Introduction
In solid malignancies, the PI3K-AKT-mTOR and
Ras-Raf-MEK-ERK pathways have been identified as the most important
oncogenic pathways. Considering the central role of the pathways in
transmitting upstream oncogenic signals, their inhibition could be
an effective therapy as regards various cancer genotypes (1).
The clinical efficiency of single pathway inhibitors
of PI3K-AKT-mTOR and Ras-Raf-MEK-ERK has generally been
disappointing, with some exceptions such as their use in cases of
B-Raf mutant melanoma. Cancers can be de
novo-dependent concurrently on these parallel pathways and
cross-signaling of the pathways is also evident (2,3).
Many in vivo and in vitro studies have shown that the
PI3K-AKT-mTOR and Ras-Raf-MEK-ERK pathways regulate each other's
activity through feedback mechanisms and have shared downstream
targets (4,5). Interaction of the PI3K-AKT-mTOR and
Ras-Raf-MEK-ERK signaling pathways is thought to explain the
inefficiency of single agent treatments and provide a rationale for
the concurrent targeting of both pathways.
Preclinical studies have shown that dual targeting
with PI3K and MEK inhibitors has antitumor activity in various
cancer models and genotypes (6–8).
Many preclinical studies have concerned predictive factors as
regards dual PI3K and MEK inhibitor therapy, but so far no clear
factors have been identified (8,9).
Numerous early-phase clinical studies concerning dual PI3K and MEK
targeting are ongoing and some results have recently been
presented. Generally, combined PI3K and MEK inhibitor therapy seems
to be feasible but, unfortunately, the rate of response is low
(10–12). In preclinical models, the vast
majority of cancer cell lines do not show apoptosis in response to
dual PI3K and MEK targeting, which could be a major factor behind
the limited clinical activity of the approach (8,9).
Some recent investigations have revealed that drugs affecting the
apoptotic pathways, such as BH3 mimetics, could dramatically
increase the rate of apoptosis and the efficiency of MEK and/or
PI3K/mTOR inhibitors (13,14). Bcl-2 family members have been
suggested to be important determinants of cell fate in targeted
cancer therapies. Induction of pro-apoptotic proteins such as BIM
is often linked to apoptosis, while anti-apoptotic proteins such as
Mcl-1 and Bcl-xl promote cell survival. The general balance between
anti- and pro-apoptotic mediators is crucial for the determination
of cell survival or apoptosis (15).
The current study builds upon our earlier work
(9) where we identified three cell
lines from solid tumors, which showed sensitivity to dual PI3K and
MEK blockage but with limited apoptotic response. In this study, we
investigated in vitro with these cell lines if an additional
agent could increase efficiency and apoptotic response to dual PI3K
and MEK blockage. We identified some pharmacological agents that
could enhance the cytotoxicity of dual PI3K and MEK blockage, and
more importantly, induce marked apoptosis. Furthermore, Bcl-xl
(Bcl2L1) and Mcl-1 were identified as being important determinants
of cell fate.
Materials and methods
Cell lines
The cell lines used in the current study included
the triple-negative NSCLC (non-small cell lung cancer) cell line
H1437, the basal-like breast cancer line MDA-MB231 and the K-Ras
mutant colorectal cell line HCT116. The NSCLC cell line H1437 was a
kind gift from Dr Pasi Jänne (Dana-Farber Cancer Institute, Boston,
MA, USA) and the breast and colorectal lines (MDA-MB231 and HCT116)
were from Dr Peppi Koivunen (Oulu University, Oulu, Finland). The
cell lines were cultured in RPMI-1640 supplemented with fetal
bovine serum (10%) plus penicillin and streptomycin (100 IU/ml).
All the cell culture reagents were purchased from HyClone (Logan,
UT, USA).
Inhibitors
Pharmacological agents used in the study and their
final concentrations are listed in Table I. The drugs were dissolved in DMSO
to a concentration of 10 mmol/l, except for cisplatin, which was
diluted in distilled water. All the agents were stored in aliquots
at −20°C. Further dilutions were made in the cell culture
medium.
 | Table IAgents studied in combination with
dual PI3K and MEK inhibition. |
Table I
Agents studied in combination with
dual PI3K and MEK inhibition.
| Inhibitor | Target/Class | Concentration in
μM |
|---|
| ABT-263 | BH3 | 0.1, 1 |
| Afatinib | HER2 | 1 |
| AG-1024 | IGF1R1 | 10 |
| 5-azacytidine | Methylation | 1 |
| AZD-2281 | PARP | 0.1, 1 |
| Cis-platin | Chemo | 1 |
| Crizotinib | ALK | 1 |
| Dasatinib | Multi-TKI | 1 |
| Entinostat | Chemo | 1 |
| GDC-0449 | Hedgehog | 10 |
| Gö9976 | PKC | 1 |
| IPI-504 | HSP90 | 1 |
| LY-2157229 | TGF-βR1 | 1 |
| Metformin | Diabetics | 100 |
| NVP-XAV939 | WNT | 1 |
| Paclitaxel | Chemo | 0.01 |
| Salinomycin | CSC | 0.1, 1 |
| SD208 | TGF-βR1 | 1 |
| Sorafenib | Multi-TKI | 1 |
| Sunitinib | Multi-TKI | 1 |
| DBC | γ-Secretase | 1 |
MTS cell viability/cytotoxicity
analysis
Cells were plated onto 96-well plates with three or
six parallel wells for each treatment, the experiments being
replicated at least twice. The following day the cells were drug
treated for 72 h, after which the plates were developed using an
MTS reagent mix ([3-(4,
5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium,
inner salt], Promega, Madison, WI, USA) supplemented with phenazine
methosulfate (Sigma-Aldrich, St. Louis, MO, USA) according to the
manufacturer's guidelines and the absorbance was read on a plate
reader (Athos Labtec Instruments, Salzburg, Austria) at a
wavelength of 488 nm. Graphic display of the data was carried out
using GraphPad Prism software (GraphPad Software, La Jolla, CA,
USA). Absorbance in the non-treated wells was set as the reference
value (100%).
Western blot analysis
The cells were plated onto 6-well plates and treated
with the drugs 24 h later for the desired time, after which they
were lysed in RIPA buffer [1% Igepal CA-630, 20 mM Tris-HCl pH 8.0,
137 mM NaCl, 10% glycerol, 2 mM EDTA, 1 mM sodium orthovanadate,
aprotinin (10 μg/ml), leupeptin (10 μg/ml) and pepstatin (10
μg/ml)]. Protein concentrations were measured by Bio-Rad Protein
Assay (Bio-Rad, Hercules, CA, USA) and the concentrations in
individual samples were equalized before adding 3X Laemmli buffer
to a final concentration of 1X. Equal amounts of protein were run
on 12% SDS-PAGE gels, transferred to PVDF membranes, probed with
the antibodies and developed using the ECL chemiluminescence system
(Millipore, Billerica, MA, USA) for detection on radiographic film,
which was scanned to an electronic format. Primary antibodies to
the following proteins were used in the current study: cleaved
PARP, Mcl-1, BIM, Bcl-xl and β-actin. All the primary antibodies
were used at a 1:1000 dilution in 5% BSA. Anti-mouse (β-actin) or
anti-rabbit (others) IgG HRP-linked antibody was used as the
secondary antibody. All antibodies were from Cell Signaling
Technology (Danvers, MA, USA).
Immunoprecipitation
For immunoprecipitation, cells were plated on 6-well
or 10-cm diameter plates and drug treated the following day for the
desired time, after which the cells were lysed in RIPA buffer and
protein concentrations were determined by Bio-Rad Protein Assay.
Protein (200 μg) at a concentration of 1 mg/ml was incubated
overnight at 4°C with primary antibody at a 1:100 concentration.
The next day protein A/G agarose beads (Santa Cruz Biotechnology,
Dallas, TX, USA) were added to each sample and incubated for 1–3 h
at 4°C, after which the samples were pelleted by centrifugation.
The pellets were washed with cell lysis buffer, resuspended in
Laemmli buffer and analyzed with western blotting.
siRNA knockdown
For the knockdown studies, BCL-XL (BCL2L1) and MCL-1
(MCL1) specific siRNAs (Dharmacon smart pools) were used at 25
nmol/l and transfected with DharmaFECT transfection reagent (both
from Dharmacon, Lafayette, CO, USA). The experiments were performed
according to the manufacturer's protocol. In short, the cells were
plated on 96- (for MTS) or 6-well (for western blotting) plates in
non-antibiotic media. After 24 h the medium was changed to medium
containing siRNA+lipid, or scramble siRNA+lipid (controls),
prediluted in Opti-MEM (Gibco/Invitrogen, Carlsbad, CA, USA). The
following day, the medium was changed to normal medium. Incubation
was continued for the desired time (total of 24–96 h) before
assays.
Statistical analysis
Student's two-tailed t-test was used for the
statistical analysis. For the analysis, percentage change compared
to PI3Ki+MEKi treated (Fig. 1) or
scramble siRNA treated cells (knockdown studies) was used. p-values
<0.001 and <0.01 are indicated.
Results
ABT-263, entinostat and dasatinib
increase the cytotoxicity of dual PI3K and MEK inhibition
The effect of combining dual PI3K and MEK inhibition
with other pharmacological agents was studied in HCT116 (colon
cancer), MDA-MB231 (breast cancer) and H1437 (NSCLC) cell lines,
which are known to be sensitive to dual PI3K and MEK inhibition but
to show limited apoptotic responses to the treatments (9). The cells were exposed to dual PI3K
and MEK inhibition (ZSTK474 and CI-1040) in combination with a
panel of other small-molecule inhibitors and cell viability was
analyzed by using 72-h MTS cytotoxicity assays (Table I). The concentrations chosen for
each drug were based on our preliminary experiments, where we used
MTS assay to determine the lowest concentration causing the maximal
cytotoxic effect (not shown). Of the 21 tested drugs, the BH3
mimetic ABT-263, the HDAC inhibitor entinostat and the multikinase
inhibitor dasatinib were observed to decrease cell viability
compared with dual blockage alone, excluding the H1437 line and
ABT-263 treatment combination. Of these three drugs, entinostat and
dasatinib had single agent cytotoxic activity, while ABT-263 was
active only in the MDA-MB231 line. ABT-263, entinostat and
dasatinib were chosen for further studies.
We next assessed whether concurrent dual blockage is
needed for cytotoxicity of the other agent or if single PI3K or MEK
inhibition is sufficient, using MTS cytotoxicity assays. In the
HCT116 line, ABT-263 was seen to decrease viability in combination
with PI3K inhibition (PI3Ki) but not with MEK inhibition (MEKi).
Conversely, when HCT116 cells were treated with entinostat or
dasatinib, concurrent administration of either PI3Ki or MEKi
decreased cell viability (Fig.
1A). In the MDA-MB231 line, a marked increase in cytotoxicity
was seen with concurrent administration of ABT-263 or dasatinib
with ZSTK474 or CI-1040. Synergistic cytotoxicity was also seen
with entinostat co-administration with PI3Ki but not with MEKi
(Fig. 1B). In the H1437 line,
entinostat was the only agent noted to increase cytotoxicity in
combination with either PI3Ki or MEKi, while no marked change was
seen with ABT-263 or dasatinib combinations (Fig. 1C).
ABT-263, entinostat and dasatinib
increase apoptotic responses to dual PI3K and MEK inhibition
Next we investigated whether the cytotoxic responses
seen in MTS assays are accompanied by apoptosis. Western blot
analysis of PARP cleavage was used to assess apoptosis after 6–72 h
of drug treatments. Based on our preliminary experiments, the rate
of the apoptotic response varied dramatically in the tested lines.
MDA-MB231 responded with apoptosis within a few hours of the
initiation of treatment, while the two other lines responded in
days. Therefore, a specific time point for each line was selected.
In the HCT116 and MDA-M231 cell lines, marked PARP cleavage was
seen when ABT-263 was combined with PI3Ki, MEKi, or their
combination. In the H1437 cell line, PARP cleavage was seen only
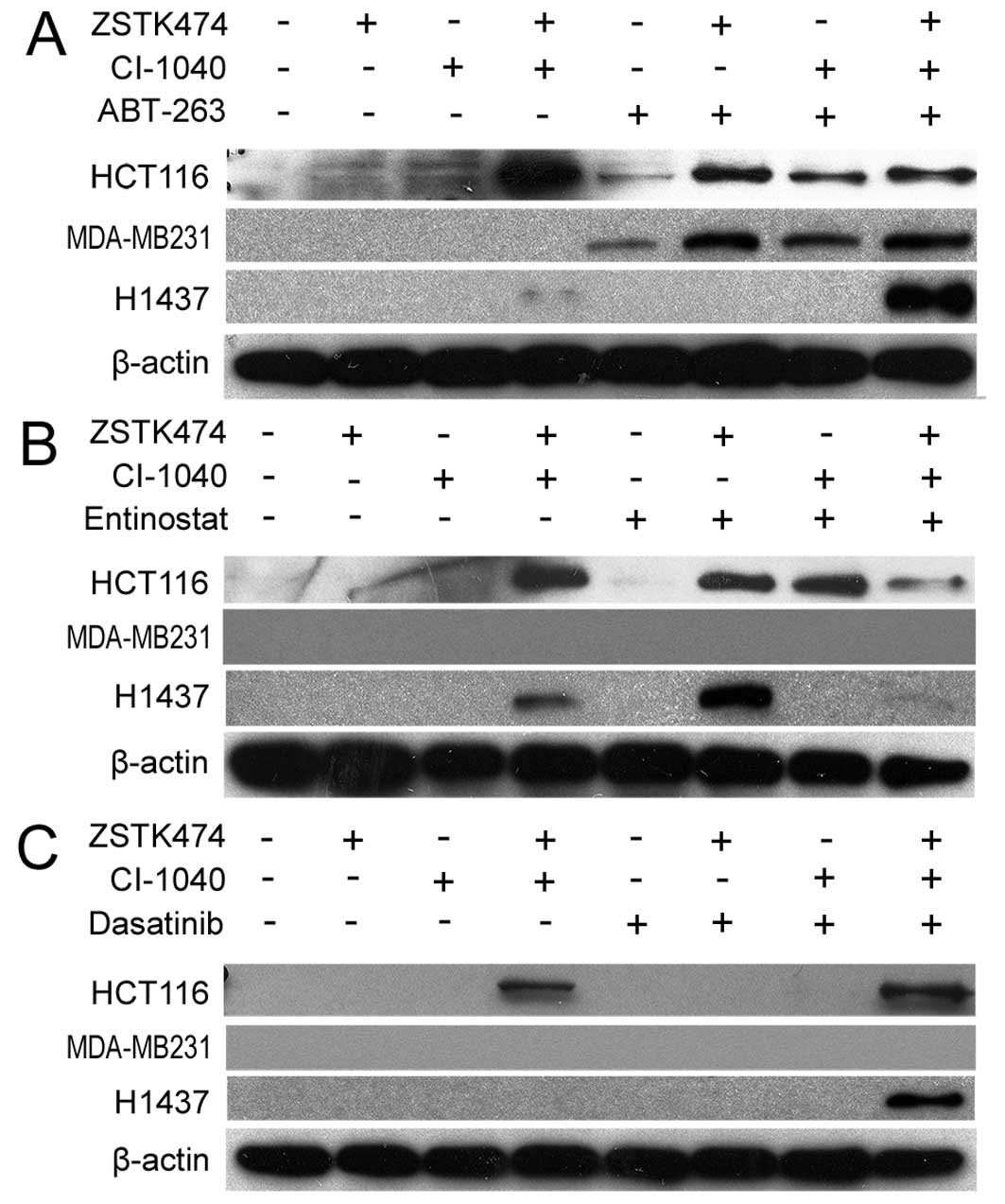
when ABT-263 was combined with dual PI3K and MEK blockage (Fig. 2A). The HCT116 line responded to
entinostat treatment analogously to ABT-263, with detectable
cleaved PARP when entinostat was co-administered with PI3Ki, MEKi,
or their combination. In the H1437 line, cleaved PARP was detected
with the entinostat and PI3Ki combination, but not with the other
treatments tested. No apoptosis was seen in the MDA-MB231 line with
entinostat combinations (Fig. 2B).
Dasatinib was able to induce marked PARP cleavage in the H1437 cell
line only when combined with dual PI3K and MEK blockage, and not in
the other combinations tested (Fig.
2C).
Downregulation of Bcl-xl and Mcl-1
correlates with apoptosis in cells treated with PI3K and MEK dual
blockage
We further analyzed the effects of the inhibitor
treatments on the pro-apoptotic protein BIM and the anti-apoptotic
proteins Mcl-1 and Bcl-xl. HCT116 and H1437 lines were selected for
the analysis since these cell lines respond to treatment within
days and are more reliably assessable as regards expression.
Conversely, the MDA-MB231 line undergoes very rapid apoptosis and
on the basis of the results of our preliminary experiments, protein
levels remain unaltered in this line and therefore we excluded it
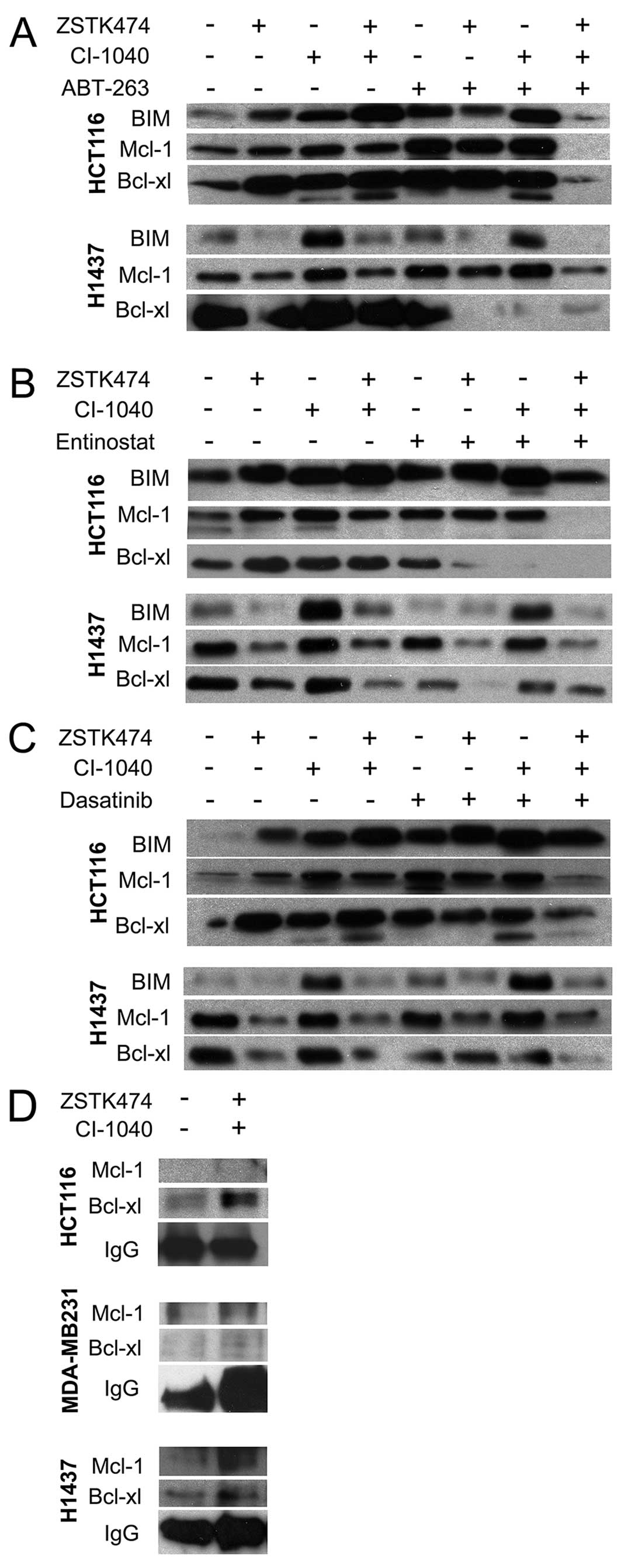
from these experiments. In HCT116 cells, BIM upregulation was
detected in response to all treatments except for co-targeting with
ABT-263 or entinostat plus dual PI3K and MEK blockage. Furthermore,
there was a tendency for BIM upregulation to occur with single
agent MEK inhibition.
In H1437 cells, BIM upregulation correlated with MEK
inhibition, but, surprisingly, this was absent when PI3Ki was
co-administered. In the HCT116 line, Mcl-1 upregulation was noted
in connection with ABT-263 and its combinations and downregulation
was seen when dual PI3K and MEK blockage was combined with ABT-263,
entinostat, or dasatinib. In the H1437 line, Mcl-1 downregulation
correlated with PI3K inhibition. In HCT116 cells, Bcl-xl
downregulation was noted in entinostat combinations, and in ABT-263
and dual PI3K and MEK blockage treatments. In the H1437 line,
Bcl-xl downregulation was seen with ABT-263 combinations, and with
entinostat+PI3Ki and dasatinib+PI3Ki+MEKi. In general, Bcl-xl
downregulation does not indicate apoptosis but is required in most
cases. As an exception, we did not see Bcl-xl downregulation when
the HCT116 line was treated with apoptosis-inducing ABT-263
combinations, but since ABT-263 is an indirect inhibitor of the
protein, this is not surprising (Fig.
3A–C).
BIM hetorodimerization with Bcl-xl and
Mcl-1 in response to dual PI3K and MEK blockage
Next, we assessed whether there is a difference in
Bcl-2 family member dimers in cells treated with PI3K and MEK
inhibitor combination. After drug treatments, the cell lysate were
immunoprecipitated with BIM antibody and detected with Mcl-1 or
Bcl-xl antibodies. In HCT116, dual PI3K and MEK targeting increased
incorporation of Bcl-xl to BIM while Mcl-1 levels remained low. In
H1437, we saw an increase in Bcl-xl and Mcl-1 incorporation to BIM,
but some increase in the Mcl-1 attachment. In MDA-MB231, we could
not detect any increase in Bcl-xl or Mcl-1 attachment to BIM in
response to dual targeting. This could be related to the short
period of drug treatment and quick apoptotic response seen with
pharmacological Bcl-2/Bcl-xl blockage in this specific cell line
(Fig. 3D).
Knockdown of Bcl-xl expression increases
the cytotoxicity of dual PI3K and MEK blockage
HCT116, MDA-MB231 and H1437 cells were subjected to
BCL-XL-specific siRNA knockdown. The cells were first
analyzed by western blotting for Bcl-xl expression. In MDA-MB231
cells we saw some downregulation of Bcl-xl after 24- and 48-h
treatment, while almost complete absence of the protein was noted
following 72- and 96-h treatments. In the HCT116 and H1437 lines,
72- or 96-h treatment with BCL-XL-specific siRNA induced
downregulation of Bcl-xl expression, but this, however, was lower
in the H1437 line when compared with the MDA-MB231 line (Fig. 4A).
Based on the MTS cytotoxicity assay, siRNA knockdown
of BCL-XL was not cytotoxic by itself in any of the tested
lines. To evaluated whether BCL-XL knockdown would sensitize
the cells to dual PI3K and MEK blockage, the cells were pretreated
with or without BCL-XL siRNA for 24–48 h, after which they
were exposed to PI3Ki, MEKi, or their combination for an additional
72 h and analyzed by using MTS cytotoxicity assays. Increased
cytoxicity was seen in the MDA-MB231 line with PI3Ki and/or MEKi
treatment after BCL-XL knockdown. This was not observed in
the other two cell lines tested except for single agent MEKi in the
HCT116 cell line (Fig. 4B). We
then investigated whether BCL-XL knockdown would increase
apoptosis in response to PI3Ki and/or MEKi. In HCT116 cells, PARP
cleavage was evident in PI3Ki and more notably, in MEKi treated
cells after BCL-XL knockdown, but not in control cells.
Furthermore, a marked increase in the cleaved PARP signal was seen
after dual PI3K and MEK targeting when BCL-XL was
suppressed. In the MDA-MB231 line, PARP cleavage was evident in
cells treated with PI3Ki and/or MEKi combination in the cells with
BCL-XL knockdown, but not in controls (Fig. 4C). In H1437 cells, we did not see a
marked change in the PARP cleavage profile in cells with
BCL-XL knockdown as expected (not shown).
Knockdown of MCl-1 expression increases
the cytotoxicity of dual PI3K and MEK blockage
Since H1437 line showed increased attachment of
Mcl-1 to BIM in response to dual PI3K and MEK targeting and was
less sensitive to pharmacological or siRNA blockage of
BCL-XL, we assessed whether knockdown of MCl-1 on
would increase the cytotoxic and apoptotic response in this cell
line. HCT116, MDA-MB231, and H1437 lines were exposed to
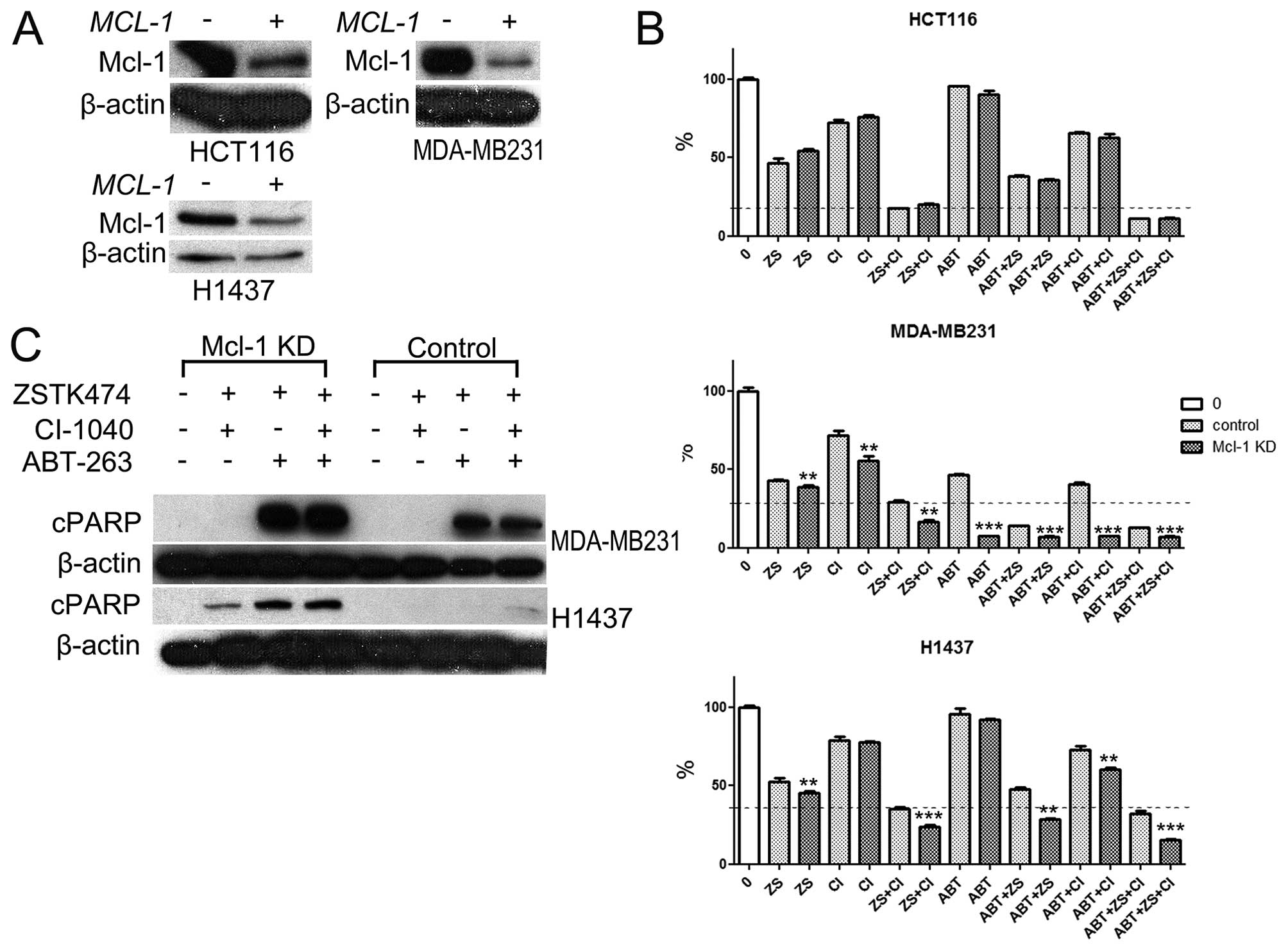
siRNA-mediated knockdown of MCl-1. Marked decrease of Mcl-1
expression was noted in cells treated with MCl-1-specific
siRNA but not with scramble siRNA (Fig. 5A). Based on MTS cytotoxicity assay,
siRNA knockdown of MCl-1 was not cytotoxic by itself in any
of the tested cell lines. In the HCT116 line, knockdown of
MCl-1 had no synergistic cytotoxic effect with the drugs
tested. In the MDA-MB231 cell line, MCl-1 knockdown was able
to provoke increased cytotoxicity with all the tested agents. The
most dramatic difference was seen in MDA-MB231 cells treated with
ABT-263 as a single agent where control cells showed some
cytotoxicity, which was highly promoted by MCl-1 knockdown.
In the H1437 cell line, siRNA knockdown of MCl-1 was able to
induce significant cytotoxicity in treatment regimens containing
PI3Ki and with MEKi+ABT-263 combination (Fig. 5B).
Next, we analyzed whether MCl-1 knockdown
would lead to apoptotic response in H1437 and MDA-MB231 lines. In
MDA-MB231 cells apoptosis was evident in the cells treated with
ABT-263 containing treatment regimens but expression of cleaved
PARP was increased after MCl-1 knockdown. In the H1437 cell
line, a trace of PARP cleavage was evident only in cells treated
with PI3Ki+MEKi+ABT-263 after scramble siRNA treatment. Conversely,
when MCl-1 was knocked down, marked apoptosis was evident in
cells treated with PI3Ki+MEKi, PI3Ki+ABT-263, and
PI3Ki+MEKi+ABT-263 (Fig. 5C).
Discussion
The PI3K-AKT-mTOR and Ras-Raf-MEK-ERK pathways are
central transmitters of oncogenic signals in solid malignancies.
Considering the central role of the pathways, their inhibition
could be an effective therapy in various cancer genotypes. Even
though PI3K-AKT-mTOR and Ras-Raf-MEK-ERK are the most commonly
altered signaling pathways in solid malignancies, the clinical
efficiency of single pathway inhibitors has generally been
disappointing and combinatorial approaches have been applied.
Preclinical models have shown that dual targeting with PI3K and MEK
inhibitors has antitumor activity in various cancer models
(3,8,16,17).
Numerous early-phase clinical studies concerning dual PI3K and MEK
targeting are ongoing and some results have already been presented.
Generally, combined PI3K and MEK inhibitor therapy seems to be
feasible, but, unfortunately, the rate of response seems to be low
(10–12). In preclinical models, the vast
majority of cancer cell lines do not show apoptosis in response to
dual PI3K and MEK targeting, which could be the major factor behind
the limited clinical activity of the approach (8,9).
In the current work, we employed cell lines
identified in our earlier study (9) to evaluate whether the apoptotic
response to dual PI3K and MEK inhibition could be augmented by
pharmacological means in vitro. In the screen, we found that
the Bcl-2/Bcl-xl inhibitor ABT-263, the HDAC inhibitor entinostat
and the multikinase inhibitor dasatinib increased the cytotoxic
response and apoptosis in combination with PI3K and MEK dual
blockage. Analogously to our results, some recent publications have
described how Bcl-2/Bcl-xl targeting can enhance the cytotoxicity
of MEK and mTOR inhibitors, and dual PI3K and MEK blockage
(13,14). Furthermore, HDAC inhibition has
earlier been shown to increase the efficiency of both MEK and
PI3K-AKT-mTOR targeting agents (16,18).
Moreover, earlier research has proposed HDAC inhibitors to act
through Mcl-1 (19). To our
knowledge, however, previous work provided only limited evidence on
the use of dasatinib in combination with PI3K and MEK
inhibitors.
It is challenging to include three investigational
agents concurrently in a clinical trial, and therefore we were also
interested to see if either PI3K or MEK inhibitor efficiency could
be enhanced by combining ABT-263, entinostat or dasatinib to them.
All three of these agents were found to increase the cytotoxicity
of either the PI3K inhibitor or the MEK inhibitor, excluding
ABT-263 in the H1437 line. Apoptotic response was also seen with
dual ABT-263 and PI3K or MEK therapy in the HCT116 and MDA-MB231
lines, with entinostat and PI3K or MEK therapy in the HCT116 line,
and with entinostat and PI3K therapy in H1437 cells. Based on the
results of our preclinical models, it would be appealing to test
Bcl-xl/Bcl-2 or HDAC inhibitors in combination with PI3K or MEK
inhibitors in animal models and clinical trials. Some of these,
such as Bcl-2/Bcl-xl and MEK (NCT02079740), and HDAC and mTOR
(NCT01087554, NCT01174199) inhibitor combinations, are currently
tested in ongoing early phase clinical trials.
Previous studies have identified the apoptotic
proteins BIM, Puma, Mcl-1, Bcl-2 and Bcl-xl as major determinants
of cell fate in response to PI3K-AKT-mTOR and/or MEK inhibitors.
Bcl-xl and Mcl-1 have been suggested to be the most important
anti-apoptotic mediators in solid malignancies (13,14).
Our results provide similar proof, since drug treatments enabling
downregulation of Bcl-xl expression, blockage of its function by
the BH3 mimetic or siRNA gene knockdown resulted in apoptosis in
most cases. However, downregulation or blockage of Bcl-xl did not
indicate apoptosis but is required in most cases. One of the tested
lines (H1437) showed more dependency on both anti-apoptotic
proteins Bcl-xl and Mcl-1, since downregulation, blockage, or
knockdown of BCL-XL itself was insufficient to cause
apoptosis if not accompanied by Mcl-1 downregulation. Furthermore,
MCl-1 knockdown in this cell line was able to produce
prominent increase in cytotoxicity and apoptosis after treatment
with PI3K inhibitor or its combinations. It is likely that many
cancer cells are able to circumvent PI3K and/or MEK
inhibition-mediated apoptosis. Drug treatments inhibiting Bcl-xl
and/or Mcl-1 would, therefore, increase the apoptotic response to
PI3K and/or MEK inhibitors. In our study, BIM expression showed no
correlation to apoptosis.
In the current study, we found that combining
Bcl-2/Bcl-xl, HDAC and multikinase inhibitors to PI3K and MEK dual
blockage can increase cytotoxicity and apoptosis in vitro.
Furthermore, these agents were also able to enhance cytotoxicity
and apoptosis of single-agent PI3K and MEK drugs. More importantly,
we found Bcl-xl and Mcl-1 to be major determinants of cell fate in
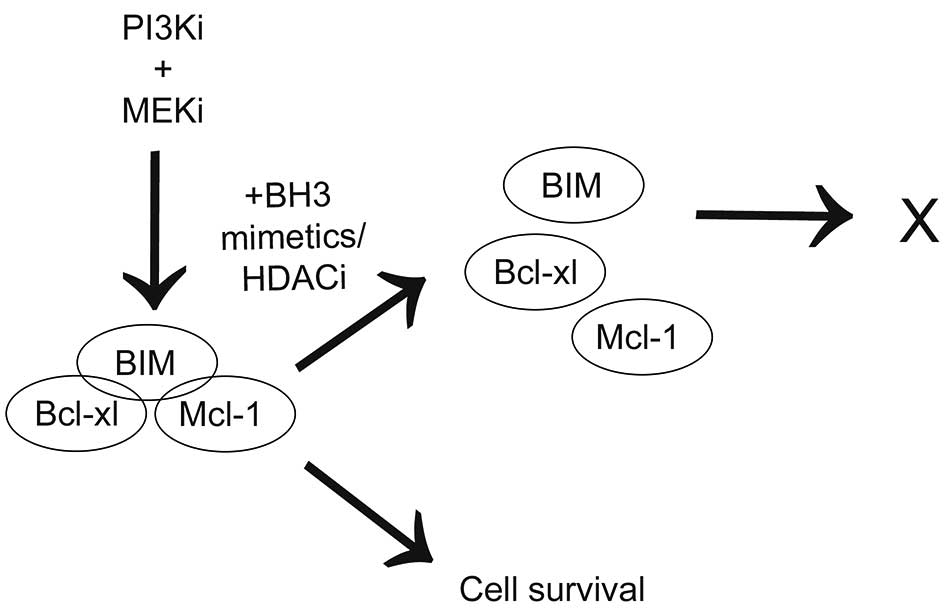
connection with PI3K and/or MEK inhibitor treatment (Fig. 6). We conclude that understanding
the molecular mechanism of the anti-apoptotic response to dual PI3K
and MEK treatment could provide new and smarter pharmacological
approaches and lead to more efficient combinations to be tested in
clinical trials.
Acknowledgements
We wish to thank Anne Bisi for her technical
assistance. The study was supported by Thelma Mäkikyrö Foundation
(J.P.K.), Sigrid Juselius Foundation (J.P.K.), Cancer Society of
Northern Finland (E.J.), Ida Montin Foundation (E.J.), and Emil
Aaltonen Foundation (E.J.).
Abbreviations:
|
cPARP
|
cleaved PARP
|
|
MEKi
|
MEK inhibition
|
|
MTSassay
|
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)-based
cytotoxicity assay
|
|
NSCLC
|
non-small cell lung cancer
|
|
PI3Ki
|
PI3K inhibition
|
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chandarlapaty S, Sawai A, Scaltriti M,
Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK,
Baselga J and Rosen N: AKT inhibition relieves feedback suppression
of receptor tyrosine kinase expression and activity. Cancer Cell.
19:58–71. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Faber AC, Li D, Song Y, Liang MC, Yeap BY,
Bronson RT, Lifshits E, Chen Z, Maira SM, García-Echeverría C, et
al: Differential induction of apoptosis in HER2 and EGFR addicted
cancers following PI3K inhibition. Proc Natl Acad Sci USA.
106:19503–19508. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mendoza MC, Er EE and Blenis J: The
Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends
Biochem Sci. 36:320–328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Turke AB, Song Y, Costa C, Cook R, Arteaga
CL, Asara JM and Engelman JA: MEK inhibition leads to PI3K/AKT
activation by relieving a negative feedback on ERBB receptors.
Cancer Res. 72:3228–3237. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Engelman JA, Luo J and Cantley LC: The
evolution of phosphatidylinositol 3-kinases as regulators of growth
and metabolism. Nat Rev Genet. 7:606–619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hoeflich KP, Merchant M, Orr C, Chan J,
Den Otter D, Berry L, Kasman I, Koeppen H, Rice K, Yang NY, et al:
Intermittent administration of MEK inhibitor GDC-0973 plus PI3K
inhibitor GDC-0941 triggers robust apoptosis and tumor growth
inhibition. Cancer Res. 72:210–219. 2012. View Article : Google Scholar
|
|
8
|
Sos ML, Fischer S, Ullrich R, Peifer M,
Heuckmann JM, Koker M, Heynck S, Stückrath I, Weiss J, Fischer F,
et al: Identifying genotype-dependent efficacy of single and
combined PI3K- and MAPK-pathway inhibition in cancer. Proc Natl
Acad Sci USA. 106:18351–18356. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jokinen E, Laurila N and Koivunen JP:
Alternative dosing of dual PI3K and MEK inhibition in cancer
therapy. BMC Cancer. 12:6122012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bedard P, Tabernero J, Kurzrock R, et al:
A phase lb, open-label, multicenter, dose-escalation study of the
oral pan-PI3K inhibitor BKM120 in combination with the oral MEK1/2
inhibitor GSK1120212 in patients (pts) with selected advanced solid
tumors. J Clin Oncol (ASC Annual Meeting abstracts).
30:30032012.
|
|
11
|
Britten C, Wainberg Z, Tabernero J, et al:
A multi-arm phase 1 dose escalation study of safety,
pharmacokinetics, and pharmacodynamics of the dual PI3K/mTOR
inhibitors PF-04691502 (oral) and PF-05212384 (IV) in combination
with the MEK inhibitor PD-0325901 or irinotecan in patients with
advanced cancer. Eur J Cancer. 48:1092012. View Article : Google Scholar
|
|
12
|
LoRusso P, Shapiro G, Pandya SS, et al: A
first-in-human phase 1b study to evaluate the MEK inhibitor
GDC-0973, combined with the pan-PI3K inhibitor GDC-0941, in
patients with advanced solid tumors. J Clin Oncol (ASC Annual
Meeting abstracts). 30:25662012.
|
|
13
|
Faber AC, Coffee EM, Costa C, Dastur A,
Ebi H, Hata AN, Yeo AT, Edelman EJ, Song Y, Tam AT, et al: mTOR
inhibition specifically sensitizes colorectal cancers with KRAS or
BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1.
Cancer Discov. 4:42–52. 2014. View Article : Google Scholar :
|
|
14
|
Hata AN, Yeo A, Faber AC, Lifshits E, Chen
Z, Cheng KA, Walton Z, Sarosiek KA, Letai A, Heist RS, et al:
Failure to induce apoptosis via BCL-2 family proteins underlies
lack of efficacy of combined MEK and PI3K inhibitors for
KRAS-mutant lung cancers. Cancer Res. 74:3146–3156. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar
|
|
16
|
Morelli MP, Tentler JJ, Kulikowski GN, Tan
AC, Bradshaw-Pierce EL, Pitts TM, Brown AM, Nallapareddy S,
Arcaroli JJ, Serkova NJ, et al: Preclinical activity of the
rational combination of selumetinib (AZD6244) in combination with
vorinostat in KRAS-mutant colorectal cancer models. Clin Cancer
Res. 18:1051–1062. 2012. View Article : Google Scholar
|
|
17
|
Wee S, Jagani Z, Xiang KX, Loo A, Dorsch
M, Yao YM, Sellers WR, Lengauer C and Stegmeier F: PI3K pathway
activation mediates resistance to MEK inhibitors in KRAS mutant
cancers. Cancer Res. 69:4286–4293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ellis L, Ku SY, Ramakrishnan S, Lasorsa E,
Azabdaftari G, Godoy A and Pili R: Combinatorial antitumor effect
of HDAC and the PI3K-Akt-mTOR pathway inhibition in a Pten
defecient model of prostate cancer. Oncotarget. 4:2225–2236.
2013.PubMed/NCBI
|
|
19
|
He L, Torres-Lockhart K, Forster N,
Ramakrishnan S, Greninger P, Garnett MJ, McDermott U, Rothenberg
SM, Benes CH and Ellisen LW: Mcl-1 and FBW7 control a dominant
survival pathway underlying HDAC and Bcl-2 inhibitor synergy in
squamous cell carcinoma. Cancer Discov. 3:324–337. 2013. View Article : Google Scholar : PubMed/NCBI
|