Introduction
Endometrial cancer (EC) is the most common
gynecological malignancy of the female genital tract. For 2015, it
is estimated that ~54,870 new cancer cases of EC will be diagnosed
and 10,170 deaths are expected in the USA alone (1). Although most EC is diagnosed at
early-stage which correlates with advanced outcome, women with
late-stage EC are at highest risk of recurrence and poor prognosis
(2,3). In particular, EC cases with regional
or distal metastasis recur with limited effect of systemic
therapies (2). It is, therefore,
important to further elucidate the molecular and cellular
mechanisms responsible for tumorigenesis and progression of EC, and
to develop novel diagnostic and therapeutic strategies.
Autocrine motility factor (AMF), which is also known
as phosphoglucose isomerase (PGI), serves as tumor-secreted
cytokine, it stimulates tumor cells motility, migration, invasion
and metastasis (4,5). AMF/PGI also behaves as a housekeeping
cytosolic enzyme of sugar metabolism and plays a key role during
glycolysis and gluconeogenesis, catalyzing the interconversion of
glucose 6-phosphate and fructose 6-phosphate (6). In addition, AMF/PGI serves several
other functions as neuroleukin, which promotes growth of embryonic
spinal and sensory neurons (7);
and as a maturation factor mediating differentiation of human
myeloid leukemia cells (8).
Several researchers independently found that secreted AMF/PGI by
tumor cells is involved in regulation of oncogenesis and tumor
progression in various human cancers (9–13).
Recent studies have shown that PGI/AMF plays an
important role during epithelial-to-mesenchymal transition (EMT)
which is an essential mechanism for the development of malignant
tumors for invasion and metastasis (13–15).
EMT was well-documented as the process that produces a complete
loss of epithelial traits by the former epithelial cells
accompanied by the acquisition of mesenchymal characteristics in
vitro (16,17) and was reflected in changes from a
tightly organized cobblestone-like structure, whereby the cells
adhere to each other and manifest apical basal polarity to spindly
migratory elongated cells with disrupted cell-cell/cell-substratum
contacts (15,18). The phenomenon of EMT is associated
with acquisition of invasive phenotype by cancer cells (19,20)
and, in particular, aggressive behavior of endometrial cancer
(21). The role of PGI/AMF during
EMT has been extensively described in other types of cancer but has
been poorly studied in EC (22,23).
Therefore, it is of considerable interest to identify novel
mechanisms to better understand how EC occurs and how it is
disseminated.
In the present report, we describe that the
association between expression of AMF and EMT in EC specimens, and
we also identified a critical role of AMF/PGI in promoting EMT in
EC. These results shed light on the mechanisms by which EC occurs
and develops and provide evidence that AMF/PGI as a novel
proto-oncoprotein of EC and therefore a potential therapeutic
target.
Materials and methods
Reagents and antibodies
Purified rabbit AMF was purchased from Sigma for
exogenous AMF stimulation (cat. no P9544). Mouse monoclonal
anti-AMF (ab66340), rabbit polyclonal anti-AMF (ab86950),
E-cadherin (ab15148), vimentin (ab45939), Snail (ab180714), TGFBR1
(abab31013) and anti-β-actin (ab8227) for use in
immunohistochemistry or western blot analyses were obtained from
Abcam Ltd. (Hong Kong, China). U0126 (MAPK inhibitor) was purchased
from Selleck Chemicals.
Cell culture
Ishikawa and HEC-1B cells were purchased from the
American Type Culture Collection (ATCC; Manassas, VA, USA) and
maintained according to the provider's instruction in DMEM/F12
media (Gibco, Auckland, New Zealand) supplemented with 10% fetal
bovine serum (FBS; Gibco, Carlsbad, CA, USA). All cells were grown
until confluent and were incubated in serum-free medium for 24 h
before treatment with various experimental agents as described.
Tissue microarray slides and
immunohistochemistry
High-density tissue microarrays were constructed by
Outdo Biotech Co., Ltd., (Shanghai, China) using clinical samples
obtained from a cohort of 62 patients who underwent surgery at the
Shanghai First People's Hospital. Each array includes 31 normal and
31 malignant tissue specimens of the endometrium (various grades
and stages). Paraffin-embedded tissue sections (5 μm) were
deparaffinized by xylene and rehydrated in a graded alcohol series
(100, 95, 80 and 70%, 5 min each). Antigen retrieval was done by
boiling the slides in a small beaker filled with 1 mM EDTA (pH
8.0). Endogenous peroxidase activity was quenched by a 10-min
incubation in 3% hydrogen peroxide. After antigen retrieval, the
slides were washed two times in a 0.1% Tween/1X TBS (0.1% TBST)
bath (5 min each) at room temperature to remove non-specific
background binding. Protein blocking was performed by incubating
the specimens in 5% normal rabbit serum or normal horse serum in
0.1% TBS for 1 h. Primary antibodies against AMF, E-cadherin or
Snail and vimentin were diluted 1:200 and 1:500, respectively and
applied for 1 h at room temperature. After a series of TBST rinses
as described above, bound antibody was subsequently detected using
EnVision reagents (Wuhan Boster Biological Engineering Co., Ltd.,
Wuhan, China) according to the manufacturer's instructions.
Evaluation of immunohistochemical
staining and statistical analysis
Immunostained slides were scored under a microscope.
The staining intensity was scored as 0 (negative), 1 (weak), 2
(medium) or 3 (strong). The extent of staining was scored as 0
(0%), 1 (1–25%), 2 (26–50%), 3 (51–75%) or 4 (76–100%), according
to the percentage of the positively stained areas in relation to
the whole tumor area. The sum of the intensity score and the extent
score was used as the final staining score (0–7). Tumors with a
final staining score of 4 or higher were considered ‘positive'
(24). Results were assessed by
two pathologists in a blinded manner with respect to the tissue
source.
Stable silencing of AMF expression by
short hairpin RNA (shRNA)
AMF shRNA constructs were cloned into pLKO.1 plasmid
under the control of U6 promoter for stable expression (Sigma).
Three pairs of annealed DNA oligonucleotides were inserted into the
AgeI and EcoRI restriction sites of pLKO.1. The most
effective pairs of sequence targeted to human AMF is: sense,
5′-CGCCATGTATGAGCACAAGAT-3′ and anti-sense
5′-GCGGTACATACTCGTGTTCTA-3′ (shAMF-1), as well as sense
5′-CCTGTCTACTAACACAACCAA-3′ and antisense
5′-GGACAGATGATTGTGTTGGTT-3′ (shAMF-2), respectively. Ishikawa and
HEC-1B cells were infected with lentiviral vector pLKO.1 (mock) or
AMF-specific shRNA lentiviral particles in 6-well plates in the
presence of polybrene (6 mg/ml) and then treated with puromycin (2
mg/ml) to generate stable clones. Puromycin-resistant AMF knockdown
clones were harvested by ring selection, and AMF gene expression
and protein level were confirmed by qRT-PCR and immunoblotting.
RNA extraction and qRT-PCR (quantitative
real-time polymerase chain reaction)
Total RNA was isolated using TRIzol reagent
(Invitrogen, Life Technologies; Shanghai, China) and reverse
transcribed using a reverse transcriptase kit (Takara, Dalian,
China). Gene expression was detected with SYBR-Green Master Mix
(Takara) on an ABI Prism 700 Thermal Cycler (Applied Biosystems,
Foster City, CA, USA). Gene expression was calculated using the
2−ΔΔCt formula and normalized against β-actin. The
oligonucleotide primers were 5′-CGCCCAACCAACTCTATTG-3′ (forward)
and 5′-GAT GATGCCCTGAACGAAG-3′ (reverse) for human AMF detection;
5′-TTATGATTCTCTGCTCGTG-3′ (forward) and 5′-ATAGTCCTGGTCTTTGTCT-3′
(reverse) for human E-cadherin detection; 5′-AGGAGGAAATGGCTCGTCA-3′
(forward) and 5′-TGTAGGTGGCAATCTCAAT-3′ (reverse) for human
vimentin detection; 5′-CGGCTCCTTCGTCCT TC-3′ (forward) and
5′-GCACCCAGGCTGAGGTATT-3′ (reverse) for human Snail detection;
5′-TGTGAAGCCTTGAG AGTAA-3′ (forward) and 5′-TGTTGACTGAGTTGCGATA-3′
(reverse) for human TGFBR1 detection; 5′-CAGCCATGT
ACGTTGCTATCCAGG-3′ (forward) and 5′-AGGTCCAGA CGCAGGATGGCATG-3′
(reverse) for human β-actin detection (as a housekeeping gene). All
experiments were performed independently in triplicate.
Protein extraction and western blot
analysis
For whole-cell lysates, cells were washed twice with
PBS and collected by scraping. Cell pellets were lysed in cold
precipitation assay buffer [20 mM Tris-HCl (pH 7.4), 150 mM NaCl,
10 mM EDTA, 1% NP40, Triton X-100, sodium deoxycholate] containing
1 mmol/l DTT, 1 mmol/l phenylmethylsulfonyl fluoride, 10 μg/ml
leupeptin and 10 μg/ml aprotinin. Samples were separated by
centrifugation (15,000 rpm in 4°C for 30 min). Lysate supernatants
were 100-fold concentrated with Amicon Ultra (30,000 nominal
molecular weight limit; Millipore).
The extracted protein concentration was measured
with BCA Protein assay kit (Thermo Fisher Scientific). Equal
amounts of the proteins (20 μg of cellular protein or 80 μg of
secreted protein) were separated on 10% SDS-PAGE gels and
transferred to 0.2-Am PVDF membrane (Osmonics, Inc.). Each membrane
was then incubated overnight at 4°C with an appropriate diluted
primary antibody. HRP-secondary anti-rabbit or anti-mouse
antibodies (diluted 1:5,000 to yield 0.2 mg/ml; Abcam) were used to
detect the bound primary antibodies. Blots were visualized using
the Bioscience Odyssey Infrared Imaging System (LI-COR
Biosciences), and band density was quantitated using ImageJ imaging
analysis software (NIH, Bethesda, MD, USA). Data were normalized to
β-actin expression by densitometry and statistical data from at
least three experiments were recorded.
Immunofluorescence and imaging
For detection of E-cadherin and vimentin, cells were
seeded onto glass coverslips, fixed with 4% paraformaldehyde for 10
min and permeabilized with PBS Triton X-100 4% for 10 min. Cells
were sequentially incubated with primary antibody (rabbit
anti-E-cadherin and anti-vimentin 1:200) diluted in blocking buffer
overnight at 4°C, which was followed by incubation with secondary
antibodies conjugated with Alexa Fluor 568 (Molecular
Probes-Invitrogen, Carlsbad, CA, USA) for 1 h at RT in the dark.
After washing with PBS, the cells were stained with
4′,6′-diamidino-2-phenylindole (DAPI; targeting DNA in the cell
nucleus) for 5 min. After extensive washing, the cells were mounted
on a glass slide with 80% glycerol and fluorescent images were
analyzed in an Olympus fluorescence microscope using a ×400
lens.
Microarray and data analysis
The Agilent SurePrint G3 Human Gene Expression
microarray (8×60K) was used in the present study. This chip targets
>30,000 genes with >50,000 probes and >12,000 lincRNA
derived from a broad survey of well known sources such as RefSeq,
Ensembl, UniGene and others. The resulting view of the human genome
covers 30K unique genes and transcripts that have been verified and
optimized by alignment to the human genome assembly and by the
Agilent Empirical Validation process. Total RNA (>300 ng) was
extracted from four independent cultures of Ishikawa, HEC-1B cells,
shAMF-1 (Ishikawa) and shAMF-1 (HEC-1B) cells. Microarray
hybridization, data collection and analysis were performed at
Oebiotech Biotechnology Corp. (Shanghai, China). The threshold set
for upregulated and downregulated genes was a fold-change ≥ 2.0.
KEGG analysis was applied to determine the roles of these
differentially expressed mRNAs.
U0126 and exogenous PGI/AMF
treatment
Cells were seeded at 1.0×105 cells/well
of a 12-well plate, containing 1.2 ml regular medium. The media was
then changed to phenol-red free medium with 0.5% stripped FBS for
incubation at 37°C overnight. Immediately prior to treatment, the
medium in the culture plates was aspirated, triply washed with PBS
and replaced with fresh medium. U0126 dissolved in DMSO or PGI/AMF
dissolved in PBS was subsequently added to each well. Concurrently,
the same amount of DMSO or PBS was added to the control wells.
Statistical analysis
Continuous variables were recorded as mean ± SD and
all statistical analyses were done using Statistical Package for
the Social Sciences (SPSS) software version 17.0 (SPSS, Inc.,
Chicago, IL, USA). Volumetric data were assessed using an unpaired
Student's t-test, or one-way ANOVA analysis followed by post-hoc
LSD test or Dunnett's test for multiple comparisons. Concordance
and correlation among antibodies were accessed by calculating
Chi-square test and McNemar's statistical test. P-values <0.05
were considered statistically significant. All experiments were
repeated independently at least three times.
Results
Autocrine motility factor was highly
expressed and correlate with EMT in EC tissues (patients clinical
information)
AMF abnormal expression has been associated with
tumor progression and EMT phenotype conversions in many human
cancers (13,22,23).
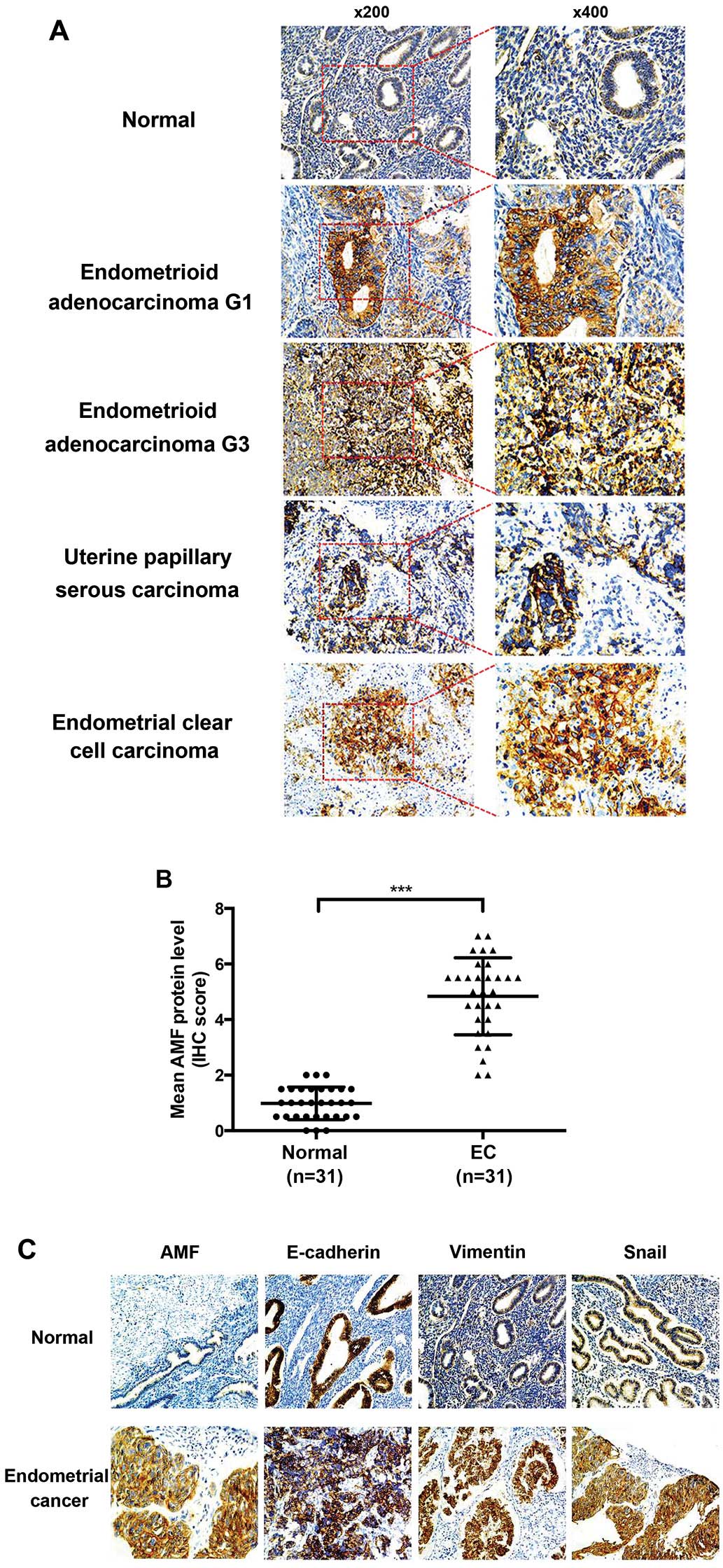
In our studies, immunochemistry staining showed that AMF protein
was predominantly localized to the cytoplasm of endometrial
epithelial cells. There was only weak or no staining in normal
endometrium, whereas strong AMF immunostaining was found in
endometrial carcinoma tissues, including endometrioid
adenocarcinoma, uterine papillary serous carcinoma and endometrial
clear cell carcinoma (Fig. 1A).
The mean scores for AMF staining were 4.5 and 0.95 for cancer
tissue and normal human endometrium, respectively (P<0.001)
(Fig. 1B). Furthermore, we used a
semi-quantitative analysis of IHC staining to evaluate AMF and EMT
markers including E-cadherin, vimentin and Snail expression in the
tissues microarrays. AMF levels positively correlated with vimentin
(P=0.012) and Snail (P=0.021) levels, inversely correlated with the
levels of E-cadherin (P=0.035) (Table
I). These data indicated that AMF expression was much higher in
EC tissue specimens and relevant to the levels of EMT marker
protein.
 | Table IExpression correlations between AMF
and E-cadherin, vimentin and Snail. |
Table I
Expression correlations between AMF
and E-cadherin, vimentin and Snail.
| |
AMF*E-cadherin/Vimentin/Snail |
|---|
| |
|
|---|
| | E-cadherin | Vimentin | Snail |
|---|
| |
|
|
|
|---|
| | − | + | − | + | − | + |
|---|
| AMF |
| − | Count | 4 | 6 | 9 | 1 | 9 | 1 |
| % of Total | 13% | 19% | 29% | 3% | 29% | 3% |
| + | Count | 17 | 4 | 10 | 11 | 9 | 12 |
| % of Total | 55% | 13% | 32% | 35% | 29% | 39% |
| Total | Count | 21 | 10 | 19 | 12 | 18 | 13 |
| % of Total | 68% | 32% | 61% | 39% | 58% | 42% |
Silencing of AMF reverses the EMT
phenotype in EC cells
Processes involved in the EMT are closely correlated
with cancer metastasis. The expression of AMF and EMT markers in EC
tissue specimens prompted us to examine whether AMF silencing could
induce morphologic changes, loss of mesenchymal and/or gain of
epithelial markers. The human EC cell lines Ishikawa and HEC-1B
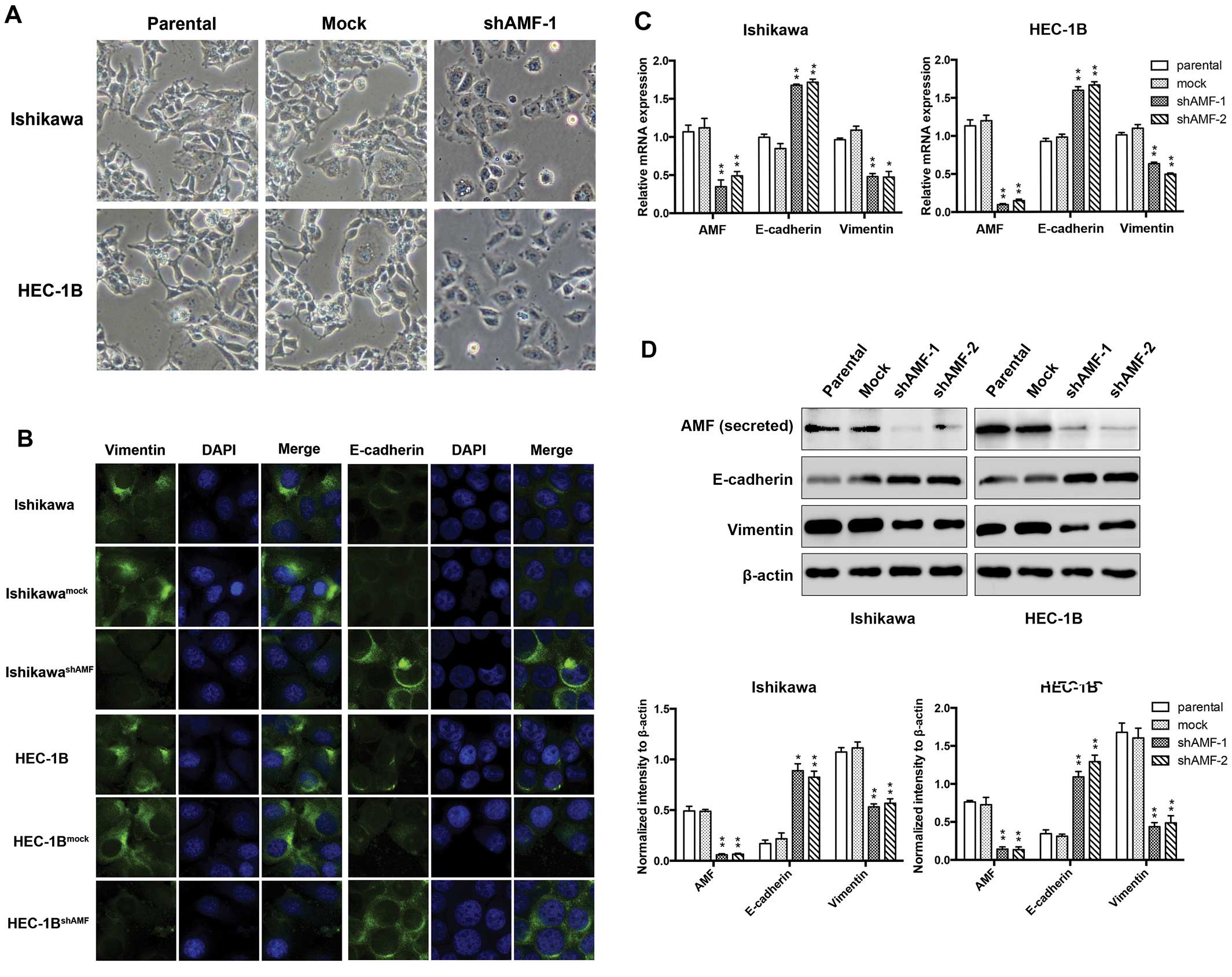
were stably expressed with either shAMF or control vectors. We
microscopically examined AMF-silencing EC cells to determine the
effects of AMF on cellular morphology. IshikawashAMF and
HEC-1BshAMF cells were morphologically transformed
toward epithelia compared with Ishikawa, HEC-1B and their control
cells (Fig. 2A). This change was
characterized a transition from the spindle-like morphology of the
parental cells to a more differentiated keratinocyte-like
morphology, suggesting a phenotypic transition from mesenchymal to
epithelial. Thus, the expression of the intermediate filaments
vimentin (a mesenchymal marker) and E-cadherin (an epithelial
marker) was examined by immunofluorescent staining (Fig. 2B). Vimentin was prominently
expressed throughout the cytoplasm of the parental Ishikawa and
HEC-1B cells, whereas a significantly weaker vimentin signal was
detected in their shAMF cell clone. Next, we tested whether the
reduced vimentin expression was associated with a gain of
epithelial markers. The cells were immunostained for the presence
of the mesenchymal intermediate filament marker (i.e., E-cadherin).
Either the parental Ishikawa or HEC-1B cells expressed E-cadherin
proteins at a low level, while E-cadherin expression increased in
the shAMF cells (Fig. 2B).
To further determine if this transformation
represented an EMT [as has been reported (23)], we analyzed the levels of AMF and
several characteristic epithelial and mesenchymal proteins by
qRT-PCR and western blotting. We observed the expression of AMF was
markedly decreased by stable transfection with shAMF (Fig. 2C and D). Furthermore, the levels of
vimentin expression were reduced, while E-cadherin expression
levels were raised in IshikawashAMF and
HEC-1BshAMF cells (Fig. 2C
and D). Taken together, these findings indicate that AMF
silencing is accompanied by the loss of mesenchymal and the gain of
epithelial markers, and show that AMF likely plays a crucial role
in the regulation of the EMT in EC cells.
Identification of downregulated genes
associated with EMT in AMF silencing cell lines
To investigate the functional significance of AMF
involved in EMT in EC cells, we compared the expression profiles of
the gene associated with EMT in Ishikawa/IshikawashAMF
and HEC-1B/HEC-1BshAMF. Pairwise comparisons identified
46 differentially expressed genes that had changed by at least
2-fold after AMF silencing in Ishikawa. In contrast, only 34
differentially expressed genes were identified following AMF
silencing of HEC-1B. We generated Venn diagrams by combining the
separate gene lists obtained from the GeneSifter analysis, and then
manually identified the common 15 genes that were regulated by AMF
silencing in both Ishikawa and HEC-1B (Fig. 3A). To validate the array data, we
performed qRT-PCR for two genes: Snail and transforming growth
factor β receptor 1 (TGFBR1) whose expression was significantly
downregulated by AMF silencing in both Ishikawa and HEC-1B. A
comparison of the fold induction of each of the two genes as
determined by microarray and qRT-PCR found that induction of gene
expression as determined by qRT-PCR was consistently with that
determined by microarrays (Fig.
3B). We further confirmed by western blotting that AMF
silencing markedly downregulated the expression of the two genes in
both Ishikawa and HEC-1B (Fig.
3C). These results demonstrated that AMF activated Snail and
TGFBR1 regulated EMT in Ishikawa and HEC-1B cells.
AMF mediated EMT by activating MAPK
pathway in EC cells
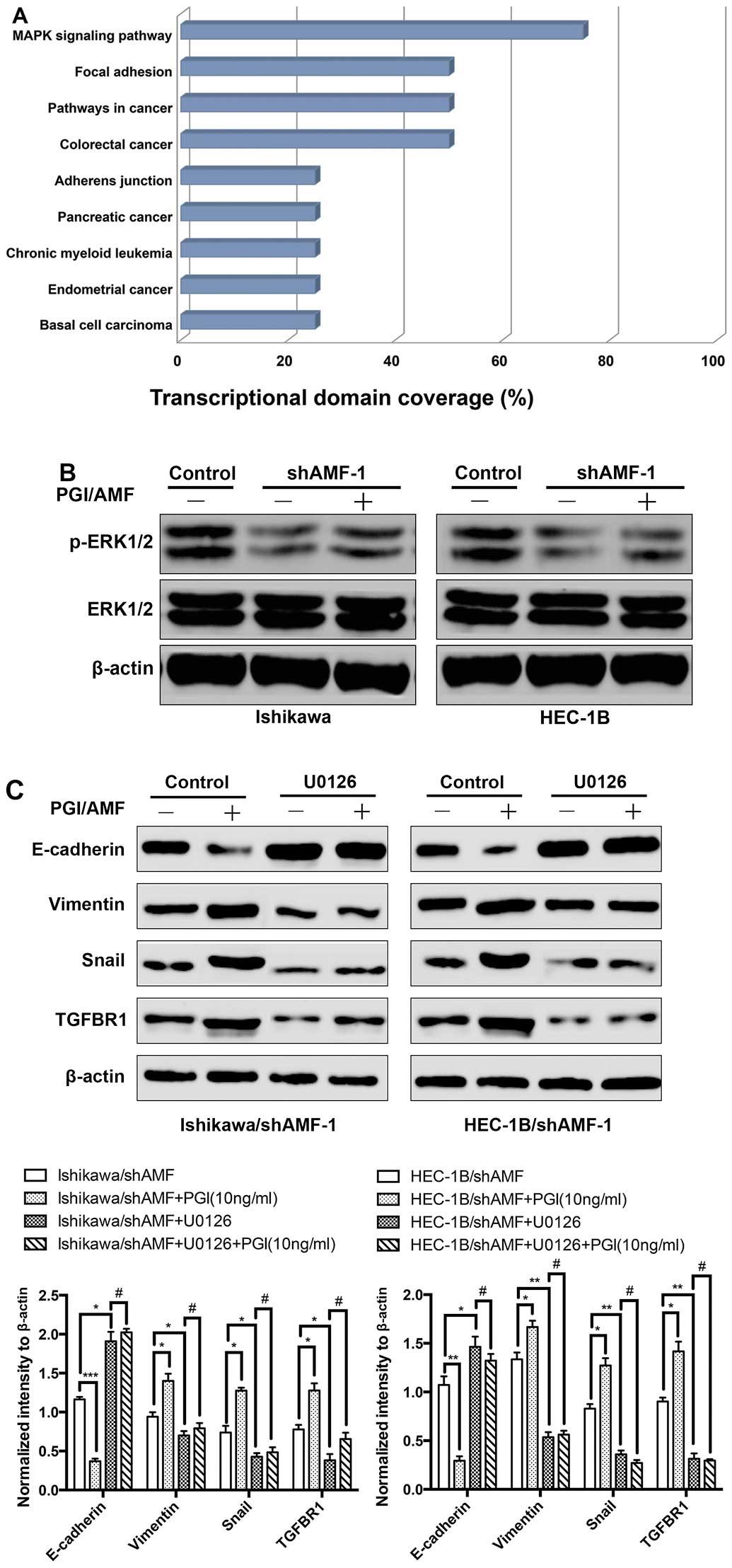
To explore which pathways were activated during EMT
mediated by AMF, we categorized the 15 differentially expressed
genes commonly regulated by AMF silencing in both Ishikawa and
HEC-1B using the KEGG pathway analysis tool provided in GeneSifter
software. As shown in Fig. 4A, the
total network comprised 9 distinct KEGG pathways, including
pathways related to EMT (mitogen-activated protein kinase pathway
and focal adhesion pathway) (19,20).
In addition, two of these 15 differentially expressed genes were
previously verified downregulated in AMF silencing cells including
Snail and TGFBR1, reside in the MAPK pathway (25). We therefore assessed levels of
phosphorylation of the MAPK pathway proteins ERK1/2 (at
Thr202/Tyr204) in Ishikawa and HEC-1B cells. Interestingly, the
phosphorylation levels of ERK were substantially inhibited by AMF
silencing, whereas, exogenous PGI/AMF increased the phosphorylation
levels of ERK in AMF silenced cells (Fig. 4B). To determine whether
AMF-mediated regulation of the EMT phenotype resulted from the AMF
ability to activate the MAPK pathway, U0126 (an inhibitor of MAPK
pathway) was used to pretreat IshikawashAMF and
HEC-1BshAMF cells. In IshikawashAMF and
HEC-1BshAMF cells, exogenous PGI/AMF decreased the level
of the epithelial marker E-cadherin, whereas not the levels of the
mesenchymal marker vimentin. The EMT inducers Snail and TGFBR1
known as TGFβ receptor type I (15,26)
were upregulated, compared with control groups (Fig. 4C and D). In contrast, the effects
of exogenous PGI/AMF on enhancing EMT phenotype were eliminated in
U0126 pretreated groups which MAPK pathway was inhibited (Fig. 4C and D). Our findings suggested
that MAPK pathway activated by MAPK induced EMT in EC cells.
Discussion
Tumor metastasis and dissemination are multistep
processes involving complex and highly coordinated interactions
between tumor cells and a constantly changing host microenvironment
(27). Like many other malignant
diseases, cancer cells that disseminate from the primary tumor and
invade distant organs are the leading causes of death in EC.
Secreted AMF/PGI functions as a growth factor as well as a motility
factor (28). However, recent work
has shown its oncogenic role in tumors, including its ability to
regulated EMT (13,22). In the present study, we observed
the correlation between AMF and EMT markers in human EC specimens
using tissue microarray. Specific silencing of AMF in endometrial
cancer cell lines led to changes in tumor cell morphology and the
level of EMT marker expression. Gene expression profile analysis
investigated the mechanism of regulating EMT by AMF, including gene
and signaling pathways.
PGI is essential for an individual cell to survive
as the second enzyme in the glycolytic pathway, which seems to be a
minimum requirement for the cell, than to secret AMF as an
extracellular form of AMF/PGI. PGI is observed in all cells
ubiquitously, whereas secretion of AMF is observed in only tumor
cells or activated T cells (5).
The extracellular form of AMF, as a growth factor, causes tumor
cells to migrate and seems to be involved in tumor invasion and
metastasis. Thus, exogenous AMF plays an important role in tumor
progression.
Although several studies have explored the oncogenic
role of AMF in solid cancers including its ability to regulate
tumor aggressiveness (2,9,14),
this is the first study to address the function of AMF in human
endometrial cancer. As shown in Fig.
1A and B, the normal endometrium produced weak levels of AMF
while AMF was extensively detected in endometrial cancer. A
correlation between AMF and EMT marker expression was also found in
endometrial cancer (Fig. 1C),
which was in accordance with reports that AMF induces EMT (13). The EMT plays an important role
during cancer progression, leading to a more invasive, metastatic
phenotype in human cancers, including EC (29,30).
In this process, reduction of E-cadherin expression is required to
lose epithelial cell-cell adhesion and to destabilize the
epithelial architecture (31).
Differentiated mesenchymal cells can spread into tissues
surrounding the original tumor as well as separate from the tumor
to new locations where they divide and form additional tumors. Our
results of the present study confirmed the former hypothesis that
AMF silencing resulted in marked changes in cell morphology,
reduction of mesenchymal cell marker proteins and detection of
epithelial cell marker proteins (32). AMF silencing induced changes of
morphology in EC cells, it formed a mesenchymal phenotype with
elongated cell shape losing apical basal polarity to the epithelial
phenotype which was bound together tightly and exhibiting polarity
(Fig. 2A). Furthermore, we
observed that the expression of E-cadherin was increased in AMF
silenced EC cells with a concomitant decrease in vimentin
expression (Fig. 2B and C). These
results may suggest that AMF can regulate, in part,
epithelial-to-mesenchymal transition during endometrial cancer
development.
Gene expression profiles suggested that silencing of
AMF in Ishikawa and HEC-1B cells resulted in downregulation of
Snail and TGFBR1 (Fig. 3A). Snail,
a transcription factor, has been described as a direct repressor of
E-cadherin expression during EMT and carcinogenesis (26,33).
Phosphorylation of TGFBR1, the type I receptor binding to TGFβ,
induced nuclear localization and transcriptional activity of SMADs
during TGFβ triggered EMT. Snail is activated by phosphorylation of
SMADs (34). Silencing of AMF
reduced the TGFBR1 production, resulting in downregulation of Snail
(Fig. 3B and C), which was
released from the basal stage of suppression of E-cadherin.
Funasaka et al (23) showed
that the knockdown of AMF induced MET in fibrosarcoma cells, in
which Snail was downregulated and E-cadherin was upregulated.
Similarly, silencing of AMF/PGI induced MET in osteosarcoma cancer
MG-63 cells, in which E-cadherin and GSK-3β were upregulated, while
Snail and TGFBR1 were downregulated (13). These accumulated data strongly
suggested that knockdown of AMF might regulate the process of EMT
through down-regulation of TGFβ1 and Snail to suppression
E-cadherin.
Multiple signaling pathways cooperate in the
initiation and progression of EMT. For example, Snail cooperates
with the transcription regulator ETS1 to activate MAPK signaling
pathway and TGFBR1 phosphorylates induced by MAPK pathway. Snail
cooperates with other transcription regulators to control gene
expression (35). Silencing of AMF
in endometrial cancer Ishikawa and HEC-1B cells commonly inhibited
MAPK pathway activation (Fig. 4A and
B). Moreover, the exogenous PGI/AMF eliminated the effects of
MET induced by silencing AMF, while U0126 pretreatment (MAPK
pathway inhibitor) abolished this interesting phenomenon. Our
results indicate that AMF activates the MAPK pathway and suggest
that AMF-mediated activation of MAPK signaling contributes to
promotion of EMT in endometrial cancer.
In summary, we demonstrated that AMF expression was
high in endometrial cancer tissue and positively related with
markers of EMT using tissue microarray. Furthermore, our results
with endometrial cancer cells show that AMF silencing suppressed
the EMT phenotype and that these effects involved inhibition of the
MAPK pathway. Our findings suggest for the first time that
AMF/MAPK/EMT system might play a critical role in acquisition of
malignant phenotypes in endometrial cancer. Thus, AMF/MAPK/EMT
might be a potential therapeutic or prevention strategy for
treatment of endometrial cancer.
Acknowledgements
The study was supported by the National Natural
Science Foundation of China (nos. 81172476, 81272885 and 81472427),
the Science and Technology Commission of Shanghai Municipality (no.
13JC1404501) and the Doctoral Fund of Ministry of Education of
China (no. 20120073110090).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Salvesen HB, Haldorsen IS and Trovik J:
Markers for individualised therapy in endometrial carcinoma. Lancet
Oncol. 13:e353–e361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sorosky JI: Endometrial cancer. Obstet
Gynecol. 120:383–397. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Watanabe H, Takehana K, Date M, Shinozaki
T and Raz A: Tumor cell autocrine motility factor is the
neuroleukin/phosphohexose isomerase polypeptide. Cancer Res.
56:2960–2963. 1996.PubMed/NCBI
|
|
5
|
Niinaka Y, Paku S, Haga A, Watanabe H and
Raz A: Expression and secretion of neuroleukin/phosphohexose
isomerase/maturation factor as autocrine motility factor by tumor
cells. Cancer Res. 58:2667–2674. 1998.PubMed/NCBI
|
|
6
|
Kim JW and Dang CV: Multifaceted roles of
glycolytic enzymes. Trends Biochem Sci. 30:142–150. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gurney ME, Apatoff BR, Spear GT, Baumel
MJ, Antel JP, Bania MB and Reder AT: Neuroleukin: A lymphokine
product of lectin-stimulated T cells. Science. 234:574–581. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu W, Seiter K, Feldman E, Ahmed T and
Chiao JW: The differentiation and maturation mediator for human
myeloid leukemia cells shares homology with neuroleukin or
phosphoglucose isomerase. Blood. 87:4502–4506. 1996.PubMed/NCBI
|
|
9
|
Kho DH, Zhang T, Balan V, Wang Y, Ha SW,
Xie Y and Raz A: Autocrine motility factor modulates EGF-mediated
invasion signaling. Cancer Res. 74:2229–2237. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kho DH, Nangia-Makker P, Balan V, Hogan V,
Tait L, Wang Y and Raz A: Autocrine motility factor promotes HER2
cleavage and signaling in breast cancer cells. Cancer Res.
73:1411–1419. 2013. View Article : Google Scholar :
|
|
11
|
Bayo J, Fiore E, Aquino JB, Malvicini M,
Rizzo M, Peixoto E, Andriani O, Alaniz L, Piccioni F, Bolontrade M,
et al: Increased migration of human mesenchymal stromal cells by
autocrine motility factor (AMF) resulted in enhanced recruitment
towards hepatocellular carcinoma. PLoS One. 9:e951712014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shih WL, Liao MH, Lin PY, Chang CI, Cheng
HL, Yu FL and Lee JW: PI 3-kinase/Akt and STAT3 are required for
the prevention of TGF-beta-induced Hep3B cell apoptosis by
autocrine motility factor/phosphoglucose isomerase. Cancer Lett.
290:223–237. 2010. View Article : Google Scholar
|
|
13
|
Niinaka Y, Harada K, Fujimuro M, Oda M,
Haga A, Hosoki M, Uzawa N, Arai N, Yamaguchi S, Yamashiro M, et al:
Silencing of autocrine motility factor induces
mesenchymal-to-epithelial transition and suppression of
osteosarcoma pulmonary metastasis. Cancer Res. 70:9483–9493. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Funasaka T, Hogan V and Raz A:
Phosphoglucose isomerase/autocrine motility factor mediates
epithelial and mesenchymal phenotype conversions in breast cancer.
Cancer Res. 69:5349–5356. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Greenburg G and Hay ED: Epithelia
suspended in collagen gels can lose polarity and express
characteristics of migrating mesenchymal cells. J Cell Biol.
95:333–339. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hay ED: The mesenchymal cell, its role in
the embryo, and the remarkable signaling mechanisms that create it.
Dev Dyn. 233:706–720. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: New insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dong P, Karaayvaz M, Jia N, Kaneuchi M,
Hamada J, Watari H, Sudo S, Ju J and Sakuragi N: Mutant p53
gain-of-function induces epithelial-mesenchymal transition through
modulation of the miR-130b-ZEB1 axis. Oncogene. 32:3286–3295. 2013.
View Article : Google Scholar :
|
|
22
|
Ahmad A, Aboukameel A, Kong D, Wang Z,
Sethi S, Chen W, Sarkar FH and Raz A: Phosphoglucose
isomerase/autocrine motility factor mediates epithelial-mesenchymal
transition regulated by miR-200 in breast cancer cells. Cancer Res.
71:3400–3409. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Funasaka T, Hu H, Yanagawa T, Hogan V and
Raz A: Down-regulation of phosphoglucose isomerase/autocrine
motility factor results in mesenchymal-to-epithelial transition of
human lung fibrosarcoma cells. Cancer Res. 67:4236–4243. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kyo S, Sakaguchi J, Ohno S, Mizumoto Y,
Maida Y, Hashimoto M, Nakamura M, Takakura M, Nakajima M and
Masutomi K: High Twist expression is involved in infiltrative
endometrial cancer and affects patient survival. Hum Pathol.
37:431–438. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Massagué J: TGFβ signalling in context.
Nat Rev Mol Cell Biol. 13:616–630. 2012. View Article : Google Scholar
|
|
26
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Subarsky P and Hill RP: The hypoxic tumour
microenvironment and metastatic progression. Clin Exp Metastasis.
20:237–250. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsutsumi S, Yanagawa T, Shimura T,
Fukumori T, Hogan V, Kuwano H and Raz A: Regulation of cell
proliferation by autocrine motility factor/phosphoglucose isomerase
signaling. J Biol Chem. 278:32165–32172. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: At the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: An alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Prindull G and Zipori D: Environmental
guidance of normal and tumor cell plasticity: Epithelial
mesenchymal transitions as a paradigm. Blood. 103:2892–2899. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Otto T, Birchmeier W, Schmidt U, Hinke A,
Schipper J, Rübben H and Raz A: Inverse relation of E-cadherin and
autocrine motility factor receptor expression as a prognostic
factor in patients with bladder carcinomas. Cancer Res.
54:3120–3123. 1994.PubMed/NCBI
|
|
33
|
Batlle E, Sancho E, Francí C, Domínguez D,
Monfar M, Baulida J and García De Herreros A: The transcription
factor snail is a repressor of E-cadherin gene expression in
epithelial tumour cells. Nat Cell Biol. 2:84–89. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pickup M, Novitskiy S and Moses HL: The
roles of TGFβ in the tumour microenvironment. Nat Rev Cancer.
13:788–799. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|