Introduction
Cholangiocellular carcinoma (CCC) is the second most
common type of primary liver cancer, accounting for 5–10% of
primary liver cancer (1–3). CCC is an aggressive malignancy, and
has one of the worst prognosis of the gastrointestinal cancers
(4). The incidence and mortality
rates of biliary tract cancers, including CCC are increasing
worldwide (1,2,4).
Since CCC is an enigmatic malignancy of the biliary tract and is
highly chemoresistant (6), there
is currently no curative treatment other than surgical resection
(5).
Recently, gemcitabine (2′,2′-difluorocytidine
monohydrochloride), a pyrimidine analogue, has been clinically
utilized to treat patients with CCC, either as a single agent or in
combination with cisplatin (GC) or S-1 (GS). Many studies have
demonstrated that gemcitabine is somewhat effective in patients
with advanced CCC (1,2,7,8).
However, the growth inhibitory effect and gemcitabine resistance of
CCC cells are poorly understood (1). Our study aims to investigate the
mechanisms underlying the antitumor effect of gemcitabine as well
as CCC cell resistance to gemcitabine.
Micro-RNAs (miRNAs) are small, endogenous, noncoding
RNAs that can modulate protein expression by regulating
translational efficiency or the cleavage of target mRNAs (6). Aberrant miRNA expression is a common
feature of various human malignancies (9), and some studies have demonstrated
that specific miRNAs are expressed in CCC cells compared with
non-malignant cells (10). In
addition, many studies, including our own, have reported that
miRNAs play an important role in the antitumor effect of anticancer
therapeutics (9,11,12).
Therefore, the present study was undertaken to identify the miRNAs
associated with the antitumor effects of gemcitabine in CCC.
Materials and methods
Chemicals
Gemcitabine was purchased from Eli Lilly Japan
(Hyogo, Japan). The Cell Counting Kit (CCK-8) was purchased from
Dojindo Laboratories (Kumamoto, Japan), and all other chemicals
were obtained from Sigma Chemical Co., Ltd. (Tokyo, Japan).
Antibodies
In this study, the following antibodies were used:
anti-β-actin monoclonal antibody (Sigma-Aldrich; A5441, used at
1:3,000); cyclin D1 (Thermo Fisher Scientific, Waltham, MA, USA;
RB-9041, used at 1:1,000); Cdk4 (Cell Signaling Technology,
Danvers, MA, USA; no 2906, used at 1:1,000); Cdk6 (Santa Cruz
Biotechnology, Santa Cruz, CA, USA; sc-177, used at 1:1,000); and
secondary horseradish peroxidase (HRP)-linked anti-mouse and
anti-rabbit IgG antibodies (GE Healthcare, Buckinghamshire, UK;
used at 1:2,000).
Cell lines and cultures
The human CCC cell lines HuCCT-1, Huh28 and TKKK
were studied. HuCCT-1 and Huh28 were obtained from the Japanese
Cancer Research Resources Bank, and TKKK was provided by the RIKEN
BRC through the National Bio-Resource Project of the MEXT, Japan.
The cells were passaged in our laboratory for fewer than 6 months,
and the cell lines were authenticated by the cell bank using short
tandem repeat PCR. HuCCT-1 cells were grown in RPMI-1640 (Gibco,
Invitrogen, USA); TKKK was grown in DMEM (High Glucose with
L-Glutamine and Phenol Red, Wako, Osaka, Japan), and Huh28 was
grown in MEM (Gibco, Invitrogen). These media were supplemented
with 10% fetal bovine serum (Wako, 533-69545, Japan) and
penicillin-streptomycin (100 mg/l, Invitrogen), and the cells were
cultured in a humidified atmosphere of 5% CO2 at
37°C.
Cell proliferation assay
Cell proliferation assays were conducted using CCK-8
according to the manufacturer's instructions, as in our previous
studies (9,11,12).
Each cell line (1×104) was seeded into a well of a
96-well plate and cultured in 100 μl of each culture medium. After
24 h, cells were treated with 0.001, 0.01, 0.1 or 1 μg/ml
gemcitabine or left untreated. At the indicated time points, the
medium was changed to 110 μl of culture medium supplemented with
CCK-8 reagent (10 μl CCK-8 and 100 μl of each culture medium), and
the cells were incubated for 2 h. Absorbance was measured at a
wavelength of 450 nm using an auto-microplate reader.
Preparation of cell lysates
Lysates were collected according to the methods
described in our previous studies (9,11,12).
All steps were performed at 4°C. Protein concentrations were
measured using a dye-binding protein assay based on the Bradford
method (13).
Gel electrophoresis and western
blotting
Samples were electrophoresed using 7.5–10% SDS-PAGE
according to the Laemmli method (14), and the proteins were transferred to
nitro-cellulose membranes. Western blots were performed according
to Towbin et al (15).
Briefly, the membranes were incubated with primary antibodies after
blocking and then with HRP-conjugated secondary antibodies.
Immunoreactive proteins were visualized with an enhanced
chemiluminescence detection system (Perkin Elmer Co.) on X-ray
film, as described in our previous studies (16–18).
Flow cytometric analysis
To evaluate the mechanism of growth inhibition by
gemcitabine, the cell cycle profile was analyzed following
treatment. HuCCT-1 cells (1.0×106 cells in a 6-well
plate dish) were treated with 0.1 μg/ml gemcitabine or without
gemcitabine for 24–72 h. After treatment, the cells were harvested
and fixed in 80% ethanol, washed with PBS, and stored at −20°C
until flow cytometric analysis was conducted as in our previous
studies (9,11,12).
Prior to analysis, the cells were washed in cold PBS and
resuspended in 100 μl of PBS and 10 μl of RNase A solution (250
μg/ml), followed by incubation for 30 min at 37°C. A total of 110
μl of propidium iodide (PI) stain (100 μg/ml) was added to each
tube, which was then incubated at 4°C for at least 30 min prior to
analysis. Flow cytometric analysis was conducted using a Cytomics
FC 500 flow cytometer (Beckman Coulter) with an argon laser (488
nm). The percentage of cells in different phases of the cell cycle
was analyzed using FlowJo software (Tree Star). All experiments
were performed in triplicate.
Angiogenic profile analysis using an
antibody array
The RayBio™ Human Angiogenesis Antibody Array 1 kit
(catalog no. AAH-ANG-1) was purchased from RayBiotech Inc.
(Norcross, GA, USA). The assay for the array was performed
according to the manufacturer's instructions as in our previous
studies (9,11,12).
Briefly, the angiogenesis antibody membranes were incubated in
blocking buffer for 30 min. The membranes were then incubated with
1 ml of lysate prepared from cell lines after the protein
concentrations were normalized. After washing with TBS containing
0.1% v/v Tween-20 3 times for 10 min, and TBS alone 2 times for 10
min to remove unbound materials, the membranes were incubated for 2
h at room temperature with anti-phospho-tyrosine-HRP antibody. The
unbound HRP antibody was removed with TBS containing 0.1% Tween-20.
Finally, each array membrane was exposed to X-ray film using a
chemiluminescence detection system (Perkin Elmer Co.). The density
of the immunoreactive band obtained on this array was analyzed by
densitometric scanning (Tlc scanner; Shimizu Co. Ltd., Kyoto,
Japan).
Antibody arrays of phosphorylated
receptor tyrosine kinase (p-RTK)
The RayBio Human Phospho Array kit (catalog no. ARY
001) was purchased from RayBiotech Inc. The assay for p-RTK array
was performed according to the manufacturer's instructions, as in
our previous studies (9,11,12).
Briefly, p-RTK array membranes were blocked with 5% BSA/TBS (0.01 M
Tris-HCl, pH 7.6) for 1 h. Membranes were then incubated with 2 ml
of lysate prepared from cell lines after normalization with equal
amounts of protein. After washing with TBS containing 0.1% v/v
Tween-20 (3 washings for 10 min each) and TBS alone (2 washings for
10 min each) to remove unbound materials, the membranes were
incubated with anti-phospho-tyrosine-HRP antibody for 2 h at room
temperature. The unbound HRP antibody was removed with TBS
containing 0.1% Tween-20. Finally, each array membrane was exposed
to X-ray film using a chemiluminescence detection system (Perkin
Elmer Co.). The density of the immunoreactive band obtained on the
p-RTK array was analyzed by densitometric scanning (Tlc scanner;
Shimizu Co. Ltd.).
Analysis of the miRNA array
The samples from the cancer cell lines were
processed for total-RNA extraction with a miRNeasy mini kit
(Qiagen, Hilden, Germany) according to the manufacturer's
instructions as used in our previous studies (9,11,12,19).
RNA samples typically showed A260/280 ratios between 1.9 and 2.1,
as determined using an Agilent 2100 Bioanalyzer (Agilent
Technologies, Santa Clara, CA, USA).
After RNA measurement with an RNA 6000 Nano kit
(Agilent Technologies), the samples were labeled using a miRCURY
Hy3 Power labeling kit and hybridized on a human miRNA Oligo chip
(v.20.0; Toray Industries, Tokyo, Japan). Scanning was performed
with a 3D-Gene Scanner 3000 (Toray Industries). 3D-Gene extraction
version 1.2 software (Toray Industries) was used to read the raw
intensity of the image. To determine the change in miRNA expression
between gemcitabine-treated and control samples, the raw data were
analyzed via GeneSpringGX v10.0 (Agilent Technologies). Samples
were first normalized relative to 28s RNA and baseline-corrected to
the median of all samples.
Replicate data were consolidated into two groups:
those from gemcitabine-treated cells and those from control cells
and were organized using the hierarchical clustering and analysis
of variance (ANOVA) functions in the GeneSpring software.
Hierarchical clustering was conducted using the clustering function
(condition tree) and Euclidean correlation as a distance metric. To
search for the miRNAs that varied most prominently across the
different groups, two-way ANOVA and asymptotic p-value computation
were performed on the samples without any error correction. The
p-value cutoff was set to 0.05. Only changes >50% in at least
one of the time points for each sample were considered significant.
All data were scaled by global normalization, and the significance
of differentially expressed miRNAs was analyzed by Student's
t-test.
All our micoroarray data in this study were submited
as a complete data set to the NCBI Gene Expression Omnibus (GEO),
no. GSE 67257. http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ghafokcytjgnrwt&acc=GSE67257
Statistical analyses
All analyses were performed using the
computer-assisted JMP8.0 (SAS Institute, Cary, NC, USA). Paired
analysis between the groups was performed using Student's t-test. A
p-value of 0.05 was considered to indicate a significant difference
between groups.
Results
Gemcitabine inhibits human CCC cell
proliferation
To evaluate the effect of gemcitabine on human CCC
cell growth activity in vitro, we examined the effect of
gemcitabine on the proliferation of the 3 CCC cell lines HuCCT-1,
Huh28 and TKKK. The cells were grown in 10% FBS and treated with
0.001, 0.01, 0.1, 1 or, as a control, 0 μg/ml gemcitabine. The cell
proliferation assay was conducted 3 days after the addition of the
reagents. As shown in Fig. 1,
gemcitabine (0, 0.001, 0.01, 0.1 and 1 μg/ml) led to a dose- and
time-dependent decrease in cell proliferation in HuCCT-1 cells that
was not observed in Huh28 or TKKK cells.
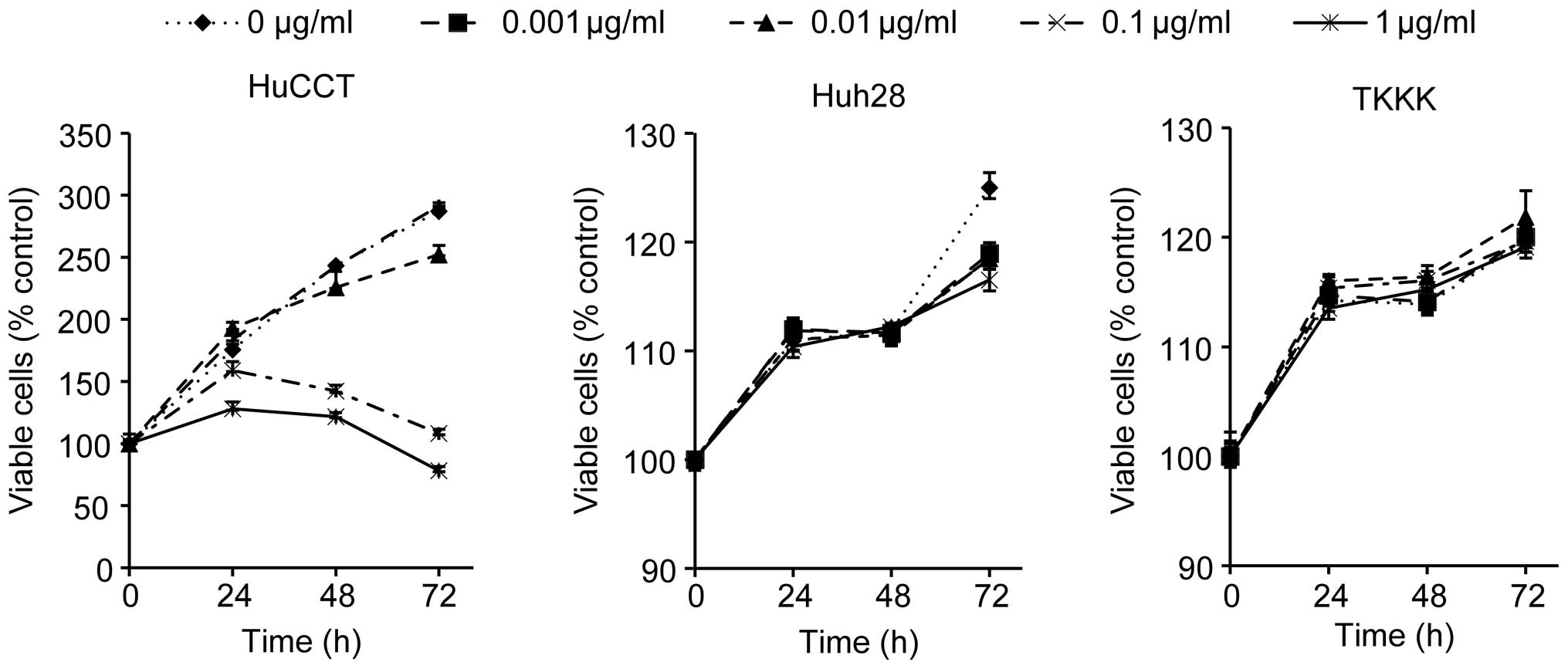 | Figure 1The effects of gemcitabine on
proliferation of cultured cholangiocellular carcinoma (CCC) cells.
HuCCT-1, Huh28 and TKKK cells were seeded at 10,000 cells per well
in 96-well plates, and gemcitabine (GEM; 0, 0.001, 0.01, 0.1 and 1
μg/ml) was added to the culture medium at 0 h. A viability assay
was conducted daily from 0 to 72 h. The data points represent the
mean cell number from 3 independent cultures. The results are
expressed as percentages of viable cells compared with the control
(0 μg/ml). GEM treatment (0, 0.001, 0.01, 0.1 and 1 μg/ml) in
HuCCT-1 cells led to a dose- and time-dependent decrease in cell
proliferation, while GEM did not inhibit cell proliferation in
Huh28 or TKKK cells. The conditions at 48 and 72 h were
significantly different in HuCCT-1 cells compared with the control
(p<0.05). |
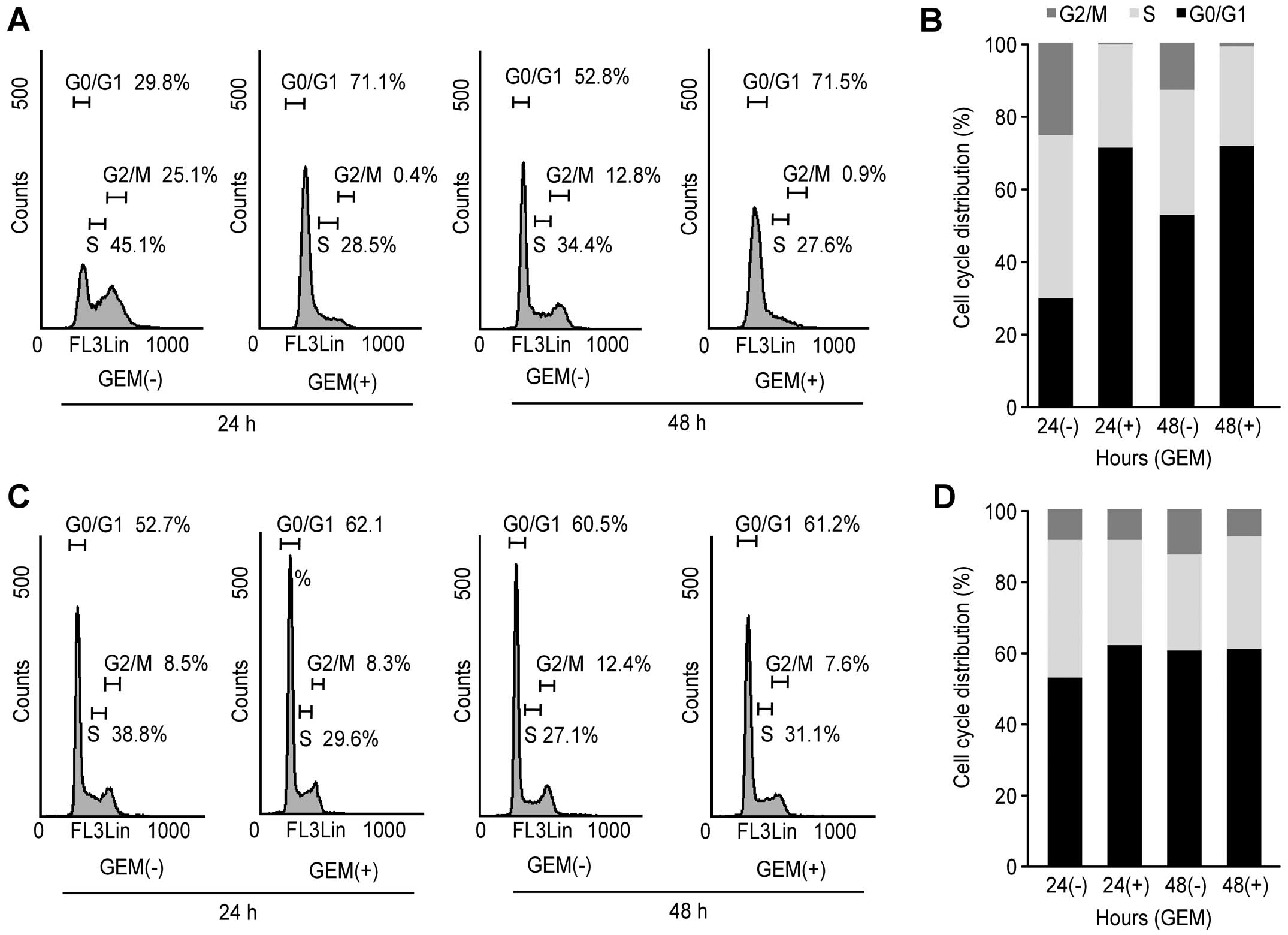
Flow cytometry analysis
To further investigate the inhibition of HuCCT-1
cell proliferation in the presence of gemcitabine, cell cycle
progression was examined by flow cytometry. We treated
proliferating HuCCT-1 cells with 0.1 μg/ml gemcitabine for
different time durations. Following the addition of 0.1 μg/ml
gemcitabine, the fraction of HuCCT-1 cells in the G0/G1 phase
increased to 71.1 and 71.5% after 24 and 48 h, respectively, and
the fraction of cells in S phase decreased to 28.5% and 27.6% after
24 and 48 h, respectively (Fig. 2A and
B). These data suggest that gemcitabine inhibits HuCCT-1
proliferation by preventing cell cycle progression from G0/G1 into
S phase, resulting in G1 cell cycle arrest. The effects of
gemcitabine in HuCCT-1 based on this flow cytometric analysis are
consistent with the cell proliferation assay, as shown in Fig. 1. In contrast, flow cytometric
analysis suggested that gemcitabine treatment of TKKK cells did not
result in G1 cell cycle arrest (Fig.
2C and D).
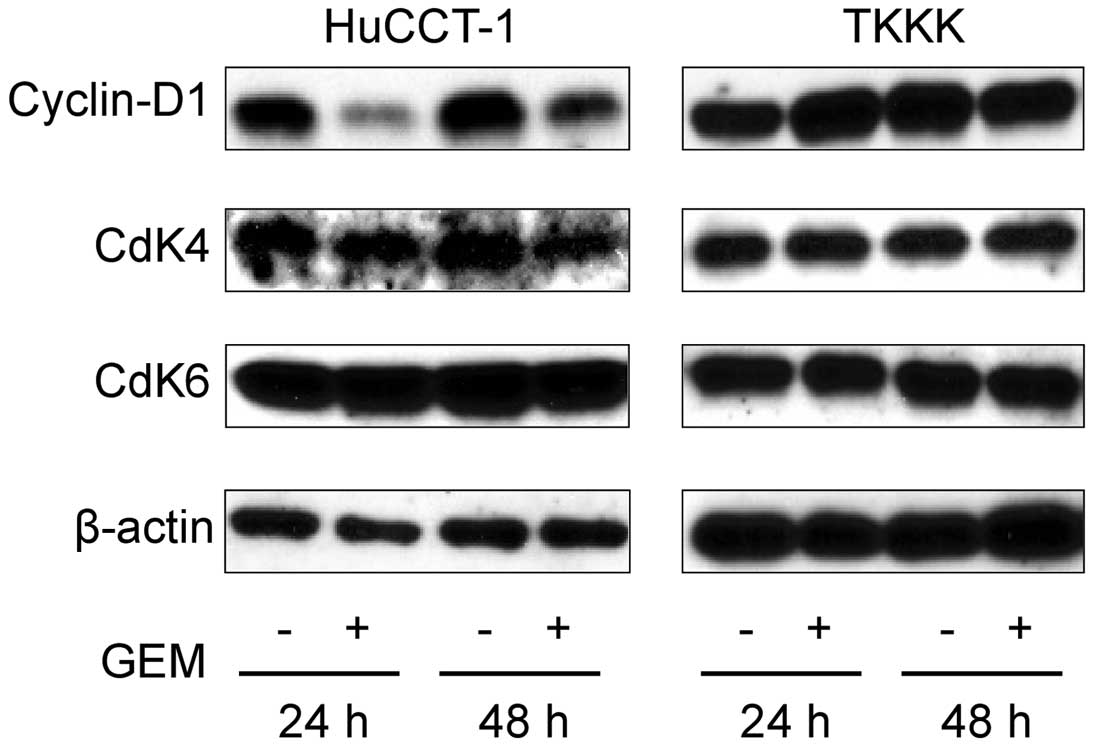
Effects of gemcitabine on cell cycle
regulatory proteins in HuCCT-1
To further study the effects of gemcitabine on the
cell cycle, the expression of cell cycle-related proteins was
studied using western blots in treated and untreated HuCCT-1 and
TKKK cells. Cells were treated with 0.1 or 0 μg/ml gemcitabine for
24 or 48 h. Cyclin D1 expression at the G0–G1 transition was
notably decreased at 24 h and slightly apparent at 48 h in treated
versus untreated HuCCT-1 cells (Fig.
3). In addition, the catalytic subunits of cyclin D1, namely,
Cdk4 and Cdk6, were not changed after 24 or 48 h with or without
0.1 μg/ml gemcitabine treatment in HuCCT-1 (Fig. 3). The expression of cyclin D1, Cdk4
and Cdk6 remained unchanged in TKKK cells, which are not sensitive
to gemcitabine. Based on these results, the gemcitabine-mediated
anti-proliferative effects and cell cycle arrest were due to the
reduction of cyclin D1.
Differences in angiogenesis-related
protein expression in HuCCT-1 and TKKK cells with or without
gemcitabine treatment
We used an angiogenesis antibody array system to
identify the key angiogenesis-related proteins in terms of the
antitumor effect of gemcitabine. Using an antibody array (Fig. 4A), we simultaneously screened the
expression of 20 angiogenic molecules in HuCCT-1 and TKKK cells
with or without 0.1 μg/ml gemcitabine treatment. In HuCCT-1 cells,
which were sensitive to gemcitabine, the expression of interleukin
(IL)-6, IL-8, ENA-78 and MCP-1 was increased after 48 h of
treatment with 0.1 μg/ml gemcitabine, as detected by the protein
array (Fig. 4B). In TKKK cells,
which are resistant to gemcitabine, there was no difference in the
expression of angiogenic molecules between gemcitabine-treated and
untreated cells (Fig. 4C). The
IL-6, IL-8, ENA-78 and MCP-1 densities obtained from the membrane
array were analyzed using the Kodak Image Station (Eastman Kodak),
and the densitometric ratios of gemcitabine-treated to non-treated
HuCCT-1 cells for IL-6, IL-8, ENA-78 and MCP-1 spots were 817, 165,
156 and 633%, respectively (Fig.
4D).
 | Figure 4(A) Template showing the location of
antibodies for angiogenesis-related protein spotted onto the Ray
Bio Human Cytokine antibody array kit. (B and C) Representative
expression of antibodies for angiogenesis-related protein in
HuCCT-1 and TKKK cells with or without gemcitabine (GEM) treatment.
In HuCCT-1 cells, the increased expression of IL-6, IL-8, ENA-78
and MCP-1 was detected in cells treated with GEM. In TKKK cells,
the expression of all angiogenesis-related proteins did not change
in treated versus untreated cells. (D) The densities of IL-6, IL-8,
ENA-78 and MCP-1 obtained from the membrane array were analyzed in
HuCCT-1. The densitometric ratios of gemcitabine-treated to
untreated cells in ENA-78, IL-6, IL-8 and MCP-1 spots were 156,
817, 165 and 633%. |
Differences in phosphorylated-receptor
tyrosine kinases p-(RTKs) in HuCCT-1 and TKKK cells with or without
gemcitabine treatment
Having established the antitumor effects of
gemcitabine in CCC cell lines, we next used a phosphorylated-RTK
array system to identify the key RTKs in terms of antitumor
effects. Using an antibody array (Fig.
5A), we simultaneously screened the expression of 42 different
RTKs in HuCCT-1 and TKKK cells after 48 h with or without 0.1 μg/ml
gemcitabine treatment. RTKs activation was not changed by
gemcitabine treatment in HuCCT-1 or TKKK cells (Fig. 5B and C).
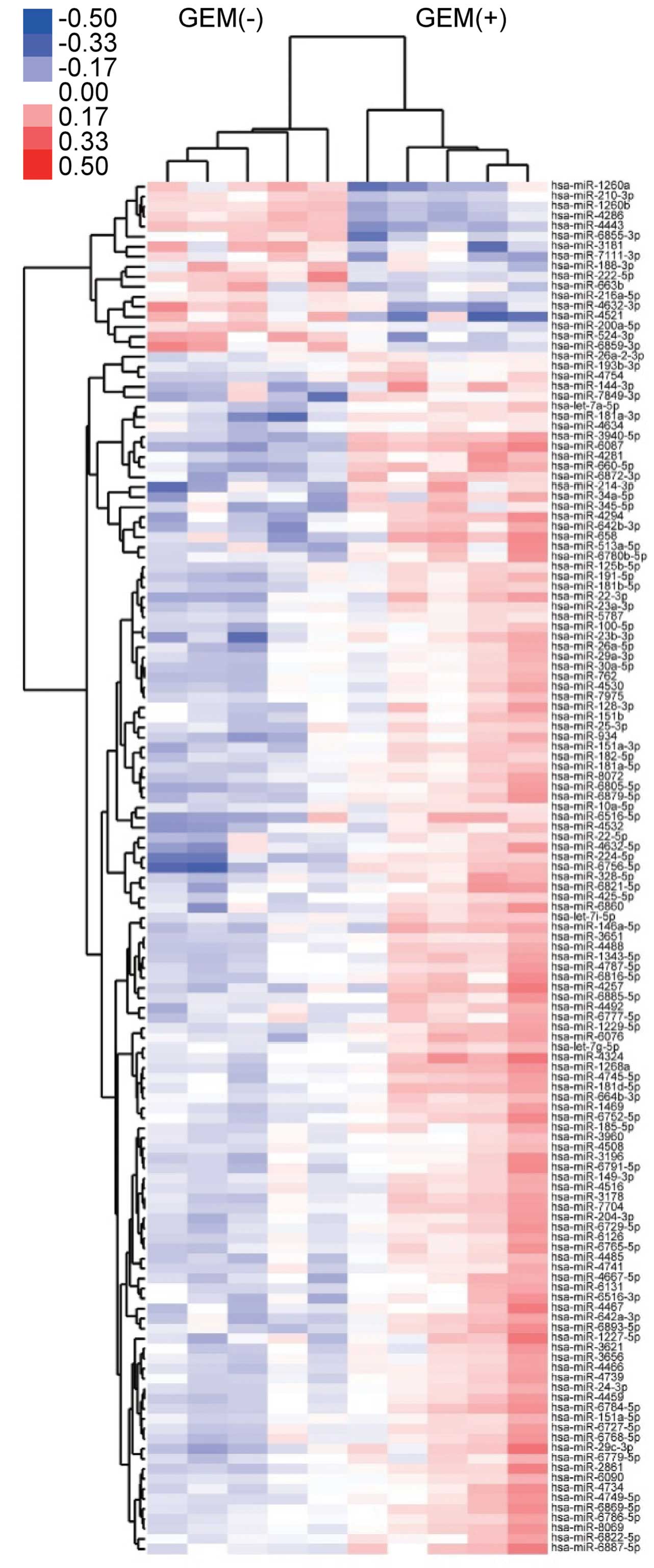
Differences in miRNA expression in
HuCCT-1 cells with or without gemcitabine treatment in vitro
Using a custom microarray platform, we studied the
in vitro expression levels of 2555 human miRNA probes in two
cell lines (gemcitabine-sensitive HuCCT-1 cells and
gemcitabine-resistant TKKK cells) with or without gemcitabine
treatment. Unsupervised hierarchical clustering analysis using
Pearson's correlation demonstrated that HuCCT-1 cell lines treated
in vitro with gemcitabine, clustered together, separate from
untreated cell lines (Fig. 6).
These subsets of 137 microRNAs in the HuCCT-1 cell lines exhibited
significantly (p<0.05) different expression levels between the
gemcitabine-treated and control groups. As shown in Table I, when in vitro miRNA
expression was studied in gemcitabine-treated and untreated HuCCT-1
cells, 95 miRNAs were significantly upregulated after 48 h, while
11 miRNAs were downregulated. These 106 HuCCT-1 miRNAs exhibited a
>1.5-fold alteration in expression levels between the
gemcitabine-treated and control groups.
 | Table IExpression changes and chromosomal
locations of miRNAs in HuCCT-1 cells (gemcitabine sensitive)
treated with gemcitabine compared with untreated cells. |
Table I
Expression changes and chromosomal
locations of miRNAs in HuCCT-1 cells (gemcitabine sensitive)
treated with gemcitabine compared with untreated cells.
| Upregulated
miRNA | Fold
(treated/untreated) | Chromosomal
localization |
|---|
|
|---|
| Mean ± SD | p-value |
|---|
| hsa-miR-6087 | 3.66±0.651 | 0.00002 | Xq22.3 |
| hsa-miR-23b-3p | 2.74±1.031 | 0.01229 | 9q22.23 |
|
hsa-miR-6756-5p | 2.33±0.847 | 0.00502 | 11q23.3 |
| hsa-miR-22-3p | 2.33±0.875 | 0.01442 | 17p13.3 |
|
hsa-miR-3940-5p | 2.21±0.685 | 0.00004 | 19p13.3 |
| hsa-miR-224-5p | 2.05±0.534 | 0.00201 | Xq28 |
| hsa-miR-29c-3p | 1.92±0.736 | 0.01513 | 1q32.2 |
|
hsa-miR-6805-5p | 1.91±0.609 | 0.00568 | 19q13.42 |
| hsa-miR-2861 | 1.91±0.832 | 0.01377 | 9 |
| hsa-miR-660-5p | 1.91±0.461 | 0.00051 | Xp11.23 |
| hsa-miR-7704 | 1.89±0.402 | 0.00804 | 2q31.1 |
| hsa-miR-4257 | 1.88±0.976 | 0.02902 | 1q21.2 |
| hsa-miR-658 | 1.88±0.082 | 0.0168 | 22q13.1 |
|
hsa-miR-6784-5p | 1.86±0.409 | 0.00642 | 17q21.31 |
| hsa-let-7a-5p | 1.85±0.945 | 0.01319 |
9q22.32/11q24.1/22q13.31 |
|
hsa-miR-146a-5p | 1.84±0.661 | 0.02201 | 5q33.3 |
| hsa-miR-8072 | 1.83±0.371 | 0.00516 | 12q24.31 |
| hsa-miR-4294 | 1.82±0.765 | 0.00554 | 10q11.23 |
| hsa-miR-4488 | 1.82±0.291 | 0.00227 | 11q12.2 |
|
hsa-miR-181a-3p | 1.81±0.666 | 0.00896 | 1q32.1 |
| hsa-miR-3196 | 1.81±0.580 | 0.0311 | 20q13.33 |
|
hsa-miR-642b-3p | 1.80±0.752 | 0.00487 | 19q13.32 |
| hsa-miR-4324 | 1.80±0.537 | 0.00874 | 19q13.33 |
| hsa-miR-214-3p | 1.78±0.966 | 0.01726 | 1q24.3 |
|
hsa-miR-6729-5p | 1.77±0.477 | 0.00946 | 1p36.22 |
| hsa-miR-3178 | 1.77±0.389 | 0.00752 | 16p13.3 |
| hsa-miR-204-3p | 1.76±0.350 | 0.01501 | 9q21.12 |
| hsa-miR-1268a | 1.73±0.355 | 0.00168 | 15q11.2 |
| hsa-miR-191-5p | 1.72±0.416 | 0.00965 | 3p21.31 |
| hsa-miR-4459 | 1.72±0.277 | 0.00785 | 5q11.2 |
| hsa-miR-100-5p | 1.72±0.367 | 0.01228 | 11q24.1 |
|
hsa-miR-513a-5p | 1.71±0.753 | 0.02639 | Xq27.3 |
|
hsa-miR-4787-5p | 1.70±0.473 | 0.02273 | 3p21.2 |
| hsa-miR-762 | 1.70±0.235 | 0.02553 | 16p11.2 |
|
hsa-miR-6879-5p | 1.70±0.563 | 0.00542 | 11q13.1 |
| hsa-miR-4281 | 1.69±0.491 | 0.00879 | 5q35.2 |
|
hsa-miR-6516-5p | 1.69±0.527 | 0.02887 | 17q25.2 |
|
hsa-miR-642a-3p | 1.69±0.467 | 0.00551 | 19q13.32 |
|
hsa-miR-1227-5p | 1.68±0.482 | 0.04756 | 19p13.3 |
| hsa-miR-328-5p | 1.68±0.390 | 0.02023 | 16q22.1 |
|
hsa-miR-6893-5p | 1.68±0.708 | 0.01314 | 8q24.3 |
| hsa-miR-29a-3p | 1.67±0.360 | 0.04206 | 7q32.3 |
| hsa-miR-6860 | 1.65±0.533 | 0.03083 | 11 |
|
hsa-miR-6869-5p | 1.65±0.490 | 0.01637 | 20p13 |
| hsa-miR-23a-3p | 1.65±0.481 | 0.02188 | 19p13.13 |
| hsa-miR-4466 | 1.65±0.309 | 0.00507 | 6q25.3 |
|
hsa-miR-151a-3p | 1.65±0.390 | 0.00371 | 8q24.3 |
| hsa-miR-24-3p | 1.64±0.231 | 0.01055 |
9q22.32/19p13.12 |
| hsa-miR-144-3p | 1.64±0.318 | 0.0193 | 17q11.2 |
| hsa-miR-4467 | 1.64±0.603 | 0.04342 | 7q22.1 |
|
hsa-miR-6816-5p | 1.64±0.463 | 0.01588 | 22q11.21 |
|
hsa-miR-6752-5p | 1.63±0.641 | 0.02179 | 11q13.2 |
|
hsa-miR-1343-5p | 1.62±0.342 | 0.01088 | 11p13 |
| hsa-miR-26a-5p | 1.62±0.412 | 0.03867 | 3p22.2/12q14.1 |
| hsa-miR-1469 | 1.62±0.425 | 0.00392 | 15q26.2 |
|
hsa-miR-6768-5p | 1.62±0.190 | 0.04194 | 16p13.3 |
| hsa-miR-4739 | 1.61±0.440 | 0.01531 | 17q25.3 |
| hsa-miR-6076 | 1.61±0.569 | 0.00643 | 14q21.3 |
|
hsa-miR-4632-5p | 1.61±0.444 | 0.04771 | 1p36.22 |
|
hsa-miR-125b-5p | 1.61±0.273 | 0.00715 |
11q24.1/21q21.1 |
| hsa-miR-3621 | 1.60±0.421 | 0.03484 | 9q34.3 |
|
hsa-miR-1229-5p | 1.60±0.378 | 0.00114 | 5q35.3 |
|
hsa-miR-6727-5p | 1.60±0.289 | 0.02885 | 1p36.33 |
| hsa-miR-345-5p | 1.60±0.334 | 0.01108 | 14q32.2 |
|
hsa-miR-6887-5p | 1.59±0.489 | 0.00986 | 19q13.12 |
| hsa-miR-934 | 1.59±0.475 | 0.00804 | Xq26.3 |
|
hsa-miR-6872-3p | 1.58±0.407 | 0.00243 | 3p21.31 |
|
hsa-miR-6821-5p | 1.58±0.417 | 0.02227 | 22q13.33 |
| hsa-miR-4530 | 1.58±0.331 | 0.04933 | 19q13.2 |
| hsa-miR-7975 | 1.58±0.386 | 0.04948 | 19q13.42 |
| hsa-miR-4492 | 1.57±0.391 | 0.02715 | 11q23.3 |
| hsa-miR-8069 | 1.57±0.430 | 0.01413 | 21 |
| hsa-miR-4516 | 1.57±0.282 | 0.03403 | 16p13.3 |
| hsa-miR-4532 | 1.56±0.456 | 0.04442 | 20q13.32 |
| hsa-miR-4734 | 1.56±0.534 | 0.03834 | 17q12 |
| hsa-miR-3656 | 1.56±0.295 | 0.02295 | 11q23.3 |
|
hsa-miR-6765-5p | 1.56±0.331 | 0.02757 | 14q32.33 |
|
hsa-miR-181d-5p | 1.56±0.381 | 0.00426 | 19p13.13 |
|
hsa-miR-6516-3p | 1.56±0.498 | 0.03436 | 17q25.2 |
|
hsa-miR-4667-5p | 1.55±0.507 | 0.01382 | 9p13.3 |
|
hsa-miR-6885-5p | 1.55±0.498 | 0.01662 | 19p13.3 |
|
hsa-miR-6791-5p | 1.54±0.329 | 0.0392 | 19p13.3 |
|
hsa-miR-6780b-5p | 1.54±0.458 | 0.02333 | 6p21.1 |
|
hsa-miR-181b-5p | 1.54±0.176 | 0.00073 | 1q32.1/9q33.3 |
| hsa-miR-4485 | 1.54±0.215 | 0.00333 | 11 |
| hsa-miR-30a-5p | 1.54±0.198 | 0.03159 | 6q13 |
| hsa-miR-34a-5p | 1.54±0.490 | 0.02875 | 1p36.22 |
| hsa-miR-3651 | 1.53±0.240 | 0.00232 | 9q22.31 |
|
hsa-miR-7849-3p | 1.53±0.373 | 0.02978 | 4q31.22 |
|
hsa-miR-6786-5p | 1.52±0.416 | 0.02395 | 17q25.3 |
|
hsa-miR-4745-5p | 1.51±0.365 | 0.01286 | 19p13.3 |
|
hsa-miR-4749-5p | 1.50±0.467 | 0.02088 | 19q13.33 |
| hsa-miR-128-3p | 1.50±0.597 | 0.03967 | 2q21.3/3p22.3 |
|
hsa-miR-664b-3p | 1.50±0.376 | 0.00448 | Xq28 |
| hsa-miR-6090 | 1.50±0.333 | 0.02555 | 11q24.3 |
| Downregulated
miRNA | Fold
(treated/untreated) | Chromosomal
localization |
|---|
|
|---|
| Mean ± SD | p-value |
|---|
| hsa-miR-1260b | 0.37±0.069 | 0.00014 | 11q21 |
| hsa-miR-4443 | 0.39±0.056 | 3.60E-06 | 3p21.31 |
| hsa-miR-4286 | 0.39±0.051 | 0.0002 | 8p23.1 |
| hsa-miR-1260a | 0.43±0.099 | 0.00599 | 14q24.3 |
| hsa-miR-4521 | 0.53±0.211 | 0.0102 | 17 |
| hsa-miR-222-5p | 0.62±0.170 | 0.00432 | Xp11.3 |
| hsa-miR-3181 | 0.63±0.175 | 0.0266 | 16q12.1 |
| hsa-miR-524-3p | 0.63±0.027 | 0.00748 | 19q13.42 |
|
hsa-miR-4632-3p | 0.63±0.282 | 0.01632 | 1p36.22 |
| hsa-miR-210-3p | 0.66±0.103 | 0.0008 | 11p15.5 |
|
hsa-miR-6859-3p | 0.67±0.194 | 0.01234 | 1/15/16 |
In TKKK cells, which were resistant to gemcitabine,
1 miRNA was upregulated and 16 miRNAs were down-regulated (Table II). In Tables I and II, miR-3181 was downregulated in both
cell lines (gemcitabine-sensitive HuCCT-1 and gemcitabine-resistant
TKKK). The miRNAs miR-6087, miR-3651 and miR-664b-3p were
upregulated in gemcitabine-treated HuCCT-1 cells (Tables I and II) and downregulated in
gemcitabine-treated TKKK cells (Table III).
| Table IIExpression changes and chromosomal
locations of miRNAs in TKKK cells (gemcitabine resistant) treated
with gemcitabine compared with non-treated cells. |
Table II
Expression changes and chromosomal
locations of miRNAs in TKKK cells (gemcitabine resistant) treated
with gemcitabine compared with non-treated cells.
| Upregulated
miRNA | Fold
(treated/untreated) | Chromosomal
localization |
|---|
|
|---|
| Mean ± SD | p-value |
|---|
|
hsa-miR-1238-3p | 2.03±0.337 | 0.00328 | 19p13.2 |
| Downregulated
miRNA | | | |
| hsa-miR-99a-5p | 0.42±0.191 | 0.00527 | 21q21.1 |
|
hsa-miR-664b-3p | 0.47±0.161 | 0.00611 | Xq28 |
| hsa-miR-625-3p | 0.52±0.205 | 0.02519 | 14q23.3 |
| hsa-miR-6087 | 0.54±0.232 | 0.02987 | Xq22.3 |
| hsa-miR-6070 | 0.55±0.195 | 0.00925 | 21q22.3 |
|
hsa-miR-513a-5p | 0.57±0.229 | 0.01055 | Xq27.3 |
| hsa-miR-502-3p | 0.57±0.088 | 0.00092 | Xp11.23 |
| hsa-miR-492 | 0.59±0.431 | 0.04099 | 12q22 |
| hsa-miR-484 | 0.59±0.270 | 0.02036 | 16p13.11 |
| hsa-miR-4454 | 0.60±0.231 | 0.00617 | 4q32.2 |
| hsa-miR-4417 | 0.62±0.239 | 0.0202 | 1p36.31 |
| hsa-miR-3687 | 0.62±0.337 | 0.01778 | 21p11.2 |
| hsa-miR-3652 | 0.63±0.227 | 0.00968 | 12q23.3 |
| hsa-miR-3651 | 0.64±0.244 | 0.03039 | 9q22.31 |
|
hsa-miR-3184-3p | 0.66±0.174 | 0.00723 | 17q11.2 |
| hsa-miR-3181 | 0.67±0.178 | 0.01088 | 16q12.1 |
| Table IIIExpression miR-6087, miR-3651 and
miR-664b-3p was the opposite in the cell types (HuCCT-1, sensitive;
TKKK, resistant) treated with gemcitabine. |
Table III
Expression miR-6087, miR-3651 and
miR-664b-3p was the opposite in the cell types (HuCCT-1, sensitive;
TKKK, resistant) treated with gemcitabine.
| Fold
(treated/untreated) Mean ± SD |
|---|
|
|
|---|
| miRNA | HuCCT-1 | TKKK |
|---|
| hsa-miR-6087 | 3.66±0.651 ↑ | 0.54±0.232 ↓ |
| hsa-miR-3651 | 1.53±0.240 ↑ | 0.64±0.244 ↓ |
|
hsa-miR-664b-3p | 1.50±0.376 ↑ | 0.47±0.161 ↓ |
Discussion
The incidence of CCC, the second most common tumor
of primary liver cancers in adults, is rising worldwide (1). Currently, there is no curative
treatment other than surgical resection (2,5,20).
Conventional chemotherapy is not always effective, because CCC is a
highly chemo-resistant malignancy (2,6).
Therefore, it is necessary to study the mechanism of growth
inhibitory effect and gemcitabine resistance in CCC cells.
In the present study, gemcitabine treatment in the
three human cell lines led to a strong, dose-dependent inhibition
of cell proliferation in only HuCCT-1 cells. In the
gemcitabine-sensitive HuCCT-1 cells, the anti-proliferative effect
of gemcitabine led to G1 arrest through a reduction of cyclin D1.
Although some studies have reported that gemcitabine could inhibit
cell-phase transitioning during G1 phase (1,21),
to date, there are no studies on cyclin D1 reduction as an
anticancer effect of gemcitabine in CCC cells. This study revealed
that gemcitabine induced a cell cycle arrest at the G0/G1 phase by
reducing cyclin D1 levels in HuCCT-1 cells in vitro.
Gemcitabine did not exert an anti-proliferative effect on the other
cell lines, Huh28 and TKKK. These results support that some CCCs
might be highly chemoresistant to clinical treatment.
Angiogenic profile analysis revealed that in
gemcitabine-sensitive HuCCT-1 cells, gemcitabine upregulated IL-6,
IL-8, ENA-78 and MCP-1. IL-6, IL-8, ENA-78 and MCP-1, which are not
only related to angiogenesis but are also involved in the promotion
of cell proliferation (22–25).
Studies suggest that these molecules are upregulated in various
cancers including cholangiocarcinoma (22–25).
In addition, IL-8, IL-6 and MCP-1 overexpression was associated
with a worse prognosis of patients with various cancers (22,23,25).
These events suggest that CCC patients might acquire gemcitabine
resistance, even if they are initially sensitive. Acquired
gemcitabine resistance might be due to the gemcitabine-induced
upregulation of IL-6, IL-8, ENA-78 and MCP-1. These data suggest
that the application of gemcitabine for CCC treatment might be
limited.
miRNAs are small, endogenous, noncoding RNA
sequences that can modulate protein expression by regulating
translational efficiency or the cleavage of target mRNA molecules
(6). To identify the miRNAs
associated with antitumor effect and acquired gemcitabine
resistance, we used a miRNA array to measure the variation in
HuCCT-1 cell lines cultured with or without gemcitabine. In the
cluster analysis, we demonstrated that treating HuCCT-1 with
gemcitabine affects various miRNAs. In the present study, sets of
miRNAs had significantly altered expression levels. In HuCCT-1
cells, these altered miRNAs may provide clues to the molecular
basis of the gemcitabine anticancer effects.
Of note, the following tumor suppressor miRNAs were
upregulated in gemcitabine-treated HuCCT-1 cells: miR-23b-3p,
miR-22-3p, miR-29c, miR-660, let-7a, miR-146a, miR-214, miR-204,
miR-100, miR-29a, miR-23a, miR-24 and miR-34a. These results
suggest that the inhibition of cell proliferation by gemcitabine
might be due to the induction of tumor suppressor miRNAs.
In previous studies, we demonstrated that members of
the let-7 family, altered by the antidiabetic drug metformin,
contribute to cell growth inhibition (9,11).
In the present study, gemcitabine upregulated let-7a in HuCCT-1
cells. The let-7 family contains 13 members and is recognized as a
class of miRNAs that induce tumor-suppressing effects. Reduction of
let-7 family members have been reported in various cancers,
including lung cancer (26),
breast cancer (27), colorectal
cancer (28), gastric cancer
(11), and hepatocellular
carcinoma (29). The let-7 family
members act as tumor suppressor molecules by binding target
oncogenes, such as Ras (30),
HMGA2 (31) and c-Myc (32). In addition, Liu et al
reported that let-7a reduced c-Myc and the c-Myc target gene cyclin
D1, leading to cell cycle arrest and the inhibition of
proliferation (33). These events
suggest that the suppression of cancer cell proliferation by
gemcitabine may result, in part, from the upregulation of
let-7a.
miR-214 inhibits cell growth in hepatocellular
carcinoma (HCC) through suppression of β-catenin (34). In addition, miR-214 results in the
suppression of cyclin D1, a downstream gene of the Wnt-β-catenin
pathway (34). Since cyclin D1 is
also a target of miR-34a in HCC (35), the upregulation of miR-34a reduces
cyclin D1 expression (36). The
upregulation of let-7a, miR-214 and miR-34a may reduce cyclin D1 in
gemcitabine-treated HuCCT-1 cells.
Among the miRNAs downregulated in
gemcitabine-treated HuCCT-1 cells, miR-1260b (37), miR-4286 (38), miR-222 (39–43)
and miR-210 (44) were upregulated
in several cancers. Collectively, the antitumor effects of
gemcitabine in HuCCT-1 cells might be related to the reduction of
these miRNAs. miR-1260b, in particular, has been recognized as an
onco-miRNA, because the antitumor effect molecule genistein
downregulates onco-miR-1260b and inhibits the Wnt-β-catenin
signaling pathway, which is involved in cell growth (37). The Wnt-β-catenin signaling pathway
is activated during CCC tumorigenesis (45). Therefore, the antitumor effect of
gemcitabine might be associated with miR-1260b inhibition. miR-222
was also recognized as oncogenic miRNA (39–43).
The cell cycle-dependent kinase inhibitor, p27Kip1 is the target
gene of miR-222 (39,40,43).
Therefore, the downregulation of miR-222 led to G1 arrest. Based on
previous studies, our data suggest that the altered miRNA,
particularly, the downregulation of miR-1260b and miR-222, may
result from the antitumor effect of gemcitabine.
In Tables I and
II, miR-3181 is shown as
downregulated in both HuCCT-1 and TKKK cells. These data suggest
that the change in miR-3181 expression might not be involved in the
antitumor effects of gemcitabine. In contrast, while miR-6087,
miR-3651 and miR-664b-3p were upregulated in gemcitabine-treated
HuCCT-1 cells, these miRNAs were downregulated in TKKK cells. The
results suggest that modulations in miR-6087, miR-3651 and
miR-664b-3p expression following gemcitabine treatment might be an
important factor in determining whether cancer cells are sensitive
to gemcitabine.
In conclusion, our results revealed that gemcitabine
inhibits HuCCT-1 cell proliferation by suppressing cell
cycle-related proteins, especially cyclin D1. In addition,
alterations in miRNA expression after gemcitabine treatment
contribute to gemcitabine resistance. Aberrant miRNA or target
molecule expression would provide a mechanism for the treatment of
CCC using gemcitabine.
References
|
1
|
Matsumoto K, Nagahara T, Okano J and
Murawaki Y: The growth inhibition of hepatocellular and
cholangiocellular carcinoma cells by gemcitabine and the roles of
extracellular signal-regulated and checkpoint kinases. Oncol Rep.
20:863–872. 2008.PubMed/NCBI
|
|
2
|
Nakajima Y, Takagi H, Kakizaki S,
Horiguchi N, Sato K, Sunaga N and Mori M: Gefitinib and gemcitabine
coordinately inhibited the proliferation of cholangiocarcinoma
cells. Anticancer Res. 32:5251–5262. 2012.PubMed/NCBI
|
|
3
|
Iwaki J, Kikuchi K, Mizuguchi Y,
Kawahigashi Y, Yoshida H, Uchida E and Takizawa T: MiR-376c
down-regulation accelerates EGF-dependent migration by targeting
GRB2 in the HuCCT1 human intrahepatic cholangiocarcinoma cell line.
PLoS One. 8:e694962013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Patel T: New insights into the molecular
pathogenesis of intra-hepatic cholangiocarcinoma. J Gastroenterol.
49:165–172. 2014. View Article : Google Scholar :
|
|
5
|
Jarnagin WR, Ruo L, Little SA, Klimstra D,
D'Angelica M, DeMatteo RP, Wagman R, Blumgart LH and Fong Y:
Patterns of initial disease recurrence after resection of
gallbladder carcinoma and hilar cholangiocarcinoma: Implications
for adjuvant therapeutic strategies. Cancer. 98:1689–1700. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meng F, Henson R, Lang M, Wehbe H,
Maheshwari S, Mendell JT, Jiang J, Schmittgen TD and Patel T:
Involvement of human micro-RNA in growth and response to
chemotherapy in human cholangiocarcinoma cell lines.
Gastroenterology. 130:2113–2129. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Valle J, Wasan H, Palmer DH, Cunningham D,
Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira
SP, et al; ABC-02 Trial Investigators. Cisplatin plus gemcitabine
versus gemcitabine for biliary tract cancer. N Engl J Med.
362:1273–1281. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Morizane C, Okusaka T, Mizusawa J,
Takashima A, Ueno M, Ikeda M, Hamamoto Y, Ishii H, Boku N and
Furuse J: Randomized phase II study of gemcitabine plus S-1 versus
S-1 in advanced biliary tract cancer: A Japan Clinical Oncology
Group trial (JCOG 0805). Cancer Sci. 104:1211–1216. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miyoshi H, Kato K, Iwama H, Maeda E,
Sakamoto T, Fujita K, Toyota Y, Tani J, Nomura T, Mimura S, et al:
Effect of the anti-diabetic drug metformin in hepatocellular
carcinoma in vitro and in vivo. Int J Oncol. 45:322–332.
2014.PubMed/NCBI
|
|
10
|
Haga H, Yan I, Takahashi K, Wood J and
Patel T: Emerging insights into the role of microRNAs in the
pathogenesis of cholangiocarcinoma. Gene Expr. 16:93–99. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kato K, Gong J, Iwama H, Kitanaka A, Tani
J, Miyoshi H, Nomura K, Mimura S, Kobayashi M, Aritomo Y, et al:
The anti-diabetic drug metformin inhibits gastric cancer cell
proliferation in vitro and in vivo. Mol Cancer Ther. 11:549–560.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kobayashi M, Kato K, Iwama H, Fujihara S,
Nishiyama N, Mimura S, Toyota Y, Nomura T, Nomura K, Tani J, et al:
Antitumor effect of metformin in esophageal cancer: In vitro study.
Int J Oncol. 42:517–524. 2013.
|
|
13
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Towbin H, Staehelin T and Gordon J:
Electrophoretic transfer of proteins from polyacrylamide gels to
nitrocellulose sheets: Procedure and some applications. Proc Natl
Acad Sci USA. 76:4350–4354. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Masaki T, Okada M, Shiratori Y, Rengifo W,
Matsumoto K, Maeda S, Kato N, Kanai F, Komatsu Y, Nishioka M, et
al: pp60c-src activation in hepatocellular carcinoma of humans and
LEC rats. Hepatology. 27:1257–1264. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Masaki T, Shiratori Y, Rengifo W, Igarashi
K, Matsumoto K, Nishioka M, Hatanaka Y and Omata M: Hepatocellular
carcinoma cell cycle: Study of Long-Evans cinnamon rats.
Hepatology. 32:711–720. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Morishita A, Masaki T, Yoshiji H, Nakai S,
Ogi T, Miyauchi Y, Yoshida S, Funaki T, Uchida N, Kita Y, et al:
Reduced expression of cell cycle regulator p18(INK4C) in human
hepatocellular carcinoma. Hepatology. 40:677–686. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Katsura A, Morishita A, Iwama H, Tani J,
Sakamoto T, Tatsuta M, Toyota Y, Fujita K, Kato K, Maeda E, et al:
MicroRNA profiles following metformin treatment in a mouse model of
non-alcoholic steatohepatitis. Int J Mol Med. 35:877–884.
2015.PubMed/NCBI
|
|
20
|
Anderson CD, Pinson CW, Berlin J and Chari
RS: Diagnosis and treatment of cholangiocarcinoma. Oncologist.
9:43–57. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y, Zhou Y, Zhou H, Jia G, Liu J, Han
B, Cheng Z, Jiang H, Pan S and Sun B: Pristimerin causes G1 arrest,
induces apoptosis, and enhances the chemosensitivity to gemcitabine
in pancreatic cancer cells. PLoS One. 7:e438262012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hong DS, Angelo LS and Kurzrock R:
Interleukin-6 and its receptor in cancer: Implications for
translational therapeutics. Cancer. 110:1911–1928. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Waugh DJJ and Wilson C: The interleukin-8
pathway in cancer. Clin Cancer Res. 14:6735–6741. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Okabe H, Beppu T, Ueda M, Hayashi H,
Ishiko T, Masuda T, Otao R, Horlad H, Mima K, Miyake K, et al:
Identification of CXCL5/ENA-78 as a factor involved in the
interaction between cholangiocarcinoma cells and cancer-associated
fibroblasts. Int J Cancer. 131:2234–2241. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Salcedo R, Ponce ML, Young HA, Wasserman
K, Ward JM, Kleinman HK, Oppenheim JJ and Murphy WJ: Human
endothelial cells express CCR2 and respond to MCP-1: Direct role of
MCP-1 in angiogenesis and tumor progression. Blood. 96:34–40.
2000.PubMed/NCBI
|
|
26
|
Takamizawa J, Konishi H, Yanagisawa K,
Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y,
et al: Reduced expression of the let-7 microRNAs in human lung
cancers in association with shortened postoperative survival.
Cancer Res. 64:3753–3756. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong
C, Huang Y, Hu X, Su F, Lieberman J, et al: let-7 regulates self
renewal and tumorigenicity of breast cancer cells. Cell.
131:1109–1123. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Akao Y, Nakagawa Y and Naoe T: let-7
microRNA functions as a potential growth suppressor in human colon
cancer cells. Biol Pharm Bull. 29:903–906. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu X-M, Wu L-J, Xu J, Yang R and Wu F-S:
Let-7c microRNA expression and clinical significance in
hepatocellular carcinoma. J Int Med Res. 39:2323–2329. 2011.
View Article : Google Scholar
|
|
30
|
Johnson SM, Grosshans H, Shingara J, Byrom
M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D and Slack
FJ: RAS is regulated by the let-7 microRNA family. Cell.
120:635–647. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee YS and Dutta A: The tumor suppressor
microRNA let-7 represses the HMGA2 oncogene. Genes Dev.
21:1025–1030. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Osada H and Takahashi T: let-7 and
miR-17-92: Small-sized major players in lung cancer development.
Cancer Sci. 102:9–17. 2011. View Article : Google Scholar
|
|
33
|
Liu Y, Yin B, Zhang C, Zhou L and Fan J:
Hsa-let-7a functions as a tumor suppressor in renal cell carcinoma
cell lines by targeting c-myc. Biochem Biophys Res Commun.
417:371–375. 2012. View Article : Google Scholar
|
|
34
|
Wang X, Chen J, Li F, Lin Y, Zhang X, Lv Z
and Jiang J: MiR-214 inhibits cell growth in hepatocellular
carcinoma through suppression of β-catenin. Biochem Biophys Res
Commun. 428:525–531. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xiao Z, Li CH, Chan SL, Xu F, Feng L, Wang
Y, Jiang JD, Sung JJ, Cheng CH and Chen Y: A small-molecule
modulator of the tumor-suppressor miR34a inhibits the growth of
hepato-cellular carcinoma. Cancer Res. 74:6236–6247. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guo Y, Li S, Qu J, Wang S, Dang Y, Fan J,
Yu S and Zhang J: MiR-34a inhibits lymphatic metastasis potential
of mouse hepatoma cells. Mol Cell Biochem. 354:275–282. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hirata H, Ueno K, Nakajima K, Tabatabai
ZL, Hinoda Y, Ishi N and Dahiya R: Genistein downregulates
onco-miR-1260b and inhibits Wnt-signalling in renal cancer cells.
Br J Cancer. 108:2070–2078. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sand M, Skrygan M, Sand D, Georgas D,
Gambichler T, Hahn SA, Altmeyer P and Bechara FG: Comparative
microarray analysis of microRNA expression profiles in primary
cutaneous malignant melanoma, cutaneous malignant melanoma
metastases, and benign melanocytic nevi. Cell Tissue Res.
351:85–98. 2013. View Article : Google Scholar
|
|
39
|
Yang Y-F, Wang F, Xiao J-J, Song Y, Zhao
YY, Cao Y, Bei YH and Yang CQ: MiR-222 overexpression promotes
proliferation of human hepatocellular carcinoma HepG2 cells by
downregulating p27. Int J Clin Exp Med. 7:893–902. 2014.PubMed/NCBI
|
|
40
|
Sun C, Li N, Zhou B, Yang Z, Ding D, Weng
D, Meng L, Wang S, Zhou J, Ma D, et al: miR-222 is upregulated in
epithelial ovarian cancer and promotes cell proliferation by
downregulating P27(kip1). Oncol Lett. 6:507–512. 2013.PubMed/NCBI
|
|
41
|
Saito Y, Suzuki H, Matsuura M, Sato A,
Kasai Y, Yamada K, Saito H and Hibi T: MicroRNAs in hepatobiliary
and pancreatic cancers. Front Genet. 2:662011. View Article : Google Scholar
|
|
42
|
Chun-Zhi Z, Lei H, An-Ling Z, Yan-Chao F,
Xiao Y, Guang-Xiu W, Zhi-Fan J, Pei-Yu P, Qing-Yu Z and Chun-Sheng
K: MicroRNA-221 and microRNA-222 regulate gastric carcinoma cell
proliferation and radioresistance by targeting PTEN. BMC Cancer.
10:3672010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Visone R, Russo L, Pallante P, De Martino
I, Ferraro A, Leone V, Borbone E, Petrocca F, Alder H, Croce CM, et
al: MicroRNAs (miR)-221 and miR-222, both overexpressed in human
thyroid papillary carcinomas, regulate p27Kip1 protein levels and
cell cycle. Endocr Relat Cancer. 14:791–798. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Võsa U, Vooder T, Kolde R, Vilo J,
Metspalu A and Annilo T: Meta-analysis of microRNA expression in
lung cancer. Int J Cancer. 132:2884–2893. 2013. View Article : Google Scholar
|
|
45
|
Sugimachi K, Aishima S, Taguchi K, Tanaka
S, Shimada M, Kajiyama K, Sugimachi K and Tsuneyoshi M: The role of
over-expression and gene amplification of cyclin D1 in intrahepatic
cholangiocarcinoma. J Hepatol. 35:74–79. 2001. View Article : Google Scholar : PubMed/NCBI
|