Introduction
Cells in vivo proliferate and survive under
the influence of adjacent cells (1–3).
Cell-cell adhesion is important for tumor cell biological behavior,
including chemosensitivity (1,2,4–7).
Intercellular adhesion is not well recapitulated in two-dimensional
(2D) cell cultures and this may explain, in part, the phenomenon
that novel anticancer drugs often offer only a small improvement
over older agents or even fail to offer improvement in clinical
trials, though they exhibit exciting anticancer effect in
vitro (1–6). Three-dimensional (3D) cell culture
models represent a better approximation of solid tumor tissue
microenvironment, including cell adhesion and chemosensitivity
in vivo than 2D cultures (1–8).
E-cadherin (Ecad), a classic member of the cadherin
family, is responsible for cell-cell adhesion (2,9–13).
On the other hand, intercellular force and E-cadherin binding to
cadherins on adjacent cells trigger certain E-cadherin signaling
cascades (9,10,12).
Decreased expression of E-cadherin is strongly correlated with
colorectal cancer, including prognosis (14,15).
Therefore, we explored the role of E-cadherin in colorectal cancer
chemosensitivity using 3D cultures. E-cadherin knockdown
significantly reduced chemosensitivity via increasing β-catenin in
3D cultures while these effects were not observed in 2D
cultures.
Materials and methods
Cell lines and cell culture
Human colorectal cancer cell lines, HCT116, HT29,
LoVo and human embryonic kidney 293T were obtained from the Cell
Bank, Chinese Academy of Science.
2D cultures were routinely grown and passaged as
previously described (4,16). In brief, cells were grown in
McCoy's 5A (Gibco, Grand Island, NY, USA) (HCT116 and HT29), F12K
(Gibco) (LoVo) or DMEM (293T) (Gibco) supplemented with 100 ml/l
fetal bovine serum (Gibco), 100,000 IU/l penicillin, and 100 mg/l
streptomycin (Gibco) under a humidified atmosphere of 5%
CO2 at 37°C.
3D cultures were prepared by using the liquid
overlay technique as previously described (2,4,17).
In brief, exponentially-growing cancer cells were seeded into
plates what were previously coated with 2% agarose. Plates were
gently horizontally swirled 10 min every 6 h for the first 24 h on
an orbital shaker in order to form multicellular spheroids. Cells
were incubated under a humidified atmosphere of 5% CO2
at 37°C. Appropriate medium was refreshed every day.
Lentiviral delivery of shRNA
E-cadherin and β-catenin were knocked down through
the use of lentiviral vector-mediated shRNA interference using The
RNAi Consortium System (Open Biosystems, Inc., Huntsville, AL, USA)
according to the manual, respectively (18). Sense sequences of shRNAs targeting
specific genes are AAGATAGGAGTTCTCTGATGC (shEcad-1) or
ATACCAGAACCTCGAACTATA (shEcad-2) for E-cadherin (18) and GCTTGGAATGAGACTGCTGAT for
β-catenin (shβ-catenin) (18).
Control shRNA (shcontrol) is targeted against green fluorescent
protein and the sense sequence of shRNA is TACAACAGCCACAACGTCTAT.
E-cadherin/β-catenin-targeting shRNA-pLKO.1 vector or a control
shRNA-pLKO.1 vector with the packaging plasmid pCMV-Dr8.91 and the
enveloping plasmid pCMV-VSV-G were co-transfected into 293T cells
with Lipofectamine® 2000 (Invitrogen, Carlsbad, CA, USA)
according to the manual. Virus-containing media was collected at 48
and 72 h post-transfection, and was filtered. Cells were infected
with lentivirus encoding shRNA targeted specific genes or control
lentivirus, respectively. Then, cells were selected using
puro-mycin (Sigma-Aldrich, St. Louis, MO, USA). Knockdown
efficiency was confirmed by western blotting.
Hematoxylin and eosin (H&E)
staining
HCT116 3D cultures were collected in a 1.5 ml
Eppendorf tube. Following centrifugation (100 g, 2 min, 4°C), the
supernatant was discarded. The pellet was fixed in 4%
paraformaldehyde for 30 min. Following centrifugation (100 g, 3
min), the pellet was placed in 100% ethanol and xylene for 10 min
at room temperature, and then in paraffin for 20 min at 65°C,
respectively. The sample was embedded in paraffin and the
paraffin-embedded sample was sectioned at a thickness of 10 μm.
Sample slides were routinely stained with H&E.
Colorectal tumors were induced in C57BL/6 mice by
azoxymethane (Sigma-Aldrich)-dextran sodium sulfate (MP
Biomedicals, molecular weight 36,000–50,000 Da., Irvine, CA, USA)
as previously described (19).
Mice were sacrificed 20 weeks after azoxymethane treatment and
colorectums were excised. Colorectal tumors were fixed in 10%
formalin/PBS and paraffin-embedded samples were sectioned at a
thickness of 6 μm. Sample slides were routinely stained with
H&E. The study was approved by the Ethics Committee of
Southwest Hospital.
Immunofluorescence staining
HCT116 cells were cultured as monolayer on cover
slides for 72 h. After fixation in 4% paraformaldehyde for 30 min,
cells were incubated in 0.2% Triton X-100 in 2% BSA/PBS for 30 min.
Antibody of β-catenin (Cell Signaling Technology, Beverly, MA, USA)
and Alexa Fluor 555 goat anti-rabbit (Invitrogen) were incubated
for 2 h and 30 min, respectively. 4′,6-diamidino-2-phenylindole
(DAPI) (1 μg/ml) (Sigma-Aldrich) was used for staining the nucleus
of cells for 30 min.
Immunohistochemical staining
HCT116 3D cultures were fixed in 4% paraformaldehyde
and OCT embedded samples were sectioned at a thickness of 10 μm.
Immunohistochemistry was performed according to protocol of the
SPlink Detection kits (ZSGB-Bio, Beijing, China) as previously
described (16).
Preparation for transmission electron
microscope slides
Sample slides were routinely prepared as previously
described (4). In brief, 3D
cultures were fixed in 2.5% glutaraldehyde, and then in 1% osmium
tetroxide. Samples were dehydrated by graded alcohol, ultrathin
sectioned. Sections were stained with uranium acetate and lead
citrate, and observed using a transmission electron microscope
(TECNAI10, Philip, The Netherlands).
Preparation of cell lysates
Cells were lysed in RIPA buffer (50 mM Tris base,
150 mM NaCl, 1% Nonidet P-40, 0.25% Na-deoxycholate, 1 mM EDTA)
with protease inhibitors and phosphatase inhibitors (1 mM PMSF, 5
μg/ml leupeptin, 2 μg/ml pepstatin, 4 μg/ml aprotinin, 10 mM NaF, 1
mM Na3VO4, 10 mM β-glycerophosphate disodium
salt pentahydrate) by incubating for 30 min on ice. Following
centrifugation (26,000 × g, 16 min, 4°C), the supernatant was
collected as total cell protein (19).
Western blot analysis
Protein was resolved by SDS/PAGE and blotted on
nitrocellulose membranes (Bio-Rad, Richmond, CA, USA) as previously
described (4,16,19).
Nitrocellulose membranes were incubated with specific primary
antibodies overnight. After incubating with secondary antibodies,
immunoreactive proteins were visualized by the enhanced
chemiluminescnet substrate (Thermo Scientific, Pittsburgh, PA,
USA).
E-cadherin antibody was from Abcam Inc. (Cambridge,
MA, USA). β-catenin antibody, phospho-mitogen-activated protein
kinase (MAPK) Family Antibody Sampler kit, α-tubulin antibody,
β-actin antibody, GAPDH antibody, histone H3, HRP-linked secondary
antibody were from Cell Signaling Technology.
Clonogenic assay
Clonogenic assay in vitro were routinely
performed as previously described (20). In brief, HCT116 3D cultures were
collected in 2 ml medium. 0.2 ml medium containing 3D cultures was
taken into a 0.5 ml Eppendorf tube. Following centrifugation (100
g, 5 min), the pellet was detached by accutase and the cells number
of the single-cell suspension were assayed. Then, the cells number
of the 3D cultures was calculated. The same amount of HCT116 3D
cultures was treated with 80 mg/l 5-FU for 24 h. Then, 3D cultures
were detached by accutase and the same ratio of single-cell
suspensions were seeded into 24-well plates duplicate. Cells were
cultured at 37°C for 7 days, then stained with crystal violet.
WST assay for sensitivity to anticancer
drugs
Cytotoxic activity in 2D cultures was determined by
tetrazolium salt-based proliferation assay (WST assay) using the
cell counting kit-8 (Dojindo Laboratories, Kumamoto, Japan)
according to the manual as previously described (4,21).
In brief, HCT116 cells were cultured in 96-well plates as
monolayer. Then, 10 μl of a graded concentration of 5-fluorouracil
(5-FU) or irinotecan (CPT-11) were added into each well and
cultured for 24 h. Control cultures received 10 μl PBS only. Each
contained 8 independent samples. After 24 h, 10 μl of WST solution
were added to each well and the plates were incubated for another 2
h. Absorbance was measured at 450 nm using a microplate reader with
reference wavelength of 650 nm. Cell viability was measured and
compared with that of control cells.
Cytotoxic activity in 3D cultures was determined by
WST using the similar way to assay cytotoxic activity in 2D
cultures as previously described (4,17,21).
Before incubating with WST, 3D cultures were detached by accutase
for 2 min. Cell viability was measured and compared with that of 3D
cultures treated with PBS.
Statistical analyses
The data shown represent the mean ± standard error.
Statistical differences between groups were analyzed by one-way
ANOVA. p<0.05 was considered statistically significant.
Results
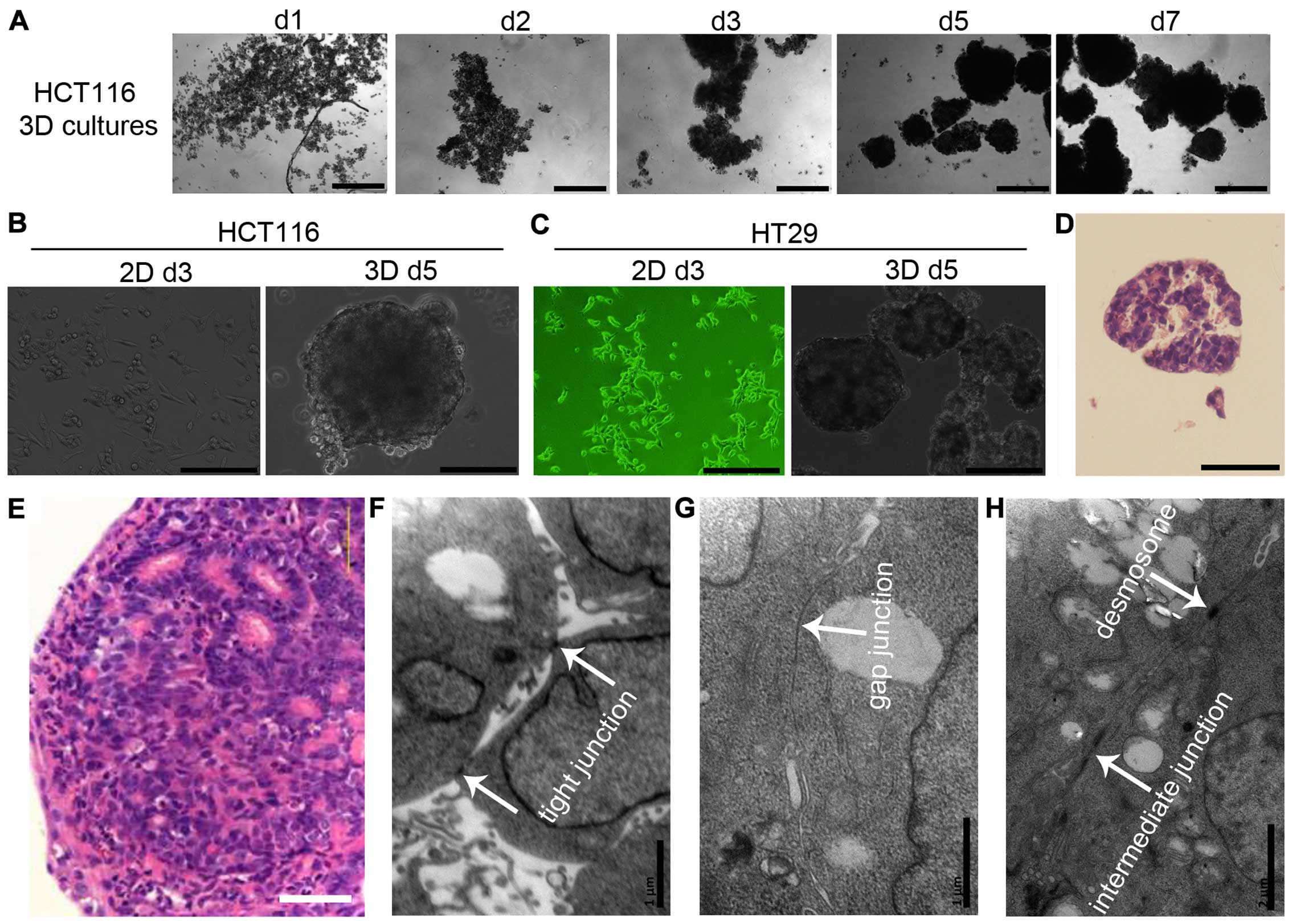
Cell-cell adhesion in 3D cultures
When seeded under non-adhesive conditions, dispersed
cells (HCT116 and HT29) aggregated automatically and formed 3D
cultures (multicellular spheroids) within 24 h. Cells adhered to
each other loosely. Three days later, cells adhered tightly to
other cells and 3D cultures could be hardly dispersed into single
cells by pipetting (Fig.
1A–C).
To further analyze cell-cell adhesive systems,
HCT116 3D cultures were stained with H&E. 3D cultures consisted
of layers of cells and the cells were packed tightly (Fig. 1D). These structures mimic tumors at
avascular stage or avascular tumor regions (Fig. 1E). HCT116 3D cultures were also
observed using a transmission electron microscope. Cell-cell
junctions, including tight junctions, gap junctions, intermediate
junctions and desmosomes, were commonly found in 3D cultures.
Cell-cell junctions were found in most of cells (Fig. 1F–H).
E-cadherin knockdown does not change the
architecture formation of 3D cultures
E-cadherin is expressed by a variety of tissues and
plays a key role in mediating cell-cell adhesive systems (9,10).
It was reported that inhibition of E-cadherin function in cell-cell
adhesion by E-cadherin neutralizing antibody (SHE78-7) disrupted
preformed colorectal cancer cell 3D cultures (6,13).
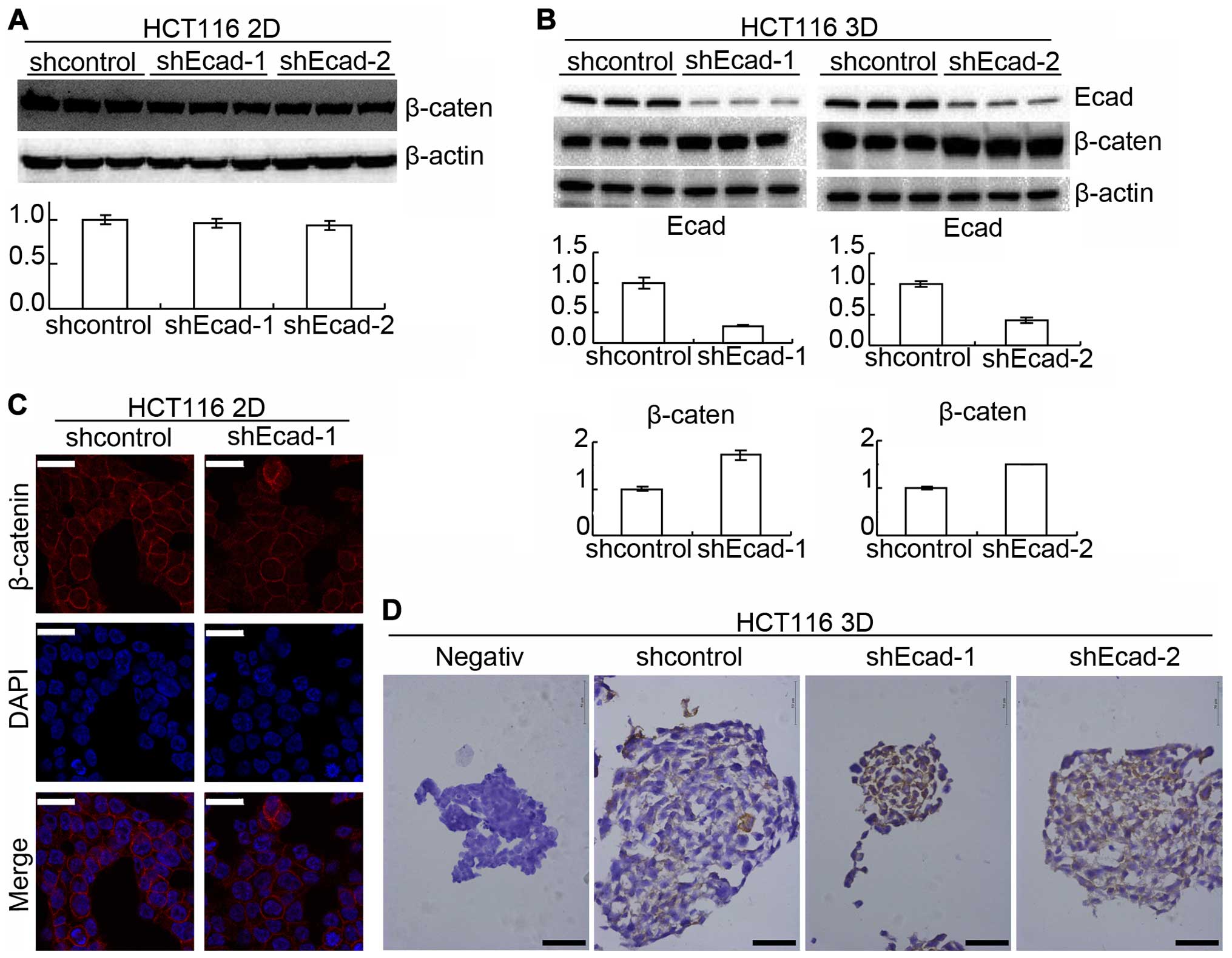
In this study, E-cadherin in HCT116 was knocked down by lentiviral
delivery of shRNA. The efficiency was confirmed by western blotting
(Fig. 2A). HCT116 with E-cadherin
knockdown (shEcad) grew as multicellular spheroids as HCT116
transfected with control lentivirus for 16 days and no difference
was observed under an invert microscope (Fig. 2B). LoVo cells were also employed
because E-cadherin expression in LoVo was too low to be detected
using western blotting (Fig. 2A).
LoVo cells also automatically formed spheroids under 3D culture
conditions (Fig. 2B). The above
suggested that E-cadherin knockdown does not change the
architecture formation of 3D cultures, as assessed under the
inverted microscope.
E-cadherin knockdown in HCT116 reduces
chemosensitivity only in 3D cultures
Intercellular force and homophilic binding of
E-cadherin on adjacent cells trigger certain E-cadherin signaling
cascades (9,10,12).
Therefore, the function of E-cadherin in 3D cultures may be
different from in 2D cultures. Thus, the role of E-cadherin in
chemosensitivity in 3D cultures and in 2D cultures was
explored.
WST assay was performed to evaluate sensitivity to
anti-cancer drugs (Fig. 3A). The
viability of parental HCT116 3D cultures treated with 80 mg/l 5-FU
for 24 h was 45.7±3.1% and that of shcontrol was 47.9±3.3%. There
was no significant difference (p≥0.05). Compare to the shcontrol,
the viability of shEcad-1 HCT116 3D cultures treated with 80 mg/l
5-FU increased to 66.0±2.9% (p<0.01) and that of shEcad-2
increased to 61.8±2.5% (p<0.01). E-cadherin knockdown also
decreased chemosensitivity to CPT-11. The viabilities of parental,
shcontrol, shEcad-1 and shEcad-2 treated with 6 mg/l CPT-11 were
39.7±3.1, 34.0±3.7 (vs parental: p≥0.05), 50.6±2.0 (vs shcontrol:
p<0.01) and 47.1±2.3% (vs shcontrol: p<0.01).
Results from the clonogenic assay were consistent
with the WST assay. E-cadherin knockdown increased the
clonogenicity (Fig. 3B). Cleaved
caspase-3 of shcontrol HCT116 3D cultures and shEcad-1 HCT116 3D
cultures treated with 20 mg/l 5-FU for 24 h was analyzed by western
blotting. Result showed that E-cadherin knockdown decreased cleaved
caspase-3 (Fig. 3C).
To explore the effect of E-cadherin knockdown on
chemosensitivity in 2D cultures, sensitivity was performed by WST
assay (Fig. 3D). The viability of
parental, shcon-trol and shEcad-1 HCT116 cells, respectively,
treated with 20 mg/l 5-FU was 35.0±2.3, 36.4±2.0 (vs parental:
p≥0.05) and 37.9±2.9% (vs shcontrol: p≥0.05). The viability of
parental, shcontrol and shEcad-1 treated with 80 mg/l 5-FU was
23.3±2.1, 21.3±2.1 (vs parental: p≥0.05) and 24.2±2.4% (vs
shcontrol: p≥0.05). The viability, respectively, of parental,
shcontrol and shEcad-1 treated with 2 mg/l CPT-11 was 40.0±1.8,
37.9±2.5 (vs parental: p≥0.05) and 41.5±3.2% (vs shcontrol:
p≥0.05). The viabilities of parental, shcontrol and shEcad-1
treated with 6 mg/l CPT-11 were 24.5±2.7, 21.0±2.5 (vs parental:
p≥0.05) and 27.2±3.1% (vs shcontrol: p≥0.05). Thus, E-cadherin
knockdown does not significantly change chemosensitivity in HCT116
2D cultures.
E-cadherin knockdown increases β-catenin
reducing chemosensitivity only in 3D cultures
Protein β-catenin was reported to interact with
E-cadherin and was also involved in cell adhesion (10,22).
Western blotting showed that E-cadherin knockdown increased
β-catenin in 3D cultures but did not detectably increased β-catenin
in 2D cultures (Fig. 4A and B). To
confirm the result, β-catenin was assayed by immunofluorescence in
2D cultures and immunohistochemistry in 3D cultures.
Immunofluorescence showed that β-catenin was not significantly
changed by E-cadherin knockdown in 2D cultures (Fig. 4C) while immunohistochemistry showed
that E-cadherin knockdown increased β-catenin in 3D cultures
(Fig. 4D).
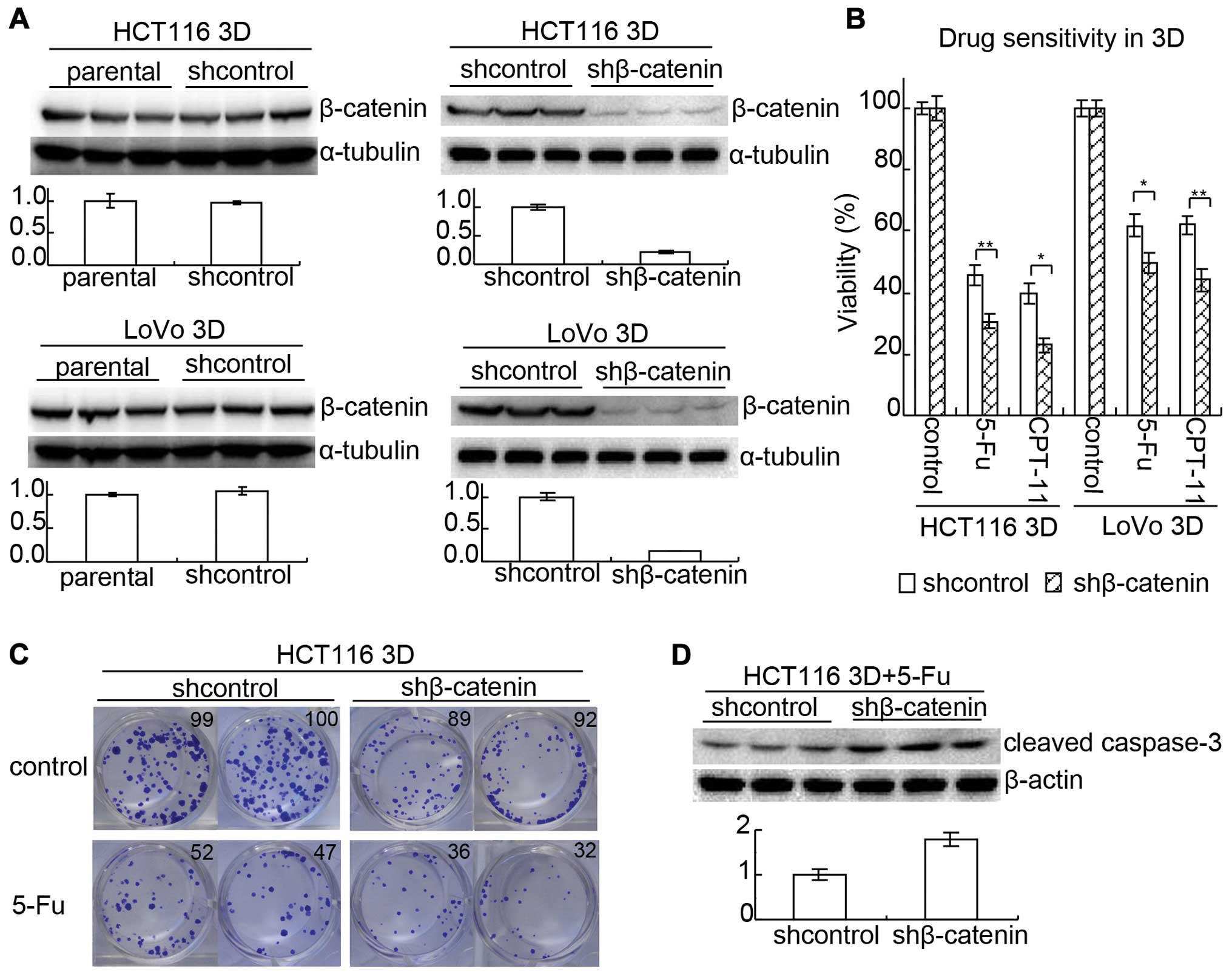
As mentioned above, E-cadherin knockdown increased
β-catenin and decreased chemosensitivity only in 3D cultures. Since
it was reported that enhancing β-catenin expression promoted
anticancer drug resistance (23),
β-catenin may be involved in the mechanism of E-cadherin knockdown
decreasing chemosensitivity in 3D cultures. To confirm this,
β-catenin in HCT116 and LoVo was knocked down by lenti-viral
delivery of shRNA, respectively. After the efficiency was confirmed
by western blotting (Fig. 5A),
chemoensitivity to 5-FU and CPT-11 in 3D cultures was evaluated by
WST assay (Fig. 5B). The viability
of shcontrol HCT116 treated with 80 mg/l 5-FU was 45.7±3.1% while
that of β-catenin knock-down (shβ-catenin) HCT116 was 30.8±2.1%
(p<0.01). The viability of shcontrol HCT116 treated with 6 mg/l
CPT-11 was 39.7±3.1% while that of shβ-catenin was 22.9±2.2%
(p<0.05). Results in LoVo 3D cultures were similar to those in
HCT116. The viability of shcontrol LoVo treated with 80 mg/l 5-FU
was 61.9±3.4% while that of shβ-catenin was 49.6±3.3% (p<0.05).
The viability of shcontrol LoVo treated with 6 mg/l CPT-11 was
62.1±3.0% while that of shβ-catenin was 44.2±3.7% (p<0.01).
Results from clonogenic assay were consistent with
WST assay. Knockdown of β-catenin decreased the clonogenicity
(Fig. 5C). It seemed that
knockdown of β-catenin decreased both the size of clones and the
clonogenicity without 5-FU treatment. The ratio of clonogenicity
with 5-FU:clonogenicity without 5-FU was lower in the shβ-catenin
group than in the shcontrol group. Cleaved caspase-3 of shcontrol
HCT116 3D cultures and shβ-catenin HCT116 3D cultures treated with
20 mg/l 5-FU for 24 h was analyzed by western blotting. Result
showed that knockdown of β-catenin increased cleaved caspase-3
(Fig. 5D).
The above suggests that E-cadherin knockdown
increases β-catenin to reduce chemosensitivity in 3D cultures.
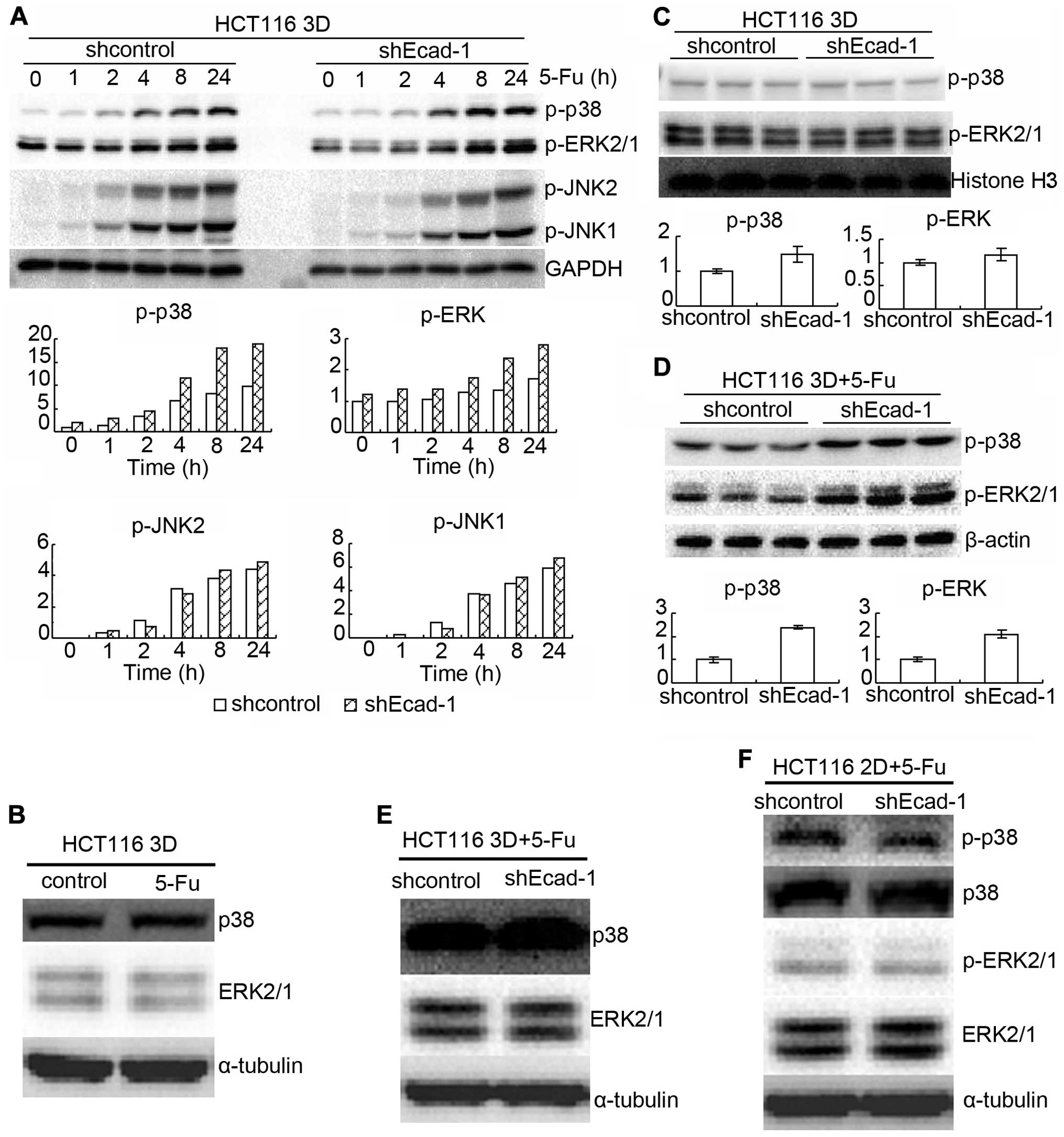
MAPK pathway is involved in mechanism of
E-cadherin knockdown increasing β-catenin to reduce
chemosensitivity
MAPK signaling plays a critical role in the
sensitivity to anticancer therapies (4,24–28).
Also, formation of E-cadherin-mediated cell-cell adhesion regulates
MAPKs (29). Thus, the role of
MAPKs in E-cadherin knockdown increasing β-catenin to reduce
chemosensitivity was explored. HCT116 3D cultures were treated with
80 mg/l 5-FU for different time points. Protein p-p38,
p-extracellular-signal-regulated kinase (ERK) 1/2 and p-c-Jun
N-terminal kinase (JNK) 1/2 were assayed by western blotting. 5-FU
treatment increased p-p38, p-ERK 1/2 and p-JNK 1/2 in a time
dependent manner in HCT116 3D cultures (Fig. 6A). E-cadherin knockdown increased
p-p38 and p-ERK 1/2, except JNK1/2 (Fig. 6A). Total p38 and ERK1/2 protein
level was not detectably changed by 80 mg/l 5-FU treatment for 8 h
(Fig. 6B). E-cadherin knockdown
mildly increased basal levels of p-p38 and p-ERK1/2 (Fig. 6A and C). When treated with 80 mg/l
5-FU for 8 h, E-cadherin knockdown increased p-p38 and p-ERK1/2 in
HCT116 3D cultures (Fig. 6A and
D). E-cadherin knockdown neither changed total p38 and ERK1/2
protein level in 2D cultures nor in 3D cultures under treatment of
5-FU (Fig. 6E and F). E-cadherin
knockdown did not detectably change p-p38 and p-ERK1/2 protein
level in 2D cultures, either (Fig.
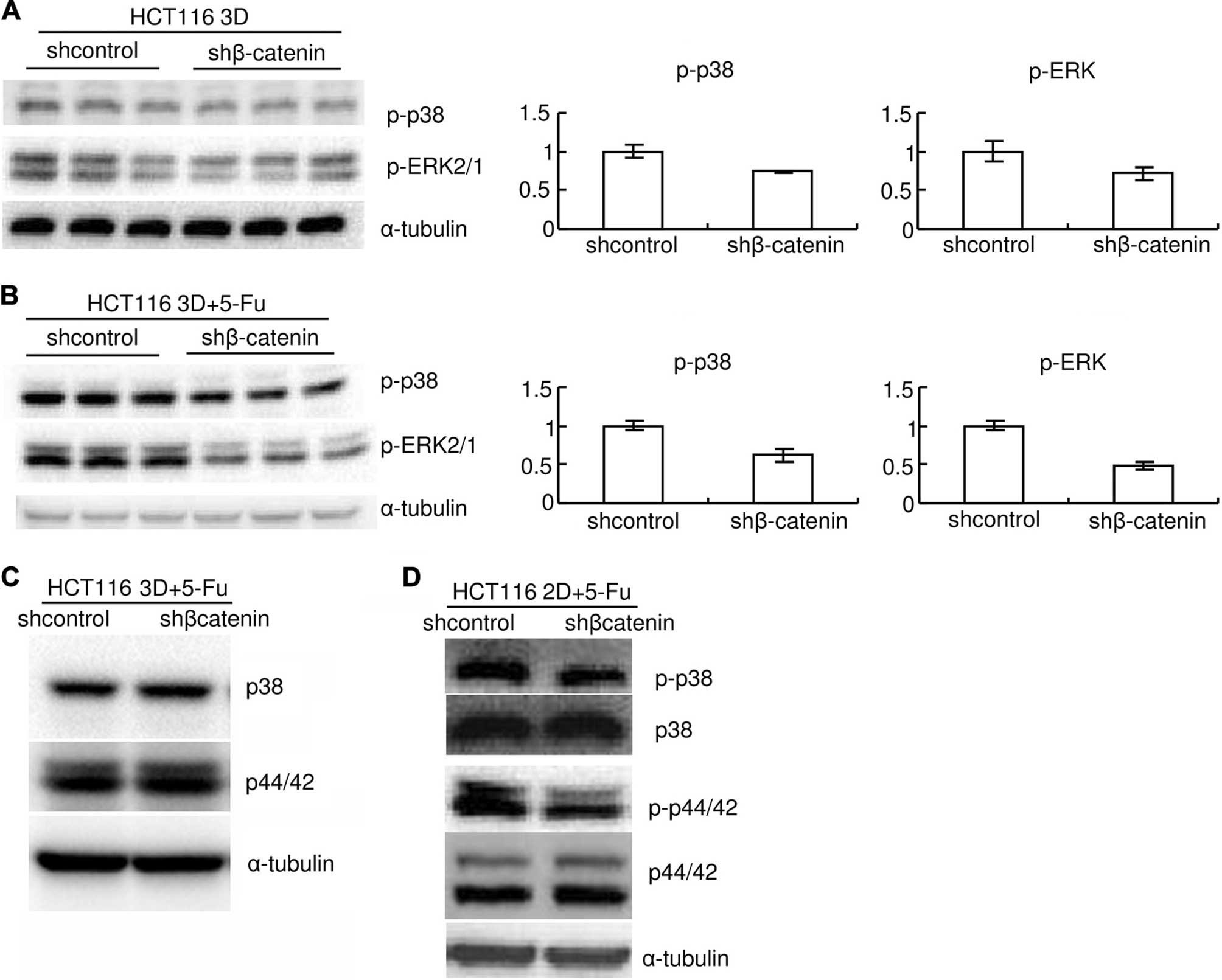
6F). Knockdown of β-catenin also mildly decreased basal levels
of p-p38 and p-ERK1/2 (Fig. 7A).
Knockdown of β-catenin decreased p-p38 and p-ERK1/2 induced by 80
mg/l 5-FU 8-h treatment both in 3D cultures and in 2D cultures
(Fig. 7B and D). Total p38 or
ERK1/2 protein level was not detectably changed by β-catenin
knockdown (Fig. 7C and D). The
results together with the above indicate that E-cadherin knockdown
increases β-catenin only in 3D cultures but β-catenin enhances
p-p38 and p-ERK1/2 both in 3D cultures and in 2D cultures.
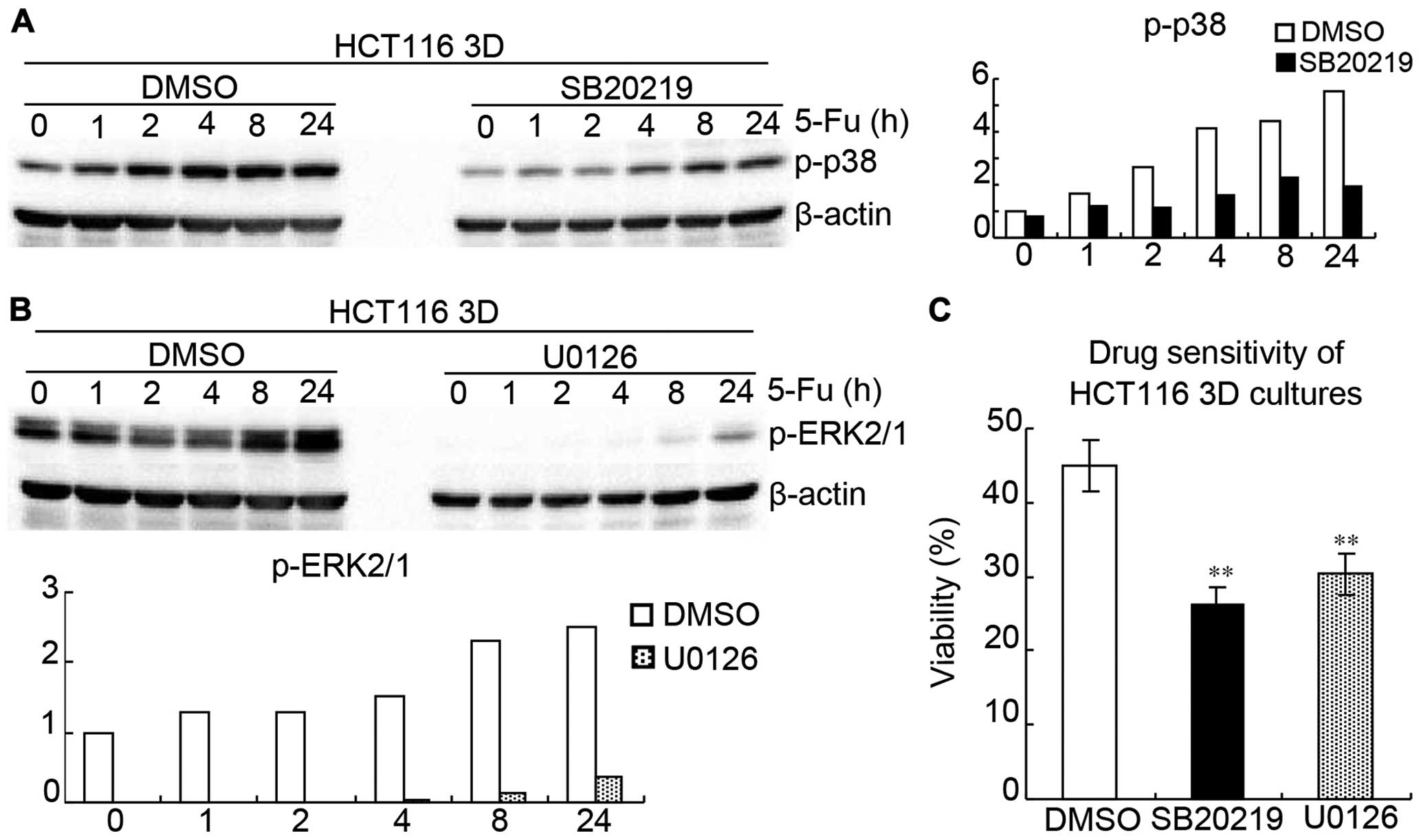
SB202190 (p38 inhibitor) (30) and U0126 (ERK1/2 inhibitor)
(31) were used to inhibit
activations of p38 and ERK1/2 in HCT116 3D cultures, respectively.
HCT116 3D cultures were treated with 20 μM SB202190, 20 μM U0126 or
DMSO 1 h following treatment of 80 mg/l 5-FU for different time
periods, respectively. Western blotting showed that SB202190
remarkably inhibited p-p38 and U0126 remarkably inhibited p-ERK1/2
(Fig. 8A and B). WST showed that
inhibition of p38 or ERK1/2 activation significantly increased
HCT116 3D cultures chemosensitivity to 5-FU, respectively (Fig. 8C). HCT116 3D cultures were treated
with 20 μM SB202190, 20 μM U0126 or DMSO 1 h following treatment of
80 mg/l 5-FU for 24 h, respectively. The viabilities of DMSO,
SB202190 and U0126 were 44.1±3.5, 26.6±2.3 (vs DMSO: p<0.01),
and 30.0±2.6% (vs DMSO: p<0.01), thus suggesting that MAPK
pathway is involved in E-cadherin knockdown increasing β-catenin to
reduce chemosensitivity.
Discussion
Colorectal cancer is one of the most lethal diseases
of all malignancies world-wide (32,33).
The incidence of colorectal cancer in developing countries is
increasing, partly attributing to lipid metabolism (19,32,33).
More than 35% of the patients die within 5 years after diagnosis
even in developed countries (32,33).
This discouraging fact is largely due to the ability of a malignant
tumor to demonstrate resistance to chemotherapies new, and old
(4,5,25).
Accumulating evidence indicates that microenvironment influences
tumor cells biological behavior, including chemosensitivity
(4,5). Among the myriad of microenvironmental
factors impacting on cancer cell chemosensitivity, cell-cell
adhesion has recently been identified as key determinant (1–2,4–6,25).
In 3D cultures, cells adhered to each other within layers of cells.
All types of cell-cell junctions, including tight junctions, gap
junctions, intermediate junctions and desmosomes, were commonly
found (Fig. 1). Their structures
are very similar to tumors at avascular stage or avascular tumor
regions (Fig. 1) (1,3,10,17),
indicating 3D cultures have the potential to bridge the gap between
monolayer cultures and xenografts for deciphering the function of
cell-cell adhesive systems (1,3,10,17).
E-cadherin plays a key role in mediating cell-cell
adhesive systems (2,6,9,10).
It was reported that inhibition of E-cadherin function in cell-cell
adhesion only by E-cadherin neutralizing antibody (SHE78-7)
disrupted preformed 3D cultures (6,13).
However, E-cadherin knockdown did not prevent suspension of HCT116
from 3D culture formation. E-cadherin protein in LoVo cells was too
low to be detected using western blotting while LoVo cells formed
3D cultures (Fig. 2). It was also
reported that prostate cancer cell line PC-3 with E-cadherin
epigenetically silenced, could form 3D cultures (13,34).
Therefore, E-cadherin may not be essential for architecture
formation of 3D cultures in vitro as tumors in
vivo.
Decreased E-cadherin expression correlates with poor
prognosis in patients with colorectal cancer (15). Intercellular force and homophilic
binding of cadherin on adjacent cells trigger E-cadherin to
interact with certain proteins, for example β-catenin, to activate
signaling cascades (9,10,12,22).
Since 3D cultures better reflect cell-cell adhesion and
chemosensitivity in vivo tumors (Fig. 1) (1,2,4–8,13,17)
the role of E-cadherin in colorectal cancer chemosensitivity was
explored in 3D cultures. E-cadherin knockdown significantly
decreased chemosensitivity to anticancer drugs in 3D cultures but
did not significantly change chemosensitivity in 2D cultures
(Fig. 3).
Under chemotherapy, the molecular mechanisms
deciding whether a tumor cell commits to cell death or survives are
complex. β-catenin plays a critical role in survival and is
involved in adhesion system (10,22,23)
E-cadherin cytoplasmic domain contains a catenin-binding domain
(10,22). E-cadherin knockdown increased
β-catenin in 3D cultures and β-catenin knockdown significantly
enhanced chemosensitivity (Figs.
3Figure 4–5). In 2D cultures, E-cadherin knockdown
did not detectably change β-catenin (Fig. 4), suggesting E-cadherin knockdown
increases β-catenin to decrease chemosensitivity only in 3D
cultures.
Mounting evidence indicates a critical role of
apoptotic pathways in determining the response of human cancers to
anticancer drugs, including 5-FU (3,25,35,36).
Caspase-3 plays a key role in apoptosis induced by 5-FU and cleaved
caspase-3 is considered as an apoptotic marker (35). E-cadherin knockdown significantly
decreased cleaved caspase-3 induced by 5-FU and β-catenin knockdown
significantly increased cleaved caspase-3 (Figs. 3C and 5D). Thus, suggesting apoptosis is
involved in the mechanism of E-cadherin knockdown increasing
β-catenin to reduce chemosensitivity.
Among the many cell signaling transduction pathways
regulating apoptosis, the MAPK signaling pathway plays an important
role in the sensitivity to anticancer therapies (27,28,30,31,37)
Also, formation of E-cadherin-mediated cell-cell adhesion regulates
MAPKs (29). However, the role of
MAPKs in cancer is as pleiotropic as cancer itself (27–31,38).
In this study, p38, ERK1/2 and JNK1/2 were dramatically activated
by chemotherapy (Fig. 6A and B).
E-cadherin knockdown enhanced chemotherapy-induced p-p38 and
p-ERK1/2, except p-JNK1/2 only in 3D cultures (Fig. 6). Knockdown of β-catenin attenuated
chemotherapy-induced p-p38 and p-ERK1/2 both in 3D cultures and in
2D cultures (Fig. 7). Treatment of
3D cultures with SB202190 to inhibit p38 activation or U0126 to
inhibit ERK1/2 activation significantly increased chemosensitivity,
respectively (Fig. 7). The above
suggests E-cadherin knockdown increases β-catenin to reduce
chemosensitivity only in 3D cultures and β-catenin increasing
p-p38/p-ERK1/2 is involved in the mechanism though the
β-catenin-MAPK pathway is not unique in 3D cultures.
In conclusion, 3D cultures consist of layers of
cells, preserving cell-cell adhesive systems. These allow a good
model to decipher the function of cell-cell adhesive systems in
cancer. Intercellular adhesion triggers certain E-cadherin
signaling cascades (9,10,12).
Data in this study indicate that E-cadherin knockdown significantly
increases β-catenin resulting in decrease of chemosensitivity in 3D
cultures but this effect was not detected in 2D cultures. β-catenin
enhancing the p-p38/p-ERK1/2 is involved in this mechanism, though
the β-catenin-MAPK pathway is not unique in 3D cultures.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81000990), the Natural
Science Foundation Project of CQ CSTC (grant no. 2009BB5339) and
the Science Foundation of Third Military Medical University for the
Young Scholar (grant no. 2009XQN32).
Abbreviations:
|
3D
|
three-dimensional
|
|
2D
|
two-dimensional
|
|
Ecad
|
E-cadherin
|
|
sh
|
small hairpin
|
|
H&E
|
hematoxylin and eosin
|
|
DAPI
|
4′,6-diamidino-2-phenylindole
|
|
MAPK
|
mitogen-activated protein kinase
|
|
WST assay
|
tetrazolium salt-based proliferation
assay
|
|
5-FU
|
5-fluorouracil
|
|
CPT-11
|
irinotecan
|
|
ERK
|
extracellular-signal-regulated
kinase
|
|
JNK
|
c-Jun N-terminal kinase
|
|
shEcad
|
knockdown E-cadherin by shRNA
|
|
shβ-catenin
|
knockdown β-catenin by shRNA
|
|
shcontrol
|
control shRNA targeted against green
fluorescent protein
|
|
d
|
day
|
|
h
|
hour
|
References
|
1
|
Shamir ER and Ewald AJ: Three-dimensional
organotypic culture: Experimental models of mammalian biology and
disease. Nat Rev Mol Cell Biol. 15:647–664. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kang HG, Jenabi JM, Zhang J, Keshelava N,
Shimada H, May WA, Ng T, Reynolds CP, Triche TJ and Sorensen PH:
E-cadherin cell-cell adhesion in ewing tumor cells mediates
suppression of anoikis through activation of the ErbB4 tyrosine
kinase. Cancer Res. 67:3094–3105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Desoize B and Jardillier J: Multicellular
resistance: A paradigm for clinical resistance? Crit Rev Oncol
Hematol. 36:193–207. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
He JM, Wang FC, Qi HB, Li Y and Liang HJ:
Down-regulation of alphav integrin by retroviral delivery of small
interfering RNA reduces multicellular resistance of HT29. Cancer
Lett. 284:182–188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kobayashi H, Man S, Graham CH, Kapitain
SJ, Teicher BA and Kerbel RS: Acquired multicellular-mediated
resistance to alkylating agents in cancer. Proc Natl Acad Sci USA.
90:3294–3298. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Green SK, Karlsson MC, Ravetch JV and
Kerbel RS: Disruption of cell-cell adhesion enhances
antibody-dependent cellular cytotoxicity: Implications for
antibody-based therapeutics of cancer. Cancer Res. 62:6891–6900.
2002.PubMed/NCBI
|
|
7
|
Yang Z and Zhao X: A 3D model of ovarian
cancer cell lines on peptide nanofiber scaffold to explore the
cell-scaffold interaction and chemotherapeutic resistance of
anticancer drugs. Int J Nanomedicine. 6:303–310. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee JM, Mhawech-Fauceglia P, Lee N,
Parsanian LC, Lin YG, Gayther SA and Lawrenson K: A
three-dimensional microenvironment alters protein expression and
chemosensitivity of epithelial ovarian cancer cells in vitro. Lab
Invest. 93:528–542. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Engl W, Arasi B, Yap LL, Thiery JP and
Viasnoff V: Actin dynamics modulate mechanosensitive immobilization
of E-cadherin at adherens junctions. Nat Cell Biol. 16:587–594.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gall TM and Frampton AE: Gene of the
month: E-cadherin (CDH1). J Clin Pathol. 66:928–932. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mattias L, Haque A, Adnan N and Akaike T:
The effects of artificial E-cadherin matrix-induced embryonic stem
cell scattering on paxillin and RhoA activation via α-catenin.
Biomaterials. 35:1797–1806. 2014. View Article : Google Scholar
|
|
12
|
Truffi M, Dubreuil V, Liang X, Vacaresse
N, Nigon F, Han SP, Yap AS, Gomez GA and Sap J: RPTPα controls
epithelial adherens junctions, linking E-cadherin engagement to
c-Src-mediated phosphorylation of cortactin. J Cell Sci.
127:2420–2432. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nakamura T, Kato Y, Fuji H, Horiuchi T,
Chiba Y and Tanaka K: E-cadherin-dependent intercellular adhesion
enhances chemoresistance. Int J Mol Med. 12:693–700.
2003.PubMed/NCBI
|
|
14
|
Govatati S, Singamsetty GK, Nallabelli N,
et al: Contribution of cyclin D1 (CCND1) and E-cadherin (CDH1)
alterations to colorectal cancer susceptibility: A case-control
study. Tumour Biol. 35:12059–12067. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yun JA, Kim SH, Hong HK, Yun SH, Kim HC,
Chun HK, Cho YB and Lee WY: Loss of E-Cadherin expression is
associated with a poor prognosis in stage III colorectal cancer.
Oncology. 86:318–328. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li Y, He J, Zhong D, Li J and Liang H:
High-mobility group box 1 protein activating nuclear factor-κB to
upregulate vascular endothelial growth factor C is involved in
lymphangiogenesis and lymphatic node metastasis in colon cancer. J
Int Med Res. 43:494–505. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Phung YT, Barbone D, Broaddus VC and Ho M:
Rapid generation of in vitro multicellular spheroids for the study
of monoclonal antibody therapy. J Cancer. 2:507–514. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Onder TT, Gupta PB, Mani SA, Yang J,
Lander ES and Weinberg RA: Loss of E-cadherin promotes metastasis
via multiple downstream transcriptional pathways. Cancer Res.
68:3645–3654. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He J, Shin H, Wei X, Kadegowda AK, Chen R
and Xie SK: NPC1L1 knockout protects against colitis-associated
tumorigenesis in mice. BMC Cancer. 15:1892015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Franken NA, Rodermond HM, Stap J, Haveman
J and van Bree C: Clonogenic assay of cells in vitro. Nat Protoc.
1:2315–2319. 2006. View Article : Google Scholar
|
|
21
|
Tanaka E, Hashimoto Y, Ito T, Kondo K,
Higashiyama M, Tsunoda S, Ortiz C, Sakai Y, Inazawa J and Shimada
Y: The suppression of aurora-A/STK15/BTAK expression enhances
chemosensitivity to docetaxel in human esophageal squamous cell
carcinoma. Clin Cancer Res. 13:1331–1340. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee DJ, Kang DH, Choi M, et al:
Peroxiredoxin-2 represses melanoma metastasis by increasing
E-cadherin/β-catenin complexes in adherens junctions. Cancer Res.
73:4744–4757. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu Y, Ye X, Zhang JB, et al: PROX1
promotes hepatocellular carcinoma proliferation and sorafenib
resistance by enhancing β-catenin expression and nuclear
translocation. Oncogene. Feb 16–2015.(Epub ahead of print).
View Article : Google Scholar
|
|
24
|
Koti M, Gooding RJ, Nuin P, Haslehurst A,
Crane C, Weberpals J, Childs T, Bryson P, Dharsee M, Evans K, et
al: Identification of the IGF1/PI3K/NF κB/ERK gene signalling
networks associated with chemotherapy resistance and treatment
response in high-grade serous epithelial ovarian cancer. BMC
Cancer. 13:5492013. View Article : Google Scholar
|
|
25
|
Liu S, Wang J, Niu W, Liu E, Wang J, Peng
C, Lin P, Wang B, Khan AQ, Gao H, et al: The β6-integrin-ERK/MAP
kinase pathway contributes to chemo resistance in colon cancer.
Cancer Lett. 328:325–334. 2013. View Article : Google Scholar
|
|
26
|
Basile KJ, Abel EV, Dadpey N, Hartsough
EJ, Fortina P and Aplin AE: In vivo MAPK reporting reveals the
heterogeneity in tumoral selection of resistance to RAF inhibitors.
Cancer Res. 73:7101–7110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: p38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar
|
|
28
|
Pritchard AL and Hayward NK: Molecular
pathways: Mitogen-activated protein kinase pathway mutations and
drug resistance. Clin Cancer Res. 19:2301–2309. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Reddy P, Liu L, Ren C, Lindgren P, Boman
K, Shen Y, Lundin E, Ottander U, Rytinki M and Liu K: Formation of
E-cadherin-mediated cell-cell adhesion activates AKT and mitogen
activated protein kinase via phosphatidylinositol 3 kinase and
ligand-independent activation of epidermal growth factor receptor
in ovarian cancer cells. Mol Endocrinol. 19:2564–2578. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tsai MS, Weng SH, Chen HJ, Chiu YF, Huang
YC, Tseng SC, Kuo YH and Lin YW: Inhibition of p38 MAPK-dependent
excision repair cross-complementing 1 expression decreases the DNA
repair capacity to sensitize lung cancer cells to etoposide. Mol
Cancer Ther. 11:561–571. 2012. View Article : Google Scholar
|
|
31
|
Csibi A, Fendt SM, Li C, Poulogiannis G,
Choo AY, Chapski DJ, Jeong SM, Dempsey JM, Parkhitko A, Morrison T,
et al: The mTORC1 pathway stimulates glutamine metabolism and cell
proliferation by repressing SIRT4. Cell. 153:840–854. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Center MM, Jemal A, Smith RA and Ward E:
Worldwide variations in colorectal cancer. CA Cancer J Clin.
59:366–378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Härmä V, Knuuttila M, Virtanen J, Mirtti
T, Kohonen P, Kovanen P, Happonen A, Kaewphan S, Ahonen I,
Kallioniemi O, et al: Lysophosphatidic acid and
sphingosine-1-phosphate promote morphogenesis and block invasion of
prostate cancer cells in three-dimensional organotypic models.
Oncogene. 31:2075–2089. 2012. View Article : Google Scholar :
|
|
35
|
Wu XX, Kakehi Y, Mizutani Y, Lu J, Terachi
T and Ogawa O: Activation of caspase-3 in renal cell carcinoma
cells by anthracyclines or 5-fluorouracil. Int J Oncol. 19:19–24.
2001.PubMed/NCBI
|
|
36
|
Samuel T, Fadlalla K, Gales DN, Putcha BD
and Manne U: Variable NF-κB pathway responses in colon cancer cells
treated with chemotherapeutic drugs. BMC Cancer. 14:5992014.
View Article : Google Scholar
|
|
37
|
Shi X, Wu S, Yang Y, Tang L, Wang Y, Dong
J, Lü B, Jiang G and Zhao W: AQP5 silencing suppresses p38 MAPK
signaling and improves drug resistance in colon cancer cells.
Tumour Biol. 35:7035–7045. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jeong S, Jing K, Kim N, Shin S, Kim S,
Song KS, Heo JY, Park JH, Seo KS, Han J, et al: Docosahexaenoic
acid-induced apoptosis is mediated by activation of
mitogen-activated protein kinases in human cancer cells. BMC
Cancer. 14:4812014. View Article : Google Scholar : PubMed/NCBI
|