635
from the Polish Ministry of Science and Higher Education,
NCN-2012/07/B/NZ1/02808 from the Polish National Science Center and
DS/8/WBBiB. Faculty of Biochemistry, Biophysics and Biotechnology
of the Jagiellonian University is a partner of the Leading National
Research Center (KNOW) supported by the Ministry of Science and
Higher Education. We thank Dr R.A. Reisfeld (Scripps Institute) for
providing us with the hybridoma cell line producing 14G2a mAb. We
are grateful to Dr M. Bzowska and Dr J. Skrzeczynska-Moncznik
(Immunology Department of the Faculty of Biochemistry, Biophysics
and Biotechnology, Jagiellonian University) for help with flow
cytometry analyses.
References
|
1
|
Modak S and Cheung N-K: Neuroblastoma:
Therapeutic strategies for a clinical enigma. Cancer Treat Rev.
36:307–317. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Iżycka-Świeszewska E, Drożyńska E, Rzepko
R, Kobierska-Gulida G, Grajkowska W, Perek D and Balcerska A:
Analysis of PI3K/AKT/mTOR signalling pathway in high risk
neuroblastic tumours. Pol J Pathol. 61:192–198. 2010.
|
|
3
|
Cheung N-KV and Dyer MA: Neuroblastoma:
Developmental biology, cancer genomics and immunotherapy. Nat Rev
Cancer. 13:397–411. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hara J: Development of treatment
strategies for advanced neuroblastoma. Int J Clin Oncol.
17:196–203. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yu AL, Gilman AL, Ozkaynak MF, London WB,
Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay
KK, et al; Children's Oncology Group. Anti-GD2 antibody with
GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J
Med. 363:1324–1334. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Aixinjueluo W and Furukawa K, Zhang Q,
Hamamura K, Tokuda N, Yoshida S, Ueda R and Furukawa K: Mechanisms
for the apoptosis of small cell lung cancer cells induced by
anti-GD2 monoclonal antibodies: Roles of anoikis. J Biol Chem.
280:29828–29836. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoshida S, Kawaguchi H, Sato S, Ueda R and
Furukawa K: An anti-GD2 monoclonal antibody enhances apoptotic
effects of anti-cancer drugs against small cell lung cancer cells
via JNK (c-Jun terminal kinase) activation. Jpn J Cancer Res.
93:816–824. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kowalczyk A, Gil M, Horwacik I, Odrowąż Z,
Kozbor D and Rokita H: The GD2-specific 14G2a monoclonal antibody
induces apoptosis and enhances cytotoxicity of chemotherapeutic
drugs in IMR-32 human neuroblastoma cells. Cancer Lett.
281:171–182. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Horwacik I, Durbas M, Boratyn E, Węgrzyn P
and Rokita H: Targeting GD2 ganglioside and Aurora A kinase as a
dual strategy leading to cell death in cultures of human
neuroblastoma cells. Cancer Lett. 341:248–264. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cochonneau D, Terme M, Michaud A,
Dorvillius M, Gautier N, Frikeche J, Alvarez-Rueda N, Bougras G,
Aubry J, Paris F, et al: Cell cycle arrest and apoptosis induced by
O-acetyl-GD2-specific monoclonal antibody 8B6 inhibits tumor growth
in vitro and in vivo. Cancer Lett. 333:194–204. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Samuels Y and Ericson K: Oncogenic PI3K
and its role in cancer. Curr Opin Oncol. 18:77–82. 2006. View Article : Google Scholar
|
|
12
|
Fruman DA and Rommel C: PI3K and cancer:
Lessons, challenges and opportunities. Nat Rev Drug Discov.
13:140–156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang L, Zhou F and ten Dijke P: Signaling
interplay between transforming growth factor-β receptor and
PI3K/AKT pathways in cancer. Trends Biochem Sci. 38:612–620. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hers I, Vincent EE and Tavaré JM: Akt
signalling in health and disease. Cell Signal. 23:1515–1527. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rodon J, Dienstmann R, Serra V and
Tabernero J: Development of PI3K inhibitors: Lessons learned from
early clinical trials. Nat Rev Clin Oncol. 10:143–153. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dienstmann R, Rodon J, Serra V and
Tabernero J: Picking the point of inhibition: A comparative review
of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 13:1021–1031.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Suzuki YJ, Mizuno M and Packer L: Signal
transduction for nuclear factor-kappa B activation. Proposed
location of antioxidant-inhibitable step. J Immunol. 153:5008–5015.
1994.PubMed/NCBI
|
|
19
|
Niehrs C: The complex world of WNT
receptor signalling. Nat Rev Mol Cell Biol. 13:767–779. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Koul HK, Pal M and Koul S: Role of p38 MAP
kinase signal transduction in solid tumors. Genes Cancer.
4:342–359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu Y, Gorospe M, Yang C and Holbrook NJ:
Role of mitogen-activated protein kinase phosphatase during the
cellular response to genotoxic stress. Inhibition of c-Jun
N-terminal kinase activity and AP-1-dependent gene activation. J
Biol Chem. 270:8377–8380. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Davison K, Mann KK, Waxman S and Miller WH
Jr: JNK activation is a mediator of arsenic trioxide-induced
apoptosis in acute promyelocytic leukemia cells. Blood.
103:3496–3502. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cuadrado A and Nebreda AR: Mechanisms and
functions of p38 MAPK signalling. Biochem J. 429:403–417. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Walsh MF, Thamilselvan V, Grotelueschen R,
Farhana L and Basson M: Absence of adhesion triggers differential
FAK and SAPKp38 signals in SW620 human colon cancer cells that may
inhibit adhesiveness and lead to cell death. Cell Physiol Biochem.
13:135–146. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Darnell JE Jr, Kerr IM and Stark GR:
Jak-STAT pathways and transcriptional activation in response to
IFNs and other extracellular signaling proteins. Science.
264:1415–1421. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Beierle EA, Ma X, Stewart J, Nyberg C,
Trujillo A, Cance WG and Golubovskaya VM: Inhibition of focal
adhesion kinase decreases tumor growth in human neuroblastoma. Cell
Cycle. 9:1005–1015. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Motti ML, De Marco C, Califano D, Fusco A
and Viglietto G: Akt-dependent T198 phosphorylation of
cyclin-dependent kinase inhibitor p27kip1 in breast cancer. Cell
Cycle. 3:1074–1080. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Viglietto G, Motti ML and Fusco A:
Understanding p27(kip1) deregulation in cancer: Down-regulation or
mislocalization. Cell Cycle. 1:394–400. 2002. View Article : Google Scholar
|
|
29
|
Pourmand G, Ziaee AA, Abedi AR, Mehrsai A,
Alavi HA, Ahmadi A and Saadati HR: Role of PTEN gene in progression
of prostate cancer. Urol J. 4:95–100. 2007.PubMed/NCBI
|
|
30
|
Guertin DA, Stevens DM, Saitoh M, Kinkel
S, Crosby K, Sheen JH, Mullholland DJ, Magnuson MA, Wu H and
Sabatini DM: mTOR complex 2 is required for the development of
prostate cancer induced by Pten loss in mice. Cancer Cell.
15:148–159. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang Z, Yuan XG, Chen J, Luo SW, Luo ZJ
and Lu NH: Reduced expression of PTEN and increased PTEN
phosphorylation at residue Ser380 in gastric cancer tissues: A
novel mechanism of PTEN inactivation. Clin Res Hepatol
Gastroenterol. 37:72–79. 2013. View Article : Google Scholar
|
|
32
|
Robertson BW, Bonsal L and Chellaiah MA:
Regulation of Erk1/2 activation by osteopontin in PC3 human
prostate cancer cells. Mol Cancer. 9:2602010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chiang GG and Abraham RT: Phosphorylation
of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by
p70S6 kinase. J Biol Chem. 280:25485–25490. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hong S, Zhao B, Lombard DB, Fingar DC and
Inoki K: Crosstalk between sirtuin and mammalian target of
rapamycin complex 1 (mTORC1) signaling in the regulation of S6
kinase 1 (S6K1) phosphorylation. J Biol Chem. 289:13132–13141.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dufner A and Thomas G: Ribosomal S6 kinase
signaling and the control of translation. Exp Cell Res.
253:100–109. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pullen N and Thomas G: The modular
phosphorylation and activation of p70s6k. FEBS Lett. 410:78–82.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pearson RB, Dennis PB, Han JW, Williamson
NA, Kozma SC, Wettenhall RE and Thomas G: The principal target of
rapamycin-induced p70s6k inactivation is a novel phosphorylation
site within a conserved hydrophobic domain. EMBO J. 14:5279–5287.
1995.PubMed/NCBI
|
|
38
|
Dowling RJ, Topisirovic I, Alain T,
Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj
A, Liu Y, et al: mTORC1-mediated cell proliferation, but not cell
growth, controlled by the 4E-BPs. Science. 328:1172–1176. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sonenberg N and Hinnebusch AG: Regulation
of translation initiation in eukaryotes: Mechanisms and biological
targets. Cell. 136:731–745. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kahn BB, Alquier T, Carling D and Hardie
DG: AMP-activated protein kinase: Ancient energy gauge provides
clues to modern understanding of metabolism. Cell Metab. 1:15–25.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mitchelhill KI, Michell BJ, House CM,
Stapleton D, Dyck J, Gamble J, Ullrich C, Witters LA and Kemp BE:
Posttranslational modifications of the 5′-AMP-activated protein
kinase β1 subunit. J Biol Chem. 272:24475–24479. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ha J, Daniel S, Broyles SS and Kim KH:
Critical phosphorylation sites for acetyl-CoA carboxylase activity.
J Biol Chem. 269:22162–22168. 1994.PubMed/NCBI
|
|
43
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Osaki M, Oshimura M and Ito H: PI3K-Akt
pathway: Its functions and alterations in human cancer. Apoptosis.
9:667–676. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Akinleye A, Avvaru P, Furqan M, Song Y and
Liu D: Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer
therapeutics. J Hematol Oncol. 6:882013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Maira SM, Stauffer F, Brueggen J, Furet P,
Schnell C, Fritsch C, Brachmann S, Chène P, De Pover A, Schoemaker
K, et al: Identification and characterization of NVP-BEZ235, a new
orally available dual phosphatidylinositol 3-kinase/mammalian
target of rapamycin inhibitor with potent in vivo antitumor
activity. Mol Cancer Ther. 7:1851–1863. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chanthery YH, Gustafson WC, Itsara M,
Persson A, Hackett CS, Grimmer M, Charron E, Yakovenko S, Kim G,
Matthay KK, et al: Paracrine signaling through MYCN enhances
tumor-vascular interactions in neuroblastoma. Sci Transl Med.
4:115ra32012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Seitz C, Hugle M, Cristofanon S,
Tchoghandjian A and Fulda S: The dual PI3K/mTOR inhibitor
NVP-BEZ235 and chloroquine synergize to trigger apoptosis via
mitochondrial-lysosomal cross-talk. Int J Cancer. 132:2682–2693.
2013. View Article : Google Scholar
|
|
49
|
Garcia-Echeverria C and Sellers WR: Drug
discovery approaches targeting the PI3K/Akt pathway in cancer.
Oncogene. 27:5511–5526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yu P, Laird AD, Du X, Wu J, Won KA,
Yamaguchi K, Hsu PP, Qian F, Jaeger CT, Zhang W, et al:
Characterization of the activity of the PI3K/mTOR inhibitor XL765
(SAR245409) in tumor models with diverse genetic alterations
impacting the PI3K pathway. Mol Cancer Ther. 13:1078–1091. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Papadopoulos KP, Tabernero J, Markman B,
Patnaik A, Tolcher AW, Baselga J, Shi W, Egile C, Ruiz-Soto R,
Laird AD, et al: Phase I safety, pharmacokinetic, and
pharmacodynamic study of SAR245409 (XL765), a novel, orally
administered PI3K/mTOR inhibitor in patients with advanced solid
tumors. Clin Cancer Res. 20:2445–2456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jänne PA, Cohen RB, Laird AD, Macé S,
Engelman JA, Ruiz-Soto R, Rockich K, Xu J, Shapiro GI, Martinez P,
et al: Phase I safety and pharmacokinetic study of the PI3K/mTOR
inhibitor SAR245409 (XL765) in combination with erlotinib in
patients with advanced solid tumors. J Thorac Oncol. 9:316–323.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Li Z, Yan S, Attayan N, Ramalingam S and
Thiele CJ: Combination of an allosteric Akt Inhibitor MK-2206 with
etoposide or rapamycin enhances the antitumor growth effect in
neuroblastoma. Clin Cancer Res. 18:3603–3615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Johnsen JI, Segerström L, Orrego A, Elfman
L, Henriksson M, Kågedal B, Eksborg S, Sveinbjörnsson B and Kogner
P: Inhibitors of mammalian target of rapamycin downregulate MYCN
protein expression and inhibit neuroblastoma growth in vitro and in
vivo. Oncogene. 27:2910–2922. 2008. View Article : Google Scholar
|
|
55
|
Lee T, Yao G, Nevins J and You L: Sensing
and integration of Erk and PI3K signals by Myc. PLOS Comput Biol.
4:e10000132008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Maris JM, Hogarty MD, Bagatell R and Cohn
SL: Neuroblastoma. Lancet. 369:2106–2120. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chesler L, Schlieve C, Goldenberg DD,
Kenney A, Kim G, McMillan A, Matthay KK, Rowitch D and Weiss WA:
Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn
protein and blocks malignant progression in neuroblastoma. Cancer
Res. 66:8139–8146. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Duffy DJ, Krstic A, Schwarzl T, Higgins DG
and Kolch W: GSK3 inhibitors regulate MYCN mRNA levels and reduce
neuroblastoma cell viability through multiple mechanisms, including
p53 and Wnt signaling. Mol Cancer Ther. 13:454–467. 2014.
View Article : Google Scholar
|
|
59
|
Otto T, Horn S, Brockmann M, Eilers U,
Schüttrumpf L, Popov N, Kenney AM, Schulte JH, Beijersbergen R,
Christiansen H, et al: Stabilization of N-Myc is a critical
function of Aurora A in human neuroblastoma. Cancer Cell. 15:67–78.
2009. View Article : Google Scholar
|
|
60
|
Yao JE, Yan M, Guan Z, Pan CB, Xia LP, Li
CX, Wang LH, Long ZJ, Zhao Y, Li MW, et al: Aurora-A down-regulates
IkappaBalpha via Akt activation and interacts with insulin-like
growth factor-1 induced phosphatidylinositol 3-kinase pathway for
cancer cell survival. Mol Cancer. 8:952009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Levine AJ: p53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Loughery J, Cox M, Smith LM and Meek DW:
Critical role for p53-serine 15 phosphorylation in stimulating
transactivation at p53-responsive promoters. Nucleic Acids Res.
42:7666–7680. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Cox ML and Meek DW: Phosphorylation of
serine 392 in p53 is a common and integral event during p53
induction by diverse stimuli. Cell Signal. 22:564–571. 2010.
View Article : Google Scholar
|














