Introduction
Neovascularization of solid tumours plays an
important role in tumour cell growth and metastasis (1). Although numerous growth factors and
cytokines stimulate angiogenesis, vascular endothelial growth
factor (VEGF) plays the predominant role in stimulating
neovascularization (1). VEGF is
overexpressed by a majority of solid tumours, and circulating
levels of VEGF are elevated in many cancer patients, including lung
cancer (2). Activation of VEGF
receptor (primarily VEGFR2) downstream signalling pathways by VEGF
increases vascular permeability and promotes endothelial cell
proliferation, survival and migration in both physiological and
pathological angiogenesis (2).
Several approaches to inhibiting tumour angiogenesis
by targeting VEGF signalling have been developed (3–6) and
are currently approved for use in the clinic against a number of
tumour types including colorectal (3), renal (5), glioblastoma (7), hepatocellular (8) and lung (9). However, identification of the patient
subsets which responds to VEGF signalling inhibition remains
elusive (10).
VEGFR2 protein has been reported to be expressed in
cells of solid tumours including breast (10), gastrointestinal (11), prostate (7), melanoma (12,13)
and non-small cell lung carcinoma (NSCLC) (14–19).
In principal, the use of VEGF-signalling inhibitors in the
treatment of these cancers might inhibit tumour angiogenesis and
additionally reduce tumour cell proliferation, invasion and
survival.
The role of VEGFR2 protein expression in NSCLC has
not yet been elucidated. The aim of this work is to investigate the
role of VEGFR2 in NSCLC cell lines and the potential impact of
signalling inhibition.
Materials and methods
Materials
Recombinant human VEGF165 (R&D Systems,
Abingdon, UK) was prepared in sterile dH2O. Cisplatin
(Sigma, Dorset, UK) was prepared at 3.3 mM in PBS. Docetaxel,
gemcitabine, pemetrexed (LC Laboratories, Woburn, UK) and the AKT
inhibitor, MK2206, MEK inhibitor, AZD6244, and VEGFR inhibitor,
Cediranib/AZD2171 (6) (Selleck,
Suffolk, UK), were prepared as 10 mM stocks in DMSO and stored at
−20°C. Formalin-fixed tumour samples were obtained from ProteoGenex
(Culver City, CA, USA). For radiation, cells were exposed to 10 Gy
(137Cs, 1.958 Gy/min) in a Gamma services GSR-D1
irradiator. Hoechst 33258 (Sigma) was prepared in dH2O
at 10 mg/ml and stored at 4°C.
Cell lines
SKBR3 (Leibniz Institute DSMZ, Braunschweig,
Germany), H3122 [National Cancer Institute (NCI), USA] and other
cell lines (all from ATCC; Manassas, VA, USA), and were cultured in
Advanced DMEM-F12 (Life Technologies, Paisley, UK) media with 5%
foetal bovine serum (Sigma), 2 mM GlutaMAX (Life Technologies) and
50 units of penicillin/50 μg/ml streptomycin (Life Technologies) at
37°C with 7.5% CO2.
Immunohistochemistry
Sections (5 μm) of formalin-fixed NSCLC cell line
pellets (n=25) or normal lung (n=4) or NSCLC tumour (n=52) were
incubated overnight with a rabbit polyclonal antibody (CST #2479)
against human VEGFR2. Formalin-fixed paraffin-embedded tumour
samples were obtained from ProteoGenex with written patient consent
and institutional review board/independent ethics committee
(IRB/IEC) approval. Sections (5 μm) were washed and incubated with
horseradish peroxidase (HRP)-linked goat anti-rabbit IgG and then
stained with diaminobenzidine (DAB). For cell lines, staining
categories (0, +, ++, +++) were defined using cell line pellets
with cells of known high (TT, +++), medium (H441, ++), low (H1792,
+) and negative (Calu3, 0) VEGFR2 expression. For evaluable tumour
samples (n=51), tumour cell VEGFR2 expression was scored as
positive (1; moderate or strong staining) or negative (0; weak or
no staining) as previously reported (15).
Quantitative PCR
Total RNA was isolated from NSCLC cell lines using
RNeasy (Qiagen, Venlo, The Netherlands) and reverse transcribed
into cDNA using iScript (Bio-Rad, Hertfordshire, UK). VEGFR2 cDNA
was amplified by RT-PCR (Applied Biosystems, Paisley, UK) and
expression quantified by the ΔΔCt method using peptidylprolyl
isomerase A (PPIA) as the control. Primer sequences were: VEGFR2
forward: TTT CGC CCG GCT CGA GG TGC, VEGFR2 reverse: CTA GGC AAA
CCC ACA GAG GCG GC; PPIA forward: CGC CAC CGC CGA GGA AAA CCG, PPIA
reverse: CTG CAA ACA GCT CAA AGG AGA CGC GG.
Immunoblotting
Immunoblotting was carried out as previously
described (20). For VEGF
stimulation, 6-well plates with cells at 70% confluency were
incubated in low serum conditions (0.2% FBS) ± cediranib (100 nM)
overnight and then treated with VEGF (0–100 ng/ml) for 0–60 min.
Protein was extracted using 50 mM Tris-HCl pH 7.6, 137 mM NaCl, 10%
glycerol, 0.1% Igepal, 0.1% SDS, 50 mM NaF, 1 mM
Na3VO4 and cocktail protease inhibitor (1 tab
per 25 ml of lysis buffer) on ice for 10 min. Immunoblots were
incubated overnight at 4°C with antibodies against total and
phosphorylated VEGFR2 (CST, Hertfordshire, UK. 1:800 dilution),
p42/44 MAPK (CST, 1:1,000 dilution), AKT (CST, 1:1,000 dilution),
PARP (CST, 1:1,000 dilution) and PPIB (Abcam, Cambridge, UK;
1:2,000). After washing, blots were incubated for 40 min with
anti-mouse or anti-rabbit LI-COR secondary antibodies (LI-COR,
Cambridge, UK; 1:50,000 dilution) or with horse-radish peroxidase
(HRP) conjugated goat anti-mouse (Thermo Scientific, Boston, MA,
USA; 1:8,000 dilution) or anti-rabbit (Thermo Scientific; 1:6,000
dilution) for 1 h. Detection was carried out using the LI-COR,
Odyssey or ECL substrate (Thermo Scientific).
Phosphorylated VEGFR2 ELISA
Phosphorylated VEGFR2 levels in cell lysates were
determined with the PathScan phospho-VEGFR-2 (Tyr1175) Sandwich
ELISA Kit Cell (CST; UK) according to manufacturer's
instructions.
Cell proliferation assay
Subconfluent cells were trypsinised, washed once
with PBS and seeded in 0.2% FBS DMEM/F12 media at a density of
1,000 to 4,000 cells per well in 96-well plates. The plates were
incubated overnight at 37°C with 7.5% CO2 to allow cells
to attach, and then treated with VEGF (0–100 ng/ml) ± drug and
incubated for a further 5 days. Cells were then stained with
crystal violet, allowed to dry and the dye eluted using glacial
acetic acid. The absorbance was read at 590 nm using the POLARstar
Omega plate reader.
VEGFR2 siRNA transfection
Small interfering RNAs (siRNA) targeting siRNA
control (ON TARGETplus control siRNA, 100 nM) and VEGFR2 (ONTARGET
plus smartpool, 100 nM) (Dharmacon, Inc., UK) smartpools were
transfected into H441 cells using DharmaFECT 2 reagent (4 μl/ml,
Dharmacon, Inc.) according to manufacturer's instructions. For the
proliferation assay end point, cells were incubated in the
transfection media for 24 h. The transfection media was removed and
replaced with 100 μl of fresh 0.2% FBS DMEM/F12. DMEM/F12 treated
with ± FBS or VEGF was added and plates were incubated for a
further 4 days. At this point the cells were stained with crystal
violet, allowed to dry and eluted with glacial acetic acid. The
absorbance was read at 590 nm using the POLARstar Omega plate
reader.
Cell death assay
Cells were trypsinised, suspended in phenol red free
10% DMEM/F12 media and seeded at a density of 10,000 to 40,000
cells/ml in 96-well plates. Plates were incubated at 37°C with 7.5%
CO2 overnight and then treated with drug ± 100 ng/ml
VEGF and incubated for a further 24 h. Hoechst 33258 (15 μM) was
added and incubation continued in the dark at 37°C with 7.5%
CO2 for 30 min. Images of each well were captured using
the In Cell Analyzer 1000 and analysed using Analyzer 1000
software. Live cells were distinguished from dead cells by the
intensity of Hoechst binding.
Migration assay
Boyden chambers were placed in a 24-well plate and
rinsed once with serum free media. Subconfluent cells were
trypsinised, wash once with PBS and seeded in 0.2% FBS DMEM/F12
media at a density of 200,000 cells per 50 μl per chamber. Into
each chamber 50 μl of 0.2% FBS media treated with of VEGF ±
cediranib was added and 250 μl of 0.2% FBS media was added below
the chamber. The plate was incubated at 37°C for 48 h, at which
point the migrated cells were stained with crystal violet. Migrated
cells were counted under an inverted microscope.
Statistics
Statistical comparisons was carried our using the
Student's t-test. Significance was set at P≤0.05.
Results
VEGFR2 expression in NSCLC tumour cells
and cell lines
VEGFR2 expression in lung cancer cells remains
unresolved (15,21–23).
Using a validated antibody (22,23),
we found VEGFR2 expression was present on the blood vessels of all
tumour samples and in the tumour cells of 20% all samples (Fig. 1A). Expression was present in both
adenocarcinoma and squamous cell carcinoma samples, but not in
normal lung epithelial cells. In cell lines, VEGFR2 expression was
seen in 9 out of 25 NSCLC cell lines by IHC and this was confirmed
in 3 NSCLC lines by immunoblotting and RT-PCR (Fig. 1D). By immunoblotting,
phosphorylation of VEGFR2 was also detectable in the H441 cells
(Fig. 1E).
VEGF stimulation of VEGFR2
phosphorylation in NSCLC cells
Immunoblotting against total VEGFR2 and quantitative
ELISA against phospho-VEGFR2 showed that, after VEGF treatment,
phosphorylation of VEGFR2 Y1175 was increased after 2 min, peaked
at 5 min and had returned to baseline levels by 10 min (Fig. 2A). Immunoblotting against
downstream proteins showed that AKT phosphorylation was reduced
after 2 min but that p42/44 MAPK was significantly increased from 2
to 10 min (Fig. 2B). This
activation corresponded with the phosphorylation of VEGFR2. The
addition of cediranib prior to VEGF stimulation prevented or
reduced the phosphorylation levels of VEGFR2 in the H441 cells
(Fig. 2C).
Activation of VEGFR2 signalling is
associated with increased NSCLC cell proliferation
VEGF-induced phenotypic effects were first evaluated
by proliferation assays (Fig. 2D and
E). VEGF treatment over a 5-day period resulted in a 20–60%
increase in H441 cell proliferation relative to the untreated
control (Fig. 2D). Noteworthy, the
level of VEGF-stimulation of H441 tumour cell proliferation was
similar to the 35–100% increase in VEGF-stimulated endothelial cell
proliferation previously reported (24,25).
The addition of cediranib (a VEGFR tyrosine kinase inhibitor)
prevented this increase in VEGF-stimulated cell proliferation
(Fig. 2D). There was no effect of
cediranib in H2009 or H1975 NSCLC cell lines which did not express
VEGFR2 (data not shown). To validate these results we used VEGFR2
siRNA (Fig. 2F) which also reduced
VEGF-stimulated H441 cell proliferation (Fig. 2E) suggesting that VEGF-induced cell
proliferation is via VEGFR2 signalling.
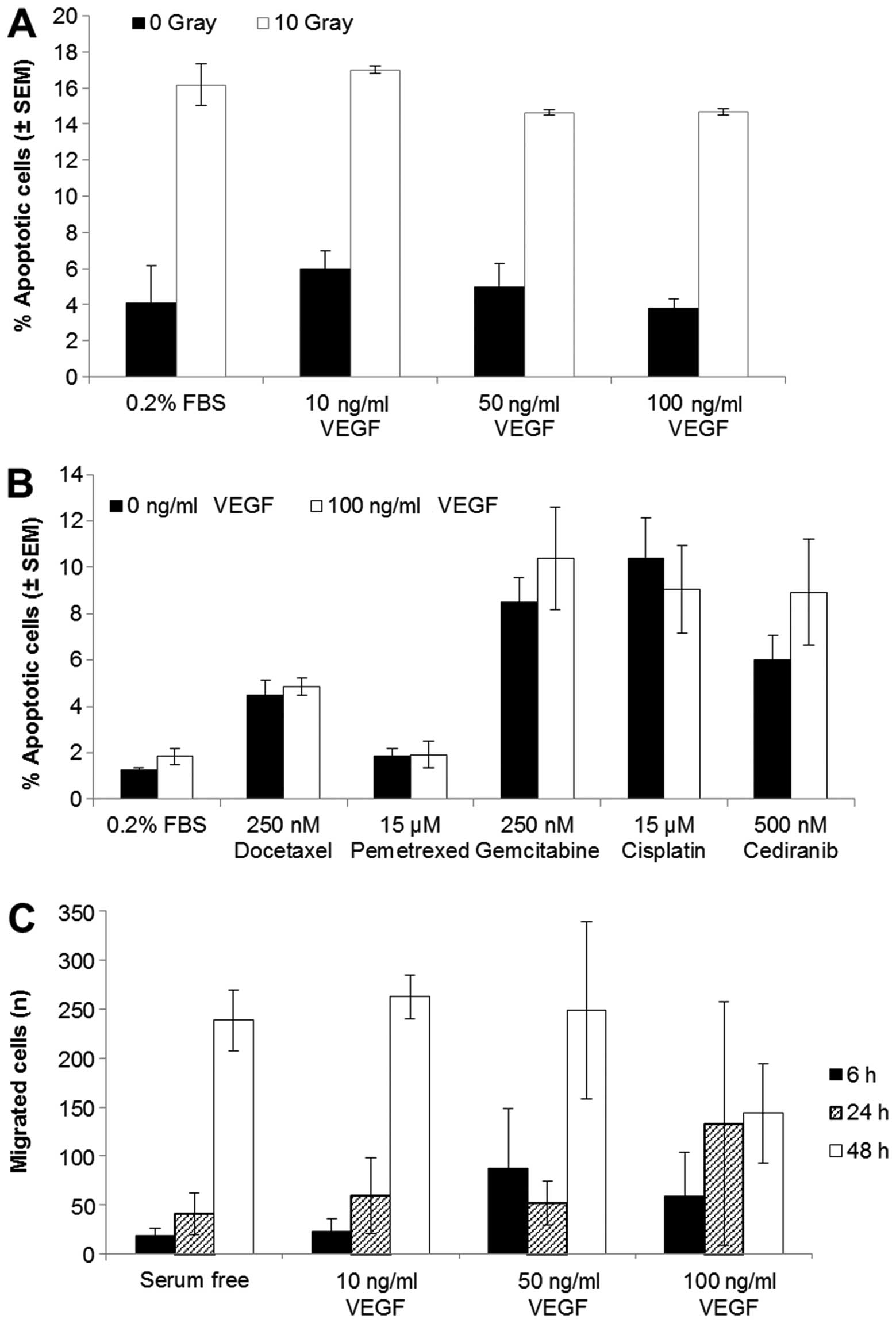
Radiation or drug-induced apoptosis are
not affected by stimulation of VEGFR2
When we exposed VEGFR2 stimulated cells to
irradiation (10 Gy, Fig. 3A) or to
docetaxel, pemetrexed, gemcitabine, cisplatin or cediranib
(Fig. 3B) there was no decrease in
cell apoptosis compared with unstimulated cells. H441 cells treated
with radiation had a 12% increase in apoptotic cells compared to
the untreated cells. The addition of 10, 50 or 100 ng/ml VEGF 4 h
prior to radiation did not reduce this induction of cell death
(Fig. 3A). The chemotherapeutic
drugs also increased the percentage of apoptotic cells relative to
the untreated control after 24 h; however, VEGF-stimulated
signalling did not prevent this increase (Fig. 3B).
Effect of VEGFR2 activation on tumour
cell migration
To determine if VEGFR2 signalling plays a role in
NSCLC cell migration, we carried out migration assays of H441 cells
in the presence or absence of VEGF (10, 50 or 100 ng/ml) (Fig. 3C). There was an increase in
migration over a 48 h period; however, the stimulation of VEGFR2
signalling did not alter the migration of these cells. Wound
healing (scratch) assays were also carried out under the same
conditions but there was no increase in wound repair in the
presence of VEGF (data not shown).
Inhibition of VEGFR2 kinase activity or
downstream signalling
Immunoblotting lysates of H441 cells treated with
cediranib, MK2206 (AKT inhibitor) or AZD6244 (MEK inhibitor) for 24
h showed that MK2206 significantly reduced AKT phosphorylation and
AZD6244 reduced p42/44 MAPK phosphorylation and, to a lesser
extent, AKT phosphorylation (Fig.
4A). Cediranib treatment did not produce a sustained reduction
in AKT or p42/44 MAPK phosphorylation after 24 h (Fig. 4A), however only cediranib
maintained a reduction in phosphorylated levels of VEGFR2 (Fig. 4B). In the proliferation assays,
cediranib was more effective than the AKT inhibitor or MEK
inhibitor (Fig. 4C) at reducing or
preventing VEGF stimulated VEGFR2 proliferation.
Discussion
It is well established that VEGFR2 activation plays
a pivotal role in increased vascular permeability and in EC
proliferation, migration and invasion (26). VEGFR2 protein expression has also
been reported in tumour cells of haematological and solid tumours
including breast, colon, prostate and melanoma, where roles in
tumour cell proliferation, survival and migration were reported
(7,10–12,27,28).
While VEGFR2 expression has been reported in lung cancer by some
groups (15,21,23,29,30);
the clinical relevance of this expression remains uncertain
(14,19,31).
Others have reported that VEGFR2 is not expressed in NSCLC
(22).
We confirmed the expression of VEGFR2 protein in
NSCLC tumour samples by IHC using a validated antibody (22,23)
and confirmed VEGFR2 mRNA and protein expression in a subset of
NSCLC cell lines. The kinetics of VEGF-dependent VEGFR2 and MAPK
phosphorylation in NSCLC cells was consistent with other studies in
ECs (32,33) suggesting VEGF activation of VEGFR2
signalling may be similar in both cell types. However, in contrast
with previous work (30,34), our data suggest that VEGFR2
activation in NSCLC cells is associated with a rapid decrease in
pAKT. Contrasting effects of VEGFR2 signalling on tumour cell
proliferation have been reported, with some studies suggesting
increased tumour cell proliferation (7,29)
whereas others suggest inhibition (35,36).
In our study, VEGFR2 TK (cediranib) fully inhibited
VEGF-dependent VEGFR2 proliferation in NSCLC cells, whereas MAPK
inhibitor (AZD6244) was not effective. AKT inhibitor (MK2206)
partially reduced VEGF-dependent proliferation although this was
not associated with a reduction in pVEGFR2. Of note, both pAKT and
pMAPK (but not pVEGFR2) levels returned to control levels within 24
h of treatment with cediranib, but not with MK2206 or AZD6244,
respectively. This suggests that the activity of MAPK and AKT
signalling pathways are not directly driving VEGFR2-dependent
proliferation in NSCLC. VEGFR2-dependent changes in pAKT have not
been reported for other solid tumour types so the importance of
this decrease in pAKT in NSCLC is not known. We also found that
cediranib significantly reduced VEGFR2 phosphorylation in untreated
NSCLC cells indicating a potential functional autocrine VEGF/VEGFR2
signalling loop as previously suggested for melanoma (27).
We were not able to demonstrate a significant role
for VEGF-dependent VEGFR2 signalling in NSCLC cell migration or
survival following treatment with radiation or cytotoxic agents.
This suggests a more restricted role for VEGFR2 signalling in NSCLC
than the roles that have been reported in ECs and other tumour
types (29). AKT is recognized as
a general mediator of survival signals following exposure to
cytotoxic agents (37). However,
in our studies on NSCLC cells, VEGF-stimulation did not increase
AKT signalling and VEGFR2 inhibition did not reduce pAKT levels,
which could underlie the lack of effect on survival.
VEGFR2-dependent increases in pMAPK have been
reported for carcinoid and SCLC, and were associated with increases
in tumour cell migration (29,35).
However, in our studies, VEGFR2 expressing NSCLC cells were poorly
migratory in both Boyden chambers and scratch wound assays and
therefore we were unable to demonstrate a significant effect of
exogenous VEGF in these assays. Although our study shows an acute
increase of pMAPK levels following VEGF stimulation, this returned
to baseline levels within 30 min suggesting MAPK pathway activity
may not be sustained sufficiently to produce a migratory
phenotype.
Inhibition of VEGF signalling markedly inhibits
tumour growth of human tumour xenografts from a broad range of
solid tumour types, including lung cancer, suggesting that
inhibition of angiogenesis is the dominant mode of action in mouse
models (30,38). Conversely, in unselected patients
in human disease, VEGFR inhibitors as single agents show only
modest activity in NSCLC (39–41)
but greater activity in other tumour settings (5,7,8),
perhaps suggesting that non-anti-angiogenic modes of action may
contribute more significantly to activity in lung cancer in certain
patients in the clinic.
Currently, there are no biomarkers that can identify
the patients that respond to VEGF-signalling inhibitors. The
majority of solid tumours express VEGF and levels are known to
increase with tumour stage (17)
and to correlate with shorter time to tumour progression and poor
prognosis (2). In addition, high
tumour cell VEGFR2 expression in NSCLC has been shown to be
associated with poor prognosis (19,31).
To our knowledge, this is the first study of a VEGF/VEGFR2
dependent cell proliferation pathway in NSCLC, with potential to
drive tumour growth through an autocrine signalling pathway. An
autocrine VEGF/VEGFR2 signalling loop has been reported in NSCLC
cells acting to stimulate VEGF production and angiogenesis
(42).
In conclusion, collectively, our results suggest
that patients with NSCLC whose tumour cells express high levels of
VEGFR2 may have tumour growth stimulated through both angiogenesis
and increased proliferation. As a consequence, VEGFR2-expressing
NSCLC tumours may be particularly sensitive to VEGFR2 TKIs through
two distinct effects of VEGF. Our data also suggest that VEGFR2
expression in tumour cells is biologically relevant, and is a
potential biomarker to identify NSCLC patients who may gain the
greatest benefit from anti-VEGFR2 therapy.
Acknowledgements
Funding was provided by the UK Medical Research
Council (MC_PC_12006).
Abbreviations:
|
VEGF
|
vascular endothelial growth factor
|
|
VEGFR2
|
vascular endothelial growth factor
receptor 2
|
|
SCLC
|
small cell lung carcinoma
|
|
NSCLC
|
non-small cell lung carcinoma
|
|
IHC
|
immunohistochemistry
|
|
TKI
|
tyrosine kinase inhibitor
|
|
EC
|
endothelial cells
|
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferrara N: Vascular endothelial growth
factor as a target for anticancer therapy. Oncologist. 9(Suppl 1):
2–10. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hurwitz H, Fehrenbacher L, Novotny W,
Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S,
Holmgren E, et al: Bevacizumab plus irinotecan, fluorouracil, and
leucovorin for metastatic colorectal cancer. N Engl J Med.
350:2335–2342. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miller K, Wang M, Gralow J, Dickler M,
Cobleigh M, Perez EA, Shenkier T, Cella D and Davidson NE:
Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic
breast cancer. N Engl J Med. 357:2666–2676. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rini BI, Halabi S, Rosenberg JE, Stadler
WM, Vaena DA, Archer L, Atkins JN, Picus J, Czaykowski P, Dutcher
J, et al: Phase III trial of bevacizumab plus interferon alfa
versus interferon alfa monotherapy in patients with metastatic
renal cell carcinoma: Final results of CALGB 90206. J Clin Oncol.
28:2137–2143. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brave SR, Ratcliffe K, Wilson Z, James NH,
Ashton S, Wainwright A, Kendrew J, Dudley P, Broadbent N, Sproat G,
et al: Assessing the activity of cediranib, a VEGFR-2/3 tyrosine
kinase inhibitor, against VEGFR-1 and members of the structurally
related PDGFR family. Mol Cancer Ther. 10:861–873. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Abdollahi A, Lipson KE, Sckell A, Zieher
H, Klenke F, Poerschke D, Roth A, Han X, Krix M, Bischof M, et al:
Combined therapy with direct and indirect angiogenesis inhibition
results in enhanced antiangiogenic and antitumor effects. Cancer
Res. 63:8890–8898. 2003.PubMed/NCBI
|
|
8
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al; SHARP Investigators Study Group. Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:378–390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cabebe E and Wakelee H: Role of
anti-angiogenesis agents in treating NSCLC: Focus on bevacizumab
and VEGFR tyrosine kinase inhibitors. Curr Treat Options Oncol.
8:15–27. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wedam SB, Low JA, Yang SX, Chow CK, Choyke
P, Danforth D, Hewitt SM, Berman A, Steinberg SM, Liewehr DJ, et
al: Antiangiogenic and antitumor effects of bevacizumab in patients
with inflammatory and locally advanced breast cancer. J Clin Oncol.
24:769–777. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morelli MP, Brown AM, Pitts TM, Tentler
JJ, Ciardiello F, Ryan A, Jürgensmeier JM and Eckhardt SG:
Targeting vascular endothelial growth factor receptor-1 and -3 with
cediranib (AZD2171): Effects on migration and invasion of
gastrointestinal cancer cell lines. Mol Cancer Ther. 8:2546–2558.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Molhoek KR, Erdag G, Rasamny JK, Murphy C,
Deacon D, Patterson JW, Slingluff CL Jr and Brautigan DL: VEGFR-2
expression in human melanoma: revised assessment. Int J Cancer.
129:2807–2815. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Adamcic U, Skowronski K, Peters C,
Morrison J and Coomber BL: The effect of bevacizumab on human
malignant melanoma cells with functional VEGF/VEGFR2 autocrine and
intracrine signaling loops. Neoplasia. 14:612–623. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Donnem T, Al-Saad S, Al-Shibli K,
Delghandi MP, Persson M, Nilsen MN, Busund LT and Bremnes RM:
Inverse prognostic impact of angiogenic marker expression in tumor
cells versus stromal cells in non small cell lung cancer. Clin
Cancer Res. 13:6649–6657. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bonnesen B, Pappot H, Holmstav J and Skov
BG: Vascular endothelial growth factor A and vascular endothelial
growth factor receptor 2 expression in non-small cell lung cancer
patients: Relation to prognosis. Lung Cancer. 66:314–318. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
An SJ, Nie Q, Chen ZH, Lin QX, Wang Z, Xie
Z, Chen SL, Huang Y, Zhang AY, Yan JF, et al: KDR expression is
associated with the stage and cigarette smoking of the patients
with lung cancer. J Cancer Res Clin Oncol. 133:635–642. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matsuyama W, Hashiguchi T, Mizoguchi A,
Iwami F, Kawabata M, Arimura K and Osame M: Serum levels of
vascular endothelial growth factor dependent on the stage
progression of lung cancer. Chest. 118:948–951. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Masood R, Cai J, Zheng T, Smith DL, Hinton
DR and Gill PS: Vascular endothelial growth factor (VEGF) is an
autocrine growth factor for VEGF receptor-positive human tumors.
Blood. 98:1904–1913. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Seto T, Higashiyama M, Funai H, Imamura F,
Uematsu K, Seki N, Eguchi K, Yamanaka T and Ichinose Y: Prognostic
value of expression of vascular endothelial growth factor and its
flt-1 and KDR receptors in stage I non-small-cell lung cancer. Lung
Cancer. 53:91–96. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bokobza SM, Jiang Y, Weber AM, Devery AM
and Ryan AJ: Short-course treatment with gefitinib enhances
curative potential of radiation therapy in a mouse model of human
non-small cell lung cancer. Int J Radiat Oncol Biol Phys.
88:947–954. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pajares MJ, Agorreta J, Larrayoz M, Vesin
A, Ezponda T, Zudaire I, Torre W, Lozano MD, Brambilla E, Brambilla
C, et al: Expression of tumor-derived vascular endothelial growth
factor and its receptors is associated with outcome in early
squamous cell carcinoma of the lung. J Clin Oncol. 30:1129–1136.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Smith NR, Baker D, James NH, Ratcliffe K,
Jenkins M, Ashton SE, Sproat G, Swann R, Gray N, Ryan A, et al:
Vascular endothelial growth factor receptors VEGFR-2 and VEGFR-3
are localized primarily to the vasculature in human primary solid
cancers. Clin Cancer Res. 16:3548–3561. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Holzer TR, Fulford AD, Nedderman DM,
Umberger TS, Hozak RR, Joshi A, Melemed SA, Benjamin LE, Plowman
GD, Schade AE, et al: Tumor cell expression of vascular endothelial
growth factor receptor 2 is an adverse prognostic factor in
patients with squamous cell carcinoma of the lung. PLoS One.
8:e802922013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zeng H, Dvorak HF and Mukhopadhyay D:
Vascular permeability factor (VPF)/vascular endothelial growth
factor (VEGF) peceptor-1 down-modulates VPF/VEGF
receptor-2-mediated endothelial cell proliferation, but not
migration, through phos-phatidylinositol 3-kinase-dependent
pathways. J Biol Chem. 276:26969–26979. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Favot L, Keravis T, Holl V, Le Bec A and
Lugnier C: VEGF-induced HUVEC migration and proliferation are
decreased by PDE2 and PDE4 inhibitors. Thromb Haemost. 90:334–343.
2003.PubMed/NCBI
|
|
26
|
Prior BM, Yang HT and Terjung RL: What
makes vessels grow with exercise training? J Appl Physiol 1985.
97:1119–1128. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Matsumori Y, Yano S, Goto H, Nakataki E,
Wedge SR, Ryan AJ and Sone S: ZD6474, an inhibitor of vascular
endothelial growth factor receptor tyrosine kinase, inhibits growth
of experimental lung metastasis and production of malignant pleural
effusions in a non-small cell lung cancer model. Oncol Res.
16:15–26. 2006.PubMed/NCBI
|
|
28
|
Liang Y, Brekken RA and Hyder SM: Vascular
endothelial growth factor induces proliferation of breast cancer
cells and inhibits the anti-proliferative activity of
anti-hormones. Endocr Relat Cancer. 13:905–919. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tanno S, Ohsaki Y, Nakanishi K, Toyoshima
E and Kikuchi K: Human small cell lung cancer cells express
functional VEGF receptors, VEGFR-2 and VEGFR-3. Lung Cancer.
46:11–19. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu W, Onn A, Isobe T, Itasaka S, Langley
RR, Shitani T, Shibuya K, Komaki R, Ryan AJ, Fidler IJ, et al:
Targeted therapy of orthotopic human lung cancer by combined
vascular endothelial growth factor and epidermal growth factor
receptor signaling blockade. Mol Cancer Ther. 6:471–483. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Carrillo de Santa Pau E, Arias FC, Caso
Peláez E, Muñoz Molina GM, Sánchez Hernández I, Muguruza Trueba I,
Moreno Balsalobre R, Sacristán López S, Gómez Pinillos A and del
Val Toledo Lobo M: Prognostic significance of the expression of
vascular endothelial growth factors A, B, C, and D and their
receptors R1, R2, and R3 in patients with nonsmall cell lung
cancer. Cancer. 115:1701–1712. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shibuya M: Differential roles of vascular
endothelial growth factor receptor-1 and receptor-2 in
angiogenesis. J Biochem Mol Biol. 39:469–478. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brekken RA, Overholser JP, Stastny VA,
Waltenberger J, Minna JD and Thorpe PE: Selective inhibition of
vascular endothelial growth factor (VEGF) receptor 2 (KDR/Flk-1)
activity by a monoclonal anti-VEGF antibody blocks tumor growth in
mice. Cancer Res. 60:5117–5124. 2000.PubMed/NCBI
|
|
34
|
Abid MR, Guo S, Minami T, Spokes KC, Ueki
K, Skurk C, Walsh K and Aird WC: Vascular endothelial growth factor
activates PI3K/Akt/forkhead signaling in endothelial cells.
Arterioscler Thromb Vasc Biol. 24:294–300. 2004. View Article : Google Scholar
|
|
35
|
Silva SR, Bowen KA, Rychahou PG, Jackson
LN, Weiss HL, Lee EY, Townsend CM Jr and Evers BM: VEGFR-2
expression in carcinoid cancer cells and its role in tumor growth
and metastasis. Int J Cancer. 128:1045–1056. 2011. View Article : Google Scholar
|
|
36
|
Adham SA, Sher I and Coomber BL: Molecular
blockade of VEGFR2 in human epithelial ovarian carcinoma cells. Lab
Invest. 90:709–723. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Datta SR, Brunet A and Greenberg ME:
Cellular survival: A play in three Akts. Genes Dev. 13:2905–2927.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dias S, Hattori K, Zhu Z, Heissig B, Choy
M, Lane W, Wu Y, Chadburn A, Hyjek E, Gill M, et al: Autocrine
stimulation of VEGFR-2 activates human leukemic cell growth and
migration. J Clin Invest. 106:511–521. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Blumenschein GR Jr, Gatzemeier U, Fossella
F, Stewart DJ, Cupit L, Cihon F, O'Leary J and Reck M: Phase II,
multicenter, uncontrolled trial of single-agent sorafenib in
patients with relapsed or refractory, advanced non-small-cell lung
cancer. J Clin Oncol. 27:4274–4280. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Socinski MA, Novello S, Brahmer JR, Rosell
R, Sanchez JM, Belani CP, Govindan R, Atkins JN, Gillenwater HH,
Pallares C, et al: Multicenter, phase II trial of sunitinib in
previously treated, advanced non-small-cell lung cancer. J Clin
Oncol. 26:650–656. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Schiller JH, Larson T, Ou SH, Limentani S,
Sandler A, Vokes E, Kim S, Liau K, Bycott P, Olszanski AJ, et al:
Efficacy and safety of axitinib in patients with advanced
non-small-cell lung cancer: Results from a phase II study. J Clin
Oncol. 27:3836–3841. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chatterjee S, Heukamp LC, Siobal M,
Schöttle J, Wieczorek C, Peifer M, Frasca D, Koker M, König K,
Meder L, et al: Tumor VEGF:VEGFR2 autocrine feed-forward loop
triggers angiogenesis in lung cancer. J Clin Invest. 123:1732–1740.
2013. View Article : Google Scholar : PubMed/NCBI
|