Introduction
Hepatocellular carcinoma (HCC) is one of the most
common cancers worldwide and the third leading cause of
cancer-related death (1–4). Major risk factors for HCC development
are cirrhosis with underlying chronic viral hepatitis, alcoholic
liver disease and non-alcoholic steatohepatitis. Despite the
introduction of targeted therapies like the multi-kinase inhibitor
sorafenib, treatment with curative intention is still limited with
unsatisfactory overall survival results (5–9).
A prerequisite for the growth and spread of
malignant solid tumors is the formation of new blood vessels to
provide oxygen and nutrient supply to the mass of tumor cells
(10). Angiogenesis is a highly
complex process involving various growth factors such as vascular
endothelial growth factor (VEGF), growth factor receptors like
fms-like tyrosine kinase-1 (FLT-1; VEGF receptor-1) or kinase
insert domain containing receptor (KDR; VEGF receptor-2) as well as
extracellular matrix components like matrix metalloproteinases,
inflammatory cells including macrophages and intracellular
signaling pathways such as the Hif-1α cascade (11–13).
VEGF is an extracellular signaling molecule stimulating
proliferation and migration of endothelial (progenitor) cells and
regulates the permeability of blood vessels via activating a family
of transmembrane tyrosine kinase receptors (14–17).
FLT-1 and especially KDR are the central mediators of VEGF
signaling in angiogenesis and have been shown to be commonly
over-expressed in malignant tumors, including HCC, and their
overexpression is associated with limited overall survival of
patients (18–22). Although various antiangiogenic
compounds like the multi-kinase inhibitor sorafenib and the
monoclonal anti-VEGF antibody bevacizumab have been used for the
therapy of HCC, the overall efficacy of anti-angiogenic treatments
is still disappointing and these treatments are associated with
considerable side effects (23–25).
The role of the surrounding stroma as well as
inflammatory cells has recently been recognized as a key feature of
tumor-driven angiogenesis (26,27).
Connective tissue growth factor (CTGF), also known as CCN2, is a
38-kDa protein and was originally identified as a growth factor
secreted by human vascular endothelial cells (28). It is a multifunctional signaling
modulator involved in several physiologic and pathologic processes
such as fibrosis in kidneys and skin, osteogenesis, angiogenesis
and tumor development (29–33).
In HCC, CTGF has been shown to contribute to tumor growth in an
autocrine loop and its overexpression is negatively correlated with
overall survival (34–38). Previously, we have shown that the
expression of CTGF can be modulated by the histone deacetylase
inhibitor (HDACi) Trichostatin A (TSA) (39).
HDACi are currently evaluated as anticancer agents
for various hematologic and solid malignancies (40). Panobinostat (LBH589) represents a
novel pan-deacetylase inhibitor that has shown excellent efficacy
against HCC growth alone or combined in preclinical models and
early clinical trials (41–44).
Besides their classical mode of action as transcriptional
regulators, HDACi are now considered to interfere with a variety of
other cellular pathways like cytosolic protein stabilization and
signaling pathways, including angiogenesis-related signaling
(45–47).
We therefore investigated if and to what extent the
pan-deacetylase inhibitor panobinostat mediates its anti-angiogenic
properties via regulation of the CTGF signaling pathway in HCC cell
lines and in a subcutaneous xenograft model in vivo. Our
study shows that panobinostat leads to a significant growth delay
with prolonged overall survival, mediated by reduced tumor cell
proliferation, increased apoptosis and reduced angiogenesis in
tumor xenografts (41).
Materials and methods
Cell culture
The human hepatoma cell lines HepG2 (p53wt) and
Hep3B (p53null) were obtained from the German collection of
microorganisms and cell cultures (DSMZ, Braunschweig, Germany) and
maintained under standard conditions as described previously
(41). Panobinostat (LBH589) was
provided by Novartis Pharma AG (Basel, Switzerland) and was
prepared as previously described (41,45).
For all in vitro experiments, 300,000 cells were seeded to
6-well tissue culture plates (Becton-Dickinson, Heidelberg,
Germany) 24 h before treatment. Cells were treated with LBH589 at
0.1 and 0.01 μM dissolved in complete growth medium. The medium was
not changed during the experiments.
HepG2 xenograft samples
Samples from previously established xenografts of
HepG2 cells to male athymic nu/nu NMRI mice were used for this
study (41).
Protein isolation and western
blotting
Total protein content was isolated after
centrifuging hepatoma cells at 1,000 rpm for 10 min, discarding the
liquid phase and adding 50 μl of Jie's protein lysis buffer,
consisting of 10 mM NaCl2, 0.5% NP-40, 20 mM Tris-HCl pH
7.4, 5 mM MgCl2, 10 μg/ml Prot-I, 1 mM PMSF. After 30
min cooling on ice with intermediate vortexing, the suspension was
divided into two portions and stored at −80°C. Samples were
subjected to 6–14% SDS-PAGE (Invitrogen, Carlsbad, CA, USA),
transferred to a nitrocellulose membrane and blocked for 1 h at
room temperature in a TBS or PBS buffer containing 0.1% Tween-20
and 5% low fat milk powder. Membranes were incubated overnight with
primary antibodies against CTGF (1:500), VEGF (1:1,000), FLT-1
(1:1,000) (all from Abcam, Cambridge, UK), KDR (1:1,000; Merck
Millipore, Darmstadt, Germany), MAPK (1:1,000) and p-MAPK (1:1,000)
(both from Cell Signaling Technology, Danvers, MA, USA). Membranes
were incubated with a peroxidase coupled secondary antibody
(1:2,000, anti-mouse or anti-rabbit IgG; Pierce, Rockford, IL, USA)
for 1 h at room temperature (RT). Reactive bands were detected with
the ECL chemiluminescnce reagent (Amersham Pharmacia Biotech,
Freiburg, Germany) and analyzed using GelScan 5 software
(BioSciTec, Frankfurt, Germany). Signals were standardized to
β-actin (1:5,000; Sigma-Aldrich, Taufkirchen, Germany) content of
each sample.
Densitometric quantification of the western blot
analyses was done using Bio 1D (Vilber Lourmat, Eberhardzell,
Germany) and normalized to untreated controls and β-actin as the
loading control.
Quantitative real-time PCR
Treated cells and controls were harvested for qPCR
after 24–72 h using TRizol® (Ambion/Life Technologies,
Vienna, Austria) and total RNA extracted using Direct-zol RNA
MiniPrep kit (Zymo Research, Irvine, CA, USA). cDNA was synthetized
with 0.15 to 0.35 μg RNA using the GoScript™ Reverse Transcription
System (Promega, Mannheim, Germany). Finally, real-time PCR was
performed with GoTaq® qPCR Master Mix (Promega) on a
ViiA7 real-time PCR system (Applied Biosystems/Life Technologies)
using the following primers (all from Qiagen, Hilden, Germany):
β-actin [internal control/reference gene, Qiagen cat. no.
QT01680476), VEGF (QT01682072), FLT-1/VEGFR-1 (QT00073640),
KDR/VEGFR-2 (QT00069818) and CTGF1 (QT00052899)]. All procedures
were performed according to the respective user manuals. Specific
amplification was verified using melt curves and gene expression
calculated using the ΔΔCt method (48). Gene expression levels are presented
as mean expression relative to their untreated controls ± standard
error of the mean (SEM).
Immunohistohemistry (IHC)
Antigen retrieval was performed by heat induced
epitope retrieval in pH 9.0 antigen retrieval buffer (Dako,
Glostrup, Denmark) at 95°C for 60 min. Endogenous peroxidase
blocking was carried out for 10 min with peroxidase blocking
reagent (Dako). Subsequently, primary antibody against CTGF (1:50;
Abcam) was applied for 30 min at RT and detected using the EnVision
Detection system (Dako). Visualisation was performed using
diaminobenzidine (DAB) as the chromogen substrate (Roche Molecular
Biochemicals, Mannheim, Germany), all according to the
manufacturers' instructions. Slides were counterstained with
hematoxylin.
Statistical analysis and scientific
graphing
Significant differences in qPCR gene expression were
calculated using the t-test for paired samples and corrected for
multiple comparisons using the Holm-Sidak post-hoc test. P<0.05
was regarded as statistically significant. Statistics and
scientific graphing were performed using Prism 6 (GraphPad Software
Inc., La Jolla, CA, USA).
Results
In vitro experiments
Treatment of the p53-wild-type HepG2 cell line with
0.1 μM panobinostat induced a strong increase of CTGF mRNA
expression to 7.7-fold after only 24 h and values dropped to
2.3-fold after 48 h. At 72 h, treatment with panobinostat led to a
drastic reduction of cells numbers, thus very few viable cells were
left for analysis, even after repeated experiments, and these cells
expressed CTGF 0.8-fold compared to the untreated controls
(Fig. 1A and B). VEGF was induced
1.3-fold after 24 h, and levels after 48 and 72 h dropped to 0.9-
and 0.8-fold, respectively. At 0.01 μM, a less dramatic change in
gene expression was observed: VEGF was upregulated to 1.3-fold at
24 h, and dropped (in a similar fashion as in 0.1 μM treated cells)
to 0.7- and 0.6-fold at 48 and 72 h. At this lower concentration,
CTGF was gradually induced 1.0-, 1.2- and 3.1-fold at 24, 48 and 72
h, respectively. The gradual time-dependent CTGF induction at 0.01
μM in the HepG2 cells was paralleled by a strong induction at 24 h
at 0.1 μM followed by a drop of CTGF in the remaining viable cells
at 48 and 72 h at this higher concentration.
The clear time- and dose-dependent change in gene
expression seen in the p53-wild-type HepG2 cells was not observable
in the p53-deficient Hep3B cells. VEGF expression remained at
0.9-fold of untreated controls at 24 and 48 h and dropped to
0.5-fold after 72 h incubation with 0.1 μM panobinostat. At the
lower concentration of 0.01 μM LBH, VEGF ranged between 0.7- and
0.4-fold during the treatment period. However, CTGF was induced
4.4-, 2.2- and 3.3-fold after 24, 48 and 72 h incubation with 0.1
μM panobinostat (and to only 0.9-, 2.0- and 1.8-fold under 0.01 μM
panobinostat) (Fig. 1C and D).
FLT-1 was not expressed in vitro at any time-point in the
HepG2 and Hep3B cells.
In western blot analysis, we could not detect VEGF
expression in HepG2 cells and neither cell line expressed FLT-1 on
protein level (Fig. 2). VEGF in
Hep3B cells was induced at 24 h by panobinostat 2.4- and 4.2-fold
at concentrations of 0.01 and 0.1 μM, respectively, as calculated
by densitometry. Longer incubation (48 h) reduced VEGF expression
to 1.6- and 0.2-fold at 0.01 and 0.1 μM. CTGF was induced most
effectively in HepG2 cells by 0.1 μM panobinostat at either 24 or
48 h to levels of 1.5- and 1.6-fold, respectively. In Hep3B cells,
24 h incubation induced CTGF most prominently, but the lower dose
of 0.01 μM was more effective than 0.1 μM and the CTGF levels were
5.3- and 2.7-fold elevated in this setting. In both cell lines,
elevated levels of MAPK were accompanied by elevated levels of
p-MAPK and vice versa. The two highest levels of MAPK in HepG2
(2.0-fold at 24 h 0.01 μM and 1.1-fold at 48 h 0.01 μM) were
accompanied by the two highest levels of p-MAPK (1.2-fold at 24 h
0.01 μM and 1.4-fold at 48 h 0.01 μM), resulting in a MAPK/p-MAPK
ratio of 1.67 and 0.79 at 24 and 48 h for 0.01 μM. At all other
time-points and concentrations, MAPK and p-MAPK were suppressed
compared to the untreated controls. The lowest expression in HepG2
was thus observed at 12 h 0.01 μM for both MAPK (0.7-fold) and
p-MAPK (0.6-fold) (ratio MAPK/p-MAPK, 1.17) and in Hep3B at 12 h
0.1 μM also for both MAPK (0.4-fold) and p-MAPK (0.2-fold) (ratio
2). KDR levels were generally suppressed at all time-points and
concentrations, with the lowest level of 0.55-fold in HepG2 and
0.51-fold in Hep3B (vs. untreated controls) at 48 h 0.1 μM in both
cell lines.
 | Figure 2Western blot analysis of expression
of connective tissue growth factor (CTGF), vascular endothelial
growth factor (VEGF), fms-like tyrosine kinase-1 (FLT-1), kinase
insert domain containing receptor (KDR), MAPK, p-MAPK in
vitro and densitometric quantification. HepG2 and Hep3B cells
were incubated with 0.1 and 0.01 μM panobinostat for 12, 24 and 48
h. Western blot results show representative examples for expression
of CTGF, VEGF, FLT-1, KDR, MAPK and p-MAPK as well as β-actin,
which served as loading control (A). Protein expression analyzed
using western blot analysis was quantified by densitometry (B).
Shown are expression values for CTGF, MAPK, p-MAPK, KDR and VEGF
after treatment of HepG2 and Hep3B cell lines with 0.01 and 0.1 μM
of panobinostat for 12–48 h. |
HepG2 xenografted nude mice
Treatment of nude mice bearing HepG2 xenografts with
daily i.p. injections of 10 mg/kg panobinostat resulted in an
increase of VEGF on mRNA level 2.0-, 2.5- and 1.5-fold after 1 day,
1 week and 4 weeks, respectively (Fig.
3). CTGF was upregulated 2.6-, 1.1- and 2.0-fold while FLT-1
reached an expression of 1.2-, 1.0- and 2.4-fold in treated vs. the
untreated mice. After 4 weeks of treatment of the HepG2 xenografts
in the nude mice, the western blot analysis revealed an relative
expression change of CTGF (1.1-fold, vs. untreated xenografts),
p-MAPK (1.2-fold) and KDR (1.3-fold) and a downregulation of MAPK
to 0.4-fold (p=0.001) of untreated controls (Fig. 4).
 | Figure 4Western blot analysis of expression
of connective tissue growth factor (CTGF), vascular endothelial
growth factor (VEGF), fms-like tyrosine kinase-1 (FLT-1), kinase
insert domain containing receptor (KDR), MAPK, p-MAPK and
densitometric analysis of protein expression in vivo.
Western blot analysis of representative HepG2 xenograft specimens
from untreated controls or animals receiving 10 mg/kg panobinostat.
Tumor samples were obtained at the end of the treatment period and
subjected to semiquantitative western blotting against CTGF, VEGF,
FLT-1, KDR, MAPK and p-MAPK. β-actin served as a loading control
(A). Protein expression was quantified by densitometry. Shown are
expression values for CTGF, MAPK, p-MAPK, KDR and after treatment
of HepG2 xenograft specimens treated with 10 mg/kg panobinostat for
4 weeks (B). |
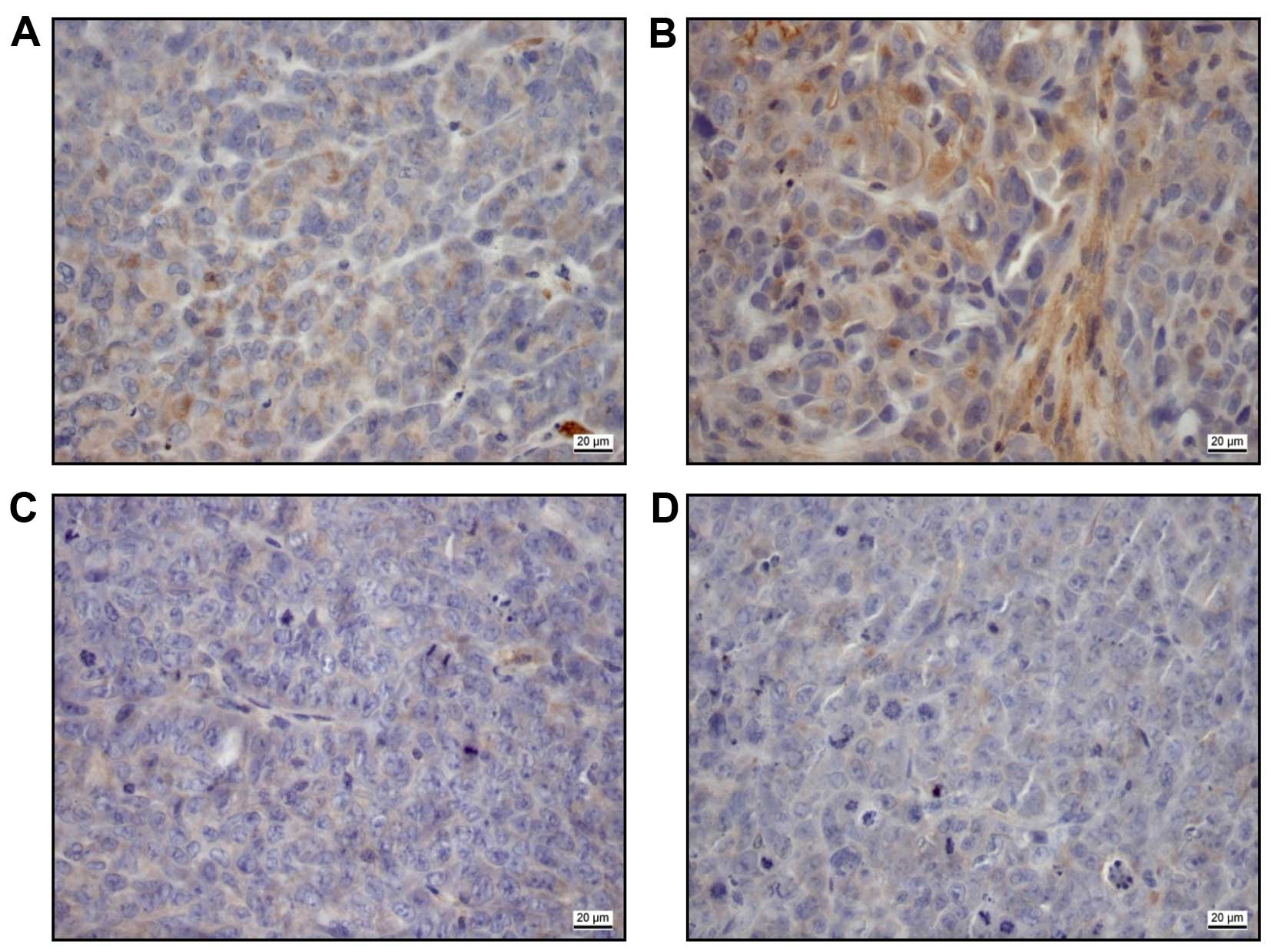
In immunohistochemical staining of the explanted
xenografts after 4 weeks (Fig. 5),
the 10 mg/kg panobinostat treated samples showed an expression of
CTGF [low (Fig. 5A), moderate
(Fig. 5B)] in contrast to 2.5 mg
panobinostat treated samples (Fig.
5C) and untreated controls (Fig.
5D).
Discussion
In the present study we investigated the
anti-angiogenic properties of the pan-deacetylase inhibitor
panobinostat via regulation of the CTGF signaling pathway in HCC
cell lines and a subcutaneous xenograft model in vivo.
Panobinostat (LBH589) plays an important role in
anti-angiogenesis (45). Recently,
Di Fazio et al showed a panobinostat-mediated significant
growth delay of a subcutaneous HCC xenograft model with prolonged
overall survival, mediated by reduced tumor cell proliferation,
increased apoptosis and especially reduced angiogenesis (41). As key message of this study,
panobinostat induces alternative apoptotic pathways dependent on
the p53 status. Furthermore, it was shown macroscopically that the
microvascular density and tumor size of panobinostat treated HCC
xenografts were significantly decreased.
Our results show that panobinostat induces a
context-dependent differential expression of CTGF in vitro
and in vivo in a subcutaneous xenograft model after daily
i.p. injections of 10 mg/kg panobinostat. The previously shown
tumor growth inhibition (41) was
associated with the inhibition of the MAPK signaling pathway and
with inhibition of tumor vascularization. For this study, in
vivo samples from those previous experiments (41) were used. Interestingly, the protein
levels of CTGF in the western blot analyses were downregulated,
while the mRNA expression of CTGF in HepG2 and Hep3B cells after 24
and 48 h were mainly upregulated. An increased expression of CTGF
was also seen in the immunohistochemical analysis of panobinostat
treated xenograft samples. Because CTGF can be both secreted and
membrane bound, western blot analysis of the cell lysate may not
reflect the total protein amount produced when compared to mRNA
expression. While in mesangial cells it is bound to the cell
surface (49), it has been shown
that in hepatocellular carcinoma cells, a fraction of the produced
protein remains cell-bound (38).
This may explain why our mRNA and protein expression data do not
necessarily correlate. One other reason may be that the CTGF mRNA
pool does not necessarily translate into a 1:1 expression on the
protein level. A further reason could be the fact that in the in
vivo xenograft model, the CTGF expression is especially
triggered by endothelial cells. This assumption is supported by
findings of Komorowsky et al (39): treatment of cultured endothelial
cells with different HDAC inhibitors upregulated CTGF mRNA and
protein. Their data indicate that the effect of HDAC inhibitors on
CTGF expression is largely cell dependent in non-tumor cells.
In this study VEGF and FLT-1 (VEGFR-1) are not
expressed and thus do not seem to play a role in treatment with
panobinostat, especially in the xenograft model. VEGF and the VEGFR
system are known to be main regulators of angiogenesis. In this
study there is absolutely no expression of VEGF and FLT-1,
especially in the controls of the xenograft samples. It could be
speculated that an alternative angiogenesis pathway is activated in
this xenograft model involving the xenograft environment (50), especially because KDR (VEGFR-2) is
expressed and induced in the xenografts after treatment.
Panobinostat is a pan-deacetylase inhibitor and,
like many other compounds targeting histone deacetylases, a
promising drug against different malignant tumors (51–55).
Especially solid malignant tumors including prostate (56,57),
breast (58,59) and lung cancers (60,61)
are supposed to be angiogenesis-dependent as shown for
hepatocellular carcinoma (62–64).
Tumor progression of HCC is associated with angiogenesis and the
increase in microvascular density is associated with a poor
prognosis. The inhibition of tumor angiogenesis and its complete
comprehension is the aim when developing a successful
anti-angiogenic tumor therapy. Angiogenesis is the formation and
growth of new blood vessels from the already existing vasculature.
It is necessary for various inflammatory, ischemic, infectious and
immune disorders and also for malignant processes (65,66).
Malignant cells are able to secrete pro-angiogenic
factors including VEGF, which induces tumor blood vessel formation
(67). The in vivo
angiogenic activity of secreted VEGF may be regulated by
extracellular inhibitors, because it is also produced in avascular
tissues such as the cartilage. CTGF can inhibit VEGF-induced
angiogenesis via complex formation of VEGF with CTGF through a
protein-to protein interaction (68,69).
On the other hand, CTGF expresses mitogenic activity in endothelial
cells to promote angiogenesis, although the potency of angiogenic
activity of CTGF is not well-evaluated (70,71).
In conclusion, panobinostat induces differential
expression of CTGF in a context-dependent manner. In this setting,
the anti-angiogenesis is obviously not mediated via the classical
VEGF-driven cascade.
Other pathways are supposed to be involved in the
CTGF mediated angiogenesis and further experiments are necessary to
explore the role of CTGF.
Acknowledgements
We thank Gabriele Krumholz for her great support in
animal care and experiments. The excellent technical assistance of
Astrid Tautand Isabel Zeitträger is gratefully acknowledged. We
also thank Professor Margarete Goppelt-Strübe (Department of
Medicine 4, University Hospital Erlangen, Erlangen, Germany) for
her support. Susanne Gahr was supported by the IZKF
(Interdisziplinäres Zentrum für Klinische Forschung der Universität
Erlangen-Nürnberg). Pietro Di Fazio is a Dame Sheila
Sherlock-Fellow of the European Association for the Study of the
Liver (EASL). Matthias Ocker received a research grant of the
University Medical Center Giessen and Marburg (UKGM). The study was
supported by the Novartis Pharma AG, Nuremberg, Germany.
References
|
1
|
Lodato F, Mazzella G, Festi D, Azzaroli F,
Colecchia A and Roda E: Hepatocellular carcinoma prevention: A
worldwide emergence between the opulence of developed countries and
the economic constraints of developing nations. World J
Gastroenterol. 12:7239–7249. 2006.PubMed/NCBI
|
|
2
|
Bosch FX, Ribes J, Díaz M and Cléries R:
Primary liver cancer: Worldwide incidence and trends.
Gastroenterology. 127(Suppl 1): S5–S16. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Peto J: Cancer epidemiology in the last
century and the next decade. Nature. 411:390–395. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Duffy A and Greten T: Developing better
treatments in hepatocellular carcinoma. Expert Rev Gastroenterol
Hepatol. 4:551–560. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Omata M, Tateishi R, Yoshida H and Shiina
S: Treatment of hepatocellular carcinoma by percutaneous tumor
ablation methods: Ethanol injection therapy and radiofrequency
ablation. Gastroenterology. 127(Suppl 1): S159–S166. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al; SHARP Investigators Study Group. Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:378–390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Okamoto K, Neureiter D and Ocker M:
Biomarkers for novel targeted therapies of hepatocellular
carcinoma. Histol Histopathol. 24:493–502. 2009.PubMed/NCBI
|
|
9
|
Chan SL, Mok T and Ma BB: Management of
hepatocellular carcinoma: Beyond sorafenib. Curr Oncol Rep.
14:257–266. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sakurai T and Kudo M: Signaling pathways
governing tumor angiogenesis. Oncology. 81(Suppl 1): 24–29. 2011.
View Article : Google Scholar
|
|
12
|
Weis SM and Cheresh DA: Tumor
angiogenesis: Molecular pathways and therapeutic targets. Nat Med.
17:1359–1370. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ichihara E, Kiura K and Tanimoto M:
Targeting angiogenesis in cancer therapy. Acta Med Okayama.
65:353–362. 2011.PubMed/NCBI
|
|
14
|
Hicklin DJ and Ellis LM: Role of the
vascular endothelial growth factor pathway in tumor growth and
angiogenesis. J Clin Oncol. 23:1011–1027. 2005. View Article : Google Scholar
|
|
15
|
Chung AS, Lee J and Ferrara N: Targeting
the tumour vasculature: Insights from physiological angiogenesis.
Nat Rev Cancer. 10:505–514. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shibuya M: Vascular endothelial growth
factor-dependent and -independent regulation of angiogenesis. BMB
Rep. 41:278–286. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Veeravagu A, Hsu AR, Cai W, Hou LC, Tse VC
and Chen X: Vascular endothelial growth factor and vascular
endothelial growth factor receptor inhibitors as anti-angiogenic
agents in cancer therapy. Recent Patents Anticancer Drug Discov.
2:59–71. 2007. View Article : Google Scholar
|
|
18
|
Amaoka N, Saio M, Nonaka K, Imai H, Tomita
H, Sakashita F, Takahashi T, Sugiyama Y, Takami T and Adachi Y:
Expression of vascular endothelial growth factor receptors is
closely related to the histological grade of hepatocellular
carcinoma. Oncol Rep. 16:3–10. 2006.PubMed/NCBI
|
|
19
|
Kanno S, Oda N, Abe M, Terai Y, Ito M,
Shitara K, Tabayashi K, Shibuya M and Sato Y: Roles of two VEGF
receptors, Flt-1 and KDR, in the signal transduction of VEGF
effects in human vascular endothelial cells. Oncogene.
19:2138–2146. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schoenleber SJ, Kurtz DM, Talwalkar JA,
Roberts LR and Gores GJ: Prognostic role of vascular endothelial
growth factor in hepatocellular carcinoma: Systematic review and
meta-analysis. Br J Cancer. 100:1385–1392. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sato Y, Kanno S, Oda N, Abe M, Ito M,
Shitara K and Shibuya M: Properties of two VEGF receptors, Flt-1
and KDR, in signal transduction. Ann NY Acad Sci. 902:201–207.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Greten TF, Korangy F, Manns MP and Malek
NP: Molecular therapy for the treatment of hepatocellular
carcinoma. Br J Cancer. 100:19–23. 2009. View Article : Google Scholar :
|
|
24
|
Carr BI, Carroll S, Muszbek N and Gondek
K: Economic evaluation of sorafenib in unresectable hepatocellular
carcinoma. J Gastroenterol Hepatol. 25:1739–1746. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Connock M, Round J, Bayliss S, Tubeuf S,
Greenheld W and Moore D: Sorafenib for the treatment of advanced
hepatocellular carcinoma. Health Technol Assess. 14(Suppl 1):
17–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bergers G and Benjamin LE: Tumorigenesis
and the angiogenic switch. Nat Rev Cancer. 3:401–410. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Baeriswyl V and Christofori G: The
angiogenic switch in carcinogenesis. Semin Cancer Biol. 19:329–337.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bradham DM, Igarashi A, Potter RL and
Grotendorst GR: Connective tissue growth factor: A cysteine-rich
mitogen secreted by human vascular endothelial cells is related to
the SRC-induced immediate early gene product CEF-10. J Cell Biol.
114:1285–1294. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shimo T, Nakanishi T, Nishida T, Asano M,
Kanyama M, Kuboki T, Tamatani T, Tezuka K, Takemura M, Matsumura T,
et al: Connective tissue growth factor induces the proliferation,
migration, and tube formation of vascular endothelial cells in
vitro, and angiogenesis in vivo. J Biochem. 126:137–145. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Perbal B: NOV (nephroblastoma
overexpressed) and the CCN family of genes: Structural and
functional issues. Mol Pathol. 54:57–79. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li MH, Sanchez T, Pappalardo A, Lynch KR,
Hla T and Ferrer F: Induction of antiproliferative connective
tissue growth factor expression in Wilms' tumor cells by
sphingosine-1-phosphate receptor 2. Mol Cancer Res. 6:1649–1656.
2008.PubMed/NCBI
|
|
32
|
Hall-Glenn F, De Young RA, Huang BL, van
Handel B, Hofmann JJ, Chen TT, Choi A, Ong JR, Benya PD, Mikkola H,
et al: CCN2/connective tissue growth factor is essential for
pericyte adhesion and endothelial basement membrane formation
during angiogenesis. PLoS One. 7:e305622012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moussad EE and Brigstock DR: Connective
tissue growth factor: What's in a name? Mol Genet Metab.
71:276–292. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Urtasun R, Latasa MU, Demartis MI, Balzani
S, Goñi S, Garcia-Irigoyen O, Elizalde M, Azcona M, Pascale RM, Feo
F, et al: Connective tissue growth factor autocriny in human
hepatocellular carcinoma: Oncogenic role and regulation by
epidermal growth factor receptor/yes-associated protein-mediated
activation. Hepatology. 54:2149–2158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang GB, Zhou XY, Yuan T, Xie J, Guo LP,
Gao N and Wang XQ: Significance of serum connective tissue growth
factor in patients with hepatocellular carcinoma and relationship
with angiogenesis. World J Surg. 34:2411–2417. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hirasaki S, Koide N, Ujike K, Shinji T and
Tsuji T: Expression of Nov, CYR61 and CTGF genes in human
hepatocellular carcinoma. Hepatol Res. 19:294–305. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zeng ZJ, Yang LY, Ding X and Wang W:
Expressions of cysteine-rich61, connective tissue growth factor and
Nov genes in hepatocellular carcinoma and their clinical
significance. World J Gastroenterol. 10:3414–3418. 2004.PubMed/NCBI
|
|
38
|
Xiu M, Liu YH, Brigstock DR, He FH, Zhang
RJ and Gao RP: Connective tissue growth factor is overexpressed in
human hepatocellular carcinoma and promotes cell invasion and
growth. World J Gastroenterol. 18:7070–7078. 2012. View Article : Google Scholar
|
|
39
|
Komorowsky C, Ocker M and Goppelt-Struebe
M: Differential regulation of connective tissue growth factor in
renal cells by histone deacetylase inhibitors. J Cell Mol Med.
13:2353–2364. 2009. View Article : Google Scholar
|
|
40
|
Schneider-Stock R and Ocker M: Epigenetic
therapy in cancer: Molecular background and clinical development of
histone deacetylase and DNA methyltransferase inhibitors. IDrugs.
10:557–561. 2007.PubMed/NCBI
|
|
41
|
Di Fazio P, Schneider-Stock R, Neureiter
D, Okamoto K, Wissniowski T, Gahr S, Quint K, Meissnitzer M,
Alinger B, Montalbano R, et al: The pan-deacetylase inhibitor
panobinostat inhibits growth of hepatocellular carcinoma models by
alternative pathways of apoptosis. Cell Oncol. 32:285–300.
2010.PubMed/NCBI
|
|
42
|
Lachenmayer A, Toffanin S, Cabellos L,
Alsinet C, Hoshida Y, Villanueva A, Minguez B, Tsai HW, Ward SC,
Thung S, et al: Combination therapy for hepatocellular carcinoma:
Additive preclinical efficacy of the HDAC inhibitor panobinostat
with sorafenib. J Hepatol. 56:1343–1350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gahr S, Wissniowski T, Zopf S, Strobel D,
Pustowka A and Ocker M: Combination of the deacetylase inhibitor
panobinostat and the multi-kinase inhibitor sorafenib for the
treatment of metastatic hepatocellular carcinoma - review of the
underlying molecular mechanisms and first case report. J Cancer.
3:158–165. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Di Fazio P, Montalbano R, Neureiter D,
Alinger B, Schmidt A, Merkel AL, Quint K and Ocker M:
Downregulation of HMGA2 by the pan-deacetylase inhibitor
panobinostat is dependent on hsa-let-7b expression in liver cancer
cell lines. Exp Cell Res. 318:1832–1843. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qian DZ, Kato Y, Shabbeer S, Wei Y,
Verheul HM, Salumbides B, Sanni T, Atadja P and Pili R: Targeting
tumor angiogenesis with histone deacetylase inhibitors: The
hydroxamic acid derivative LBH589. Clin Cancer Res. 12:634–642.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ocker M: Deacetylase inhibitors - focus on
non-histone targets and effects. World J Biol Chem. 1:55–61. 2010.
View Article : Google Scholar
|
|
47
|
Ellis L, Hammers H and Pili R: Targeting
tumor angiogenesis with histone deacetylase inhibitors. Cancer
Lett. 280:145–153. 2009. View Article : Google Scholar
|
|
48
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
49
|
Zuehlke J, Ebenau A, Krueger B and
Goppelt-Struebe M: Vectorial secretion of CTGF as a cell-type
specific response to LPA and TGF-β in human tubular epithelial
cells. Cell Commun Signal. 10:252012. View Article : Google Scholar
|
|
50
|
Saif MW: Anti-VEGF agents in metastatic
colorectal cancer (mCRC): Are they all alike? Cancer Manag Res.
5:103–115. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gahr S, Peter G, Wissniowski TT, Hahn EG,
Herold C and Ocker M: The histone-deacetylase inhibitor MS-275 and
the CDK-inhibitor CYC-202 promote anti-tumor effects in hepatoma
cell lines. Oncol Rep. 20:1249–1256. 2008.PubMed/NCBI
|
|
52
|
Gahr S, Ocker M, Ganslmayer M, Zopf S,
Okamoto K, Hartl A, Leitner S, Hahn EG and Herold C: The
combination of the histone-deacetylase inhibitor trichostatin A and
gemcitabine induces inhibition of proliferation and increased
apoptosis in pancreatic carcinoma cells. Int J Oncol. 31:567–576.
2007.PubMed/NCBI
|
|
53
|
Budman DR, Tai J, Calabro A and John V:
The histone deacetylase inhibitor panobinostat demonstrates marked
synergy with conventional chemotherapeutic agents in human ovarian
cancer cell lines. Invest New Drugs. 29:1224–1229. 2011. View Article : Google Scholar
|
|
54
|
Neri P, Bahlis NJ and Lonial S:
Panobinostat for the treatment of multiple myeloma. Expert Opin
Investig Drugs. 21:733–747. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Simmons JK, Patel J, Michalowski A, Zhang
S, Wei BR, Sullivan P, Gamache B, Felsenstein K, Kuehl WM and
Simpson RM: TORC1 and class I HDAC inhibitors synergize to suppress
mature B cell neoplasms. Mol Oncol. 8:261–272. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Antonarakis ES and Carducci MA: Targeting
angiogenesis for the treatment of prostate cancer. Expert Opin Ther
Targets. 16:365–376. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Schweizer MT and Carducci MA: From
bevacizumab to tasquinimod: angiogenesis as a therapeutic target in
prostate cancer. Cancer J. 19:99–106. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Liu TJ, Sun BC, Zhao XL, Zhao XM, Sun T,
Gu Q, Yao Z, Dong XY, Zhao N and Liu N: CD133+ cells
with cancer stem cell characteristics associates with vasculogenic
mimicry in triple-negative breast cancer. Oncogene. 32:544–553.
2013. View Article : Google Scholar
|
|
59
|
Zhang Y, Hong H, Nayak TR, Valdovinos HF,
Myklejord DV, Theuer CP, Barnhart TE and Cai W: Imaging tumor
angiogenesis in breast cancer experimental lung metastasis with
positron emission tomography, near-infrared fluorescence, and
bioluminescence. Angiogenesis. 16:663–674. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Aggarwal C, Somaiah N and Simon G:
Antiangiogenic agents in the management of non-small cell lung
cancer: Where do we stand now and where are we headed? Cancer Biol
Ther. 13:247–263. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xiao YY, Zhan P, Yuan DM, Liu HB, Lv TF,
Song Y and Shi Y: Chemotherapy plus multitargeted antiangiogenic
tyrosine kinase inhibitors or chemotherapy alone in advanced NSCLC:
A meta-analysis of randomized controlled trials. Eur J Clin
Pharmacol. 69:151–159. 2013. View Article : Google Scholar
|
|
62
|
Pang R, Tse E and Poon RT: Molecular
pathways in hepatocellular carcinoma. Cancer Lett. 240:157–169.
2006. View Article : Google Scholar
|
|
63
|
Ribatti D, Vacca A, Nico B, Sansonno D and
Dammacco F: Angiogenesis and anti-angiogenesis in hepatocellular
carcinoma. Cancer Treat Rev. 32:437–444. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhang Q, Du Y, Xue Z, Chi C, Jia X and
Tian J: Comprehensive evaluation of the anti-angiogenic and
anti-neoplastic effects of Endostar on liver cancer through optical
molecular imaging. PLoS One. 9:e855592014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Carmeliet P: Angiogenesis in life, disease
and medicine. Nature. 438:932–936. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Fischer C, Schneider M and Carmeliet P:
Principles and therapeutic implications of angiogenesis,
vasculogenesis and arteriogenesis. Handb Exp Pharmacol.
176:157–212. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lin EY and Pollard JW: Tumor-associated
macrophages press the angiogenic switch in breast cancer. Cancer
Res. 67:5064–5066. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Inoki I, Shiomi T, Hashimoto G, Enomoto H,
Nakamura H, Makino K, Ikeda E, Takata S, Kobayashi K and Okada Y:
Connective tissue growth factor binds vascular endothelial growth
factor (VEGF) and inhibits VEGF-induced angiogenesis. FASEB J.
16:219–221. 2002.
|
|
69
|
Hashimoto G, Inoki I, Fujii Y, Aoki T,
Ikeda E and Okada Y: Matrix metalloproteinases cleave connective
tissue growth factor and reactivate angiogenic activity of vascular
endothelial growth factor 165. J Biol Chem. 277:36288–36295. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Shimo T, Nakanishi T, Kimura Y, Nishida T,
Ishizeki K, Matsumura T and Takigawa M: Inhibition of endogenous
expression of connective tissue growth factor by its antisense
oligonucleotide and antisense RNA suppresses proliferation and
migration of vascular endothelial cells. J Biochem. 124:130–140.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Babic AM, Chen CC and Lau LF: Fisp12/mouse
connective tissue growth factor mediates endothelial cell adhesion
and migration through integrin alphavbeta3, promotes endothelial
cell survival, and induces angiogenesis in vivo. Mol Cell Biol.
19:2958–2966. 1999.PubMed/NCBI
|