Introduction
Ovarian cancer is often diagnosed at an advanced
stage (III or IV) and is typically treated with a combination of
cyto-reductive surgery and platinum-based chemotherapy (1–3).
Platinum-based cisplatin is a first-line therapeutic agent used in
the treatment of multiple cancers, including ovarian cancer.
Cisplatin acts by inducing DNA damage, triggering cell cycle
arrest, and initiating apoptosis (4). However, many ovarian cancer patients
develop acquired cisplatin resistance, resulting in failure of
chemotherapy (5). Several recent
studies demonstrate that adaptive responses, such as endoplasmic
reticulum (ER) stress and autophagy, can promote cisplatin
resistance in cancer cells (6,7).
Autophagy is characterized by sequestration of
cytoplasmic components within autophagosomes. These autophagosomes
subsequently fuse with lysosomes to form autolysosomes in which
cytoplasmic materials including organelles are degraded (8–10).
Many chemotherapeutic agents, including cisplatin, can induce
autophagy (8,9). This induction of autophagy can have
either pro-survival or pro-death effects to the anticancer drugs
(11). Autophagy is induced under
many physiological and pathological conditions, and it often
promotes survival of cancer cells (5). For example, treatment of HeLa cells
with ammonium chloride or chloroquine (CQ) to inhibit autophagy
disrupts autolysosome function and enhances cellular sensitivity to
cisplatin (12,13). Consistently, our previous results
demonstrate that p62/SQSTM1 maintains autophagic flux and
alleviates ER stress by clearing accumulation of ubiquitinated
proteins, thereby promoting cisplatin resistance in human ovarian
cancer cells (13,14). Autophagy is a lysosomal-dependent
process in which lysosomes are consumed over time. Therefore,
normal lysosomal numbers and function are required for maintaining
autophagic flux (15). Examining
lysosomal maintenance may further our understanding of the
mechanisms underlying autophagy-mediated cisplatin resistance.
Lysosomes can degrade major cellular macromolecules
and damaged organelles (16,17).
Within the lysosome, members of the lysosome-associated membrane
protein (LAMP) family contribute to a glycocalyx that protects the
structural integrity of lysosomal membranes from lysosomal
hydrolases (18–20). Both LAMP1 and LAMP2 are required
for successful autolysosome fusion, as LAMP1 or LAMP2 defects lead
to accumulation of autophagic vacuoles and inhibits autophagy
(21). Furthermore, the acidic
environment within the lysosome (pH 4–5) is a necessary condition
for lysosomal protease activation (22). Lysosomal acidic environment and
protease are both required for normal lysosomal function. This
acidic environment enables indirect evaluation of lysosomal
function using stains such as LysoTracker. Moreover, when lysosomal
function is activated in the course of autophagy, LysoTracker
staining is increased (23).
Investigation of the mechanisms involved in regulating lysosomal
function may also aid in understanding autophagy-mediated cisplatin
resistance.
Lysosomes are dynamic organelles within living
cells, and have both pro-survival and pro-death roles during
chemotherapeutic treatment of cancer cells. Lysosomal sequestration
of chemotherapeutic agents not only prevents drugs from reaching
their intracellular targets, but also induces lysosomal biogenesis
by triggering translocation of transcription factor EB from the
cytoplasm to the nucleus (24).
Therefore, the lysosome can determine whether a tumor cell exhibits
chemotherapeutic resistance.
The pH gradient between the lysosomal lumen and the
cytoplasm has been recently found to determine the extent of
lysosomal sequestration of chemotherapeutic agents (25). V-ATPase is a protein complex which
regulates lysosomal luminal acidity (26). ATP-sensitive sodium and potassium
channels maintain the low pH of the lysosomal lumen, consuming ATP
in the process (27). However,
lysosomes contain abundant ATP, which is sufficient for maintaining
normal lysosomal physiology (28)
and enabling protease activation (29). Accumulation of lysosomal ATP can be
impaired, leading to lysosomal cathepsin D activity inhibition,
formation of lipofuscin aggregates, and even cell death (28). Lysosomal hydrolases can also move
into the cytoplasm and retain transient activity, enabling them to
trigger mitochondria-lysosome crosstalk to promote cell death
(30,31). Precisely how the abundance of
lysosomal ATP regulates lysosome function and homeostasis remains
unclear, and requires further investigation.
We investigated cisplatin-resistance in SKOV3/DDP
cells and examined lysosomal involvement in cisplatin-induced
autophagy. Cisplatin induced a more obvious autophagic response in
cisplatin-resistant SKOV3/DDP cells than in cisplatin-resistance
SKOV3/DDP cells. Moreover, inhibiting autophagy by disturbing
autophagosome-lysosome fusion sensitized cisplatin-resistant
SKOV3/DDP cells to cisplatin. SKOV3/DDP cells possess abundant
lysosomes and cathepsin D, which facilitated clearance of
accumulated materials via autophagy. Furthermore, SKOV3/DDP
lysosomes contain abundant ATP. We inhibited lysosomal ATP
accumulation and observed impaired lysosomal function. These
findings suggest an important role for lysosomal ATP levels in
promoting resistance to chemotherapeutic agents.
Materials and methods
Cell culture
Cisplatin-sensitive ovarian carcinoma SKOV3 cells
and their cisplatin-resistant clone SKOV3/DDP were obtained from
the Chinese Academy of Medical Sciences and Peking Union Medical
College. Both cell lines were maintained at 37°C in a 5%
CO2 and 95% air atmosphere in Roswell Park Memorial
Institute (RPMI)-1640 culture medium (Gibco Life Technologies,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum
(Invitrogen, Carlsbad, CA, USA), 100 U/ml penicillin, and 100 U/ml
streptomycin. Cisplatin-resistant SKOV3/DDP cells were cultured in
the presence of 1 μg/ml cisplatin to maintain
resistance.
Cell viability assay
The MTT assay was used to determine cell viability.
Cells were seeded in 96-well plates at a density of
1×104 cells/well. Each condition was mirrored in six
wells. The following day, cells were treated with different
concentrations of cisplatin and incubated for 24 h. Next, 20
μl MTT reagent [5 mg/ml in phosphate-buffered saline (PBS);
Sigma-Aldrich, St. Louis, MO, USA] was added to each well and
incubated for 4 h. Lastly, 150 μl dimethylsulfoxide was
added to each well and absorbance at 570 nm measured using a Vmax
Microplate Reader (Molecular Devices, LLC, Sunnyvale, CA, USA).
Immunofluorescence staining and confocal
laser microscopy
Cells were seeded onto coverslips in 24-well plates
at (5×104 cells/well) and incubated overnight. Apoptotic
nuclear changes were detected using Hoechst 33258 (Sigma-Aldrich)
staining. Following cisplatin treatment, cells were washed with
cold PBS three times, fixed in 4% (w/v) paraformaldehyde/PBS for 20
min, and then washed with cold PBS three times. After fixation,
cells were subjected to proteinase K digestion for 1 min, washed
twice with PBS, permeabilized with 0.1% Triton X-100 for 5 min,
washed with cold PBS three times, and then blocked with bovine
serum albumen for 30 min. Cells were incubated with primary
antibody (LC3 or LAMP1 - all at 1:100 dilution) overnight at 4°C,
washed three times with PBS, and stained with FITC/Texas
Red-conjugated secondary antibodies (1:400 dilution; all
antibodies, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA)
for 30 min in the dark. Cells were treated with Hoechst
33342/H2O (1 μg/ml) for 2 min, and then washed
three times with PBS. Images were acquired by an Olympus FV1000
confocal laser microscope.
ATP staining
Cells were cultured onto coverslips as described
above and treated with combinations of
4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS;
Sigma-Aldrich) and cisplatin. Cells were then incubated with 10
μM quinacrine (Merck Millipore, Darmstadt, Germany) together
with 50 nM LysoTracker Red DND-99 (Invitrogen) for 30 min at 37°C.
Quinacrine binds ATP, and its fluorescence signal intensity
indicates ATP levels (32).
Following staining, cells were washed three times with PBS and
images were acquired using an Olympus FV1000 confocal laser
microscope.
Measurement of cathepsin D activity
Cathepsin D activity in cell lysates was detected
using a fluorometric cathepsin D activity assay kit (Biovision, San
Francisco, CA, USA) according to the manufacturer's instructions.
Fluorescence intensity at an excitation/emission of 328/460 nm was
measured using a Packard Bioscience Fusion™ instrument. Three
independent measurements were made for each sample.
Western blot analysis
Cells subjected to desired treatments were
harvested, washed twice with cold PBS, and then gently scraped into
120 μl of RIPA buffer. Cell lysates were sonicated for 30
sec on ice and then lysed at 4°C for 45 min. Cell lysates were
centrifuged at 3,000 × g for 15 min, and supernatant protein
concentrations were determined using the Bio-Rad kit (Pierce
Biotechnology, Inc., Rockford, IL, USA). For western blot analysis,
equivalent amounts of lysate proteins (30–50 μg) were
separated by 12% w/v SDS-polyacrylamide gel electrophoresis and
transferred onto nitrocellulose transfer membranes (Millipore
Corp., Bedford, MA, USA). Membranes were blocked with 5% (w/v) skim
milk in buffer [PBST: 10 mM Tris-HCl (pH 7.6), 100 mM NaCl, and
0.1% (v/v) Tween-20] for 1 h at room temperature, and incubated
with primary antibodies (Santa Cruz Biotechnology, Inc.) overnight
at 4°C. The following day, membranes were washed with PBST and
incubated with horseradish peroxidase-conjugated secondary
antibodies (Thermo Fisher Scientific, Waltham, MA, USA) at 1:2,000
dilution for 1 h at room temperature. After washing the membranes
with PBST, immunodetection was performed using ECL reagent (Thermo
Fisher Scientific) and visualized using a Syngene Bio Imaging
(Synoptics, Cambridge, UK). Protein levels were quantified by
densitometry using Quantity One software (Bio-Rad Laboratories,
Inc., Hercules, CA, USA).
Flow cytometry
Cells were trypsinized following desired treatments
and incubated with Muse™ Annexin V and Dead Cell reagent for 20 min
at room temperature. Apoptotic cells were then analyzed with a
Muse™ Cell Analyzer. All experiments reported in this study were
repeated three times.
Statistics
Results are representative of three independent
experiments performed in triplicate and presented as means ± SD.
One-way ANOVA was used to perform statistical analysis of the data.
P<0.05 was regarded as statistically significant.
Results
Autophagic activity and lysosome numbers
are higher in cisplatin-resistant SKOV3/DDP cells than in
cisplatin-sensitive SKOV3 cells
SKOV3/DDP cells underwent less apoptosis and exhibit
increased cisplatin resistance compared with SKOV3 cells following
treatment with cisplatin for 24 h (Fig. A–C).
 | Figure 1Cisplatin-sensitive SKOV3 and
cisplatin-resistant SKOV3/DDP cells exhibit differences in
autophagy and lysosome function. (A) SKOV3 and SKOV3/DDP cells were
treated with indicated concentrations of cisplatin for 24 h. Cell
viability was evaluated by MTT assay. Data are presented as mean ±
SD, n=3. (B and D) Western blot analysis of levels of caspase-3,
cleaved caspase-3, LC3II, LC3I, and p62 protein in SKOV3 and
SKOV3/DDP cells treated with 6 μg/ml cisplatin. (C and E)
Quantitation of cleaved caspase-3, LC3II/LC3I, and p62 protein
levels. Data are presented as mean ± SD, n=3,
**P<0.01. (F) Western blot analysis of LAMP1 levels
in SKOV3 and SKOV3/DDP cells treated with 6 μg/ml cisplatin.
(G) Quantitation of LAMP1 protein. Data are presented as mean ± SD,
n=3. **P<0.01. (H) Western blot analysis of cathepsin
D protein levels in SKOV3 and SKOV3/DDP cells treated with 6
μg/ml cisplatin. (I) Quantitation of active cathepsin D and
total cathepsin D protein levels. Data are presented as mean ± SD,
n=3. *P<0.05 **P<0.01 vs. control. |
Next, we examined the protein expression levels of
p62, LC3I, and LC3II in both cisplatin-sensitive SKOV3 cells and
cisplatin-resistant SKOV3/DDP cells. The LC3II/LC3I ratio was
significantly increased in SKOV3/DDP cells, especially at 12 h,
while p62 protein levels in SKOV3/DDP cells decreased gradually
(Fig. 1D and E).
Autophagy requires lysosomal degradation of
cytoplasmic materials. Therefore, we next investigated the change
of lysosomes in SKOV3 and SKOV3/DDP cells following exposure to
cisplatin. Compared with cisplatin-sensitive SKOV3 cells,
cisplatin-resistant SKOV3/DDP cells expressed more LAMP1 (Fig. 1F and G). Lysosomal proteases are
required for macromolecule degradation in lysosomes, and cathepsin
D is an important aspartic protease in the lysosome. Compared with
SKOV3 cells treated with cisplatin, SKOV3/DDP cells expressed both
a higher level of total cathepsin D, and a higher level of
activated cathespin D (Fig. 1H and
I). Taken together, these results indicate that abundant
lysosomes exist in cisplatin-resistant SKOV3/DDP cells treated with
cisplatin, and autophagy was induced in these cells. Furthermore,
lysosomes may sustain cellular metabolism and maintain cell
homeostasis through autophagy.
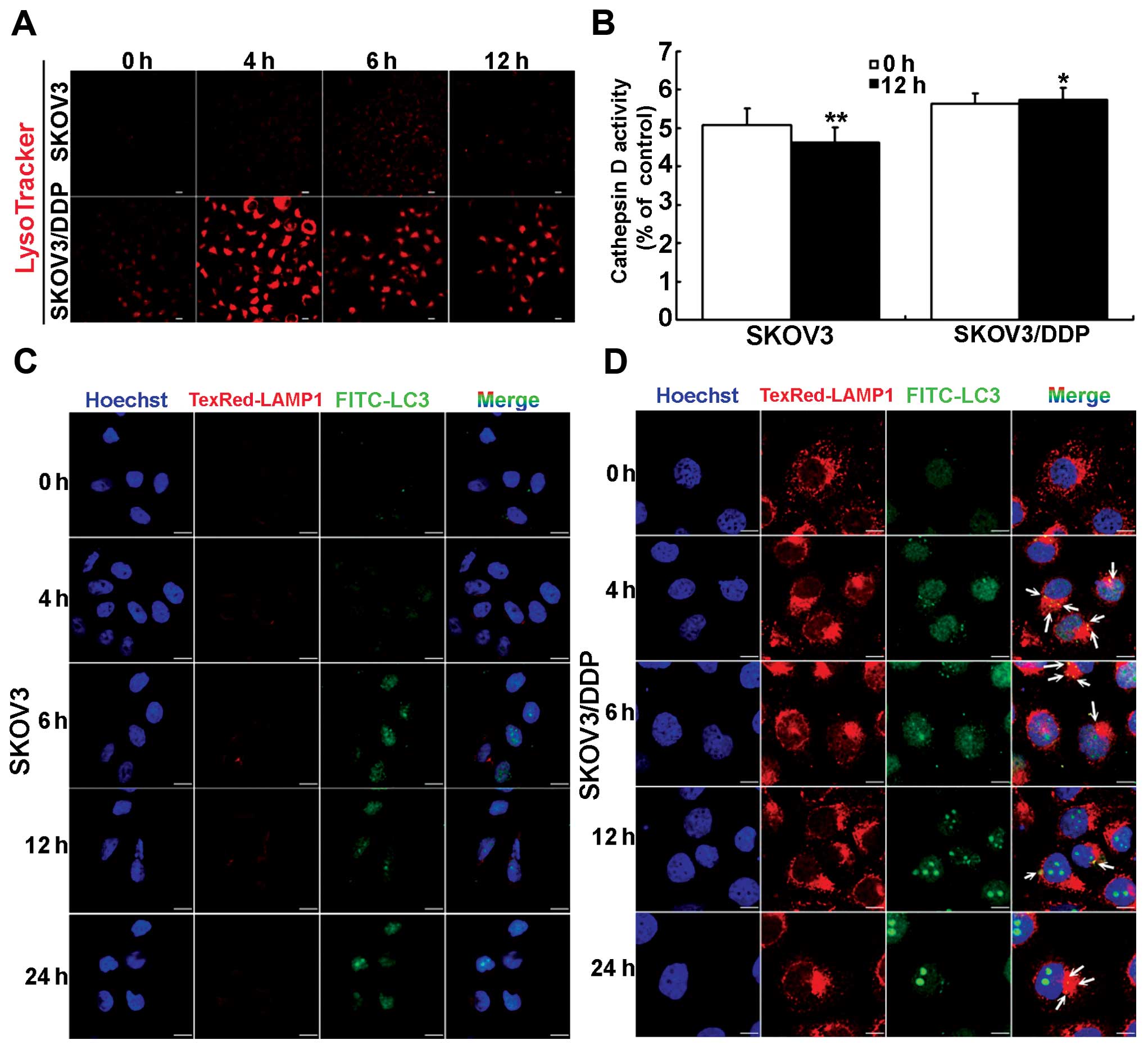
Lysosomal function increases with
cisplatin-induced autophagy in cisplatin-resistant SKOV3/DDP
cells
We next investigated changes to lysosomal activity
alongside cisplatin-induced autophagy. First, compared with SKOV3
cells, LysoTracker staining was increased in SKOV3/DDP cells
treated with cisplatin, especially at 4 h (Fig. 2A). This suggests increased
acidification of lysosomes. Second, compared with
cisplatin-sensitive SKOV3 cells, cathepsin D activity was
significantly higher under normal conditions in cisplatin-resistant
SKOV3/DDP cells. Notably, cathepsin D activity was reduced
significantly at 12 h in SKOV3 cells treated with cisplatin, but
unchanged in SKOV3/DDP cells following the same treatment (Fig. 2B). Lysosomes can be differentiated
from autolysosomes on the basis of LC3 staining: lysosomes are
LAMP1+ and LC3−, while autolysosomes are
LAMP1+ and LC3+ (33). SKOV3/DDP cells treated with
cisplatin had increased LC3 staining, and therefore more
autolysosomes, at 4 h, but decreased staining at 6, 12 and 24 h
(Fig. 2C and D). Furthermore, the
size and number of lysosomes in SKOV3/DDP cells slightly decreased
at 6 h, but was restored to an almost normal level at 12 h
following treatment with cisplatin (Fig. 2D). These results indicate that
lysosomal function is increased during cisplatin-induced autophagy
in SKOV3/DDP cells, and abundant lysosomes contribute to
autolysosome formation.
Inhibiting autophagy enhances
cisplatin-mediated cytotoxicity in cisplatin-resistant SKOV3/DDP
cells, but targeting the lysosome to disturb autolysosome formation
is more effective
We next examined whether inhibition of a specific
stage of autophagy could protect SKOV3/DDP cells from
cisplatin-induced autophagy. Cisplatin-resistant SKOV3/DDP cells
were treated with 3-methyladenine (3-MA), which inhibits autophagy
at an early stage by interfering with recruitment of the class III
PI3K Vps34, or antimalarial CQ, a late-stage autophagy inhibitor
that impairs lysosome acidification, to disrupt autolysosome
formation.
No significant toxic effect was observed following
treatment of SKOV3/DDP cells with 10 mM 3-MA or 50 μM CQ.
MTT assays revealed that neither 3-MA or CQ alone had a significant
impact on cell viability. However, combining either inhibitor with
cisplatin significantly decreased the growth of SKOV3/DDP cells,
with the CQ-cisplatin combination being slightly more effective
(Fig. 3A). Autolysosomes were
rarely observed in SKOV3/DDP cells treated with a combination of
3-MA and cisplatin (Fig. 3B).
However, SKOV3/DDP cells possessed enlarged autolysosomes following
co-treatment with CQ and cisplatin (Fig. 3B). Moreover, the number of
autolysosomes was increased following CQ-cisplatin treatment.
Inhibition of autophagy and/or cisplatin could abolish lysosomal
activity in SKOV3/DDP cells (Fig.
3C). Moreover, cathepsin D activity was significantly lowered
following treatment with CQ and/or cisplatin (Fig. 3D).
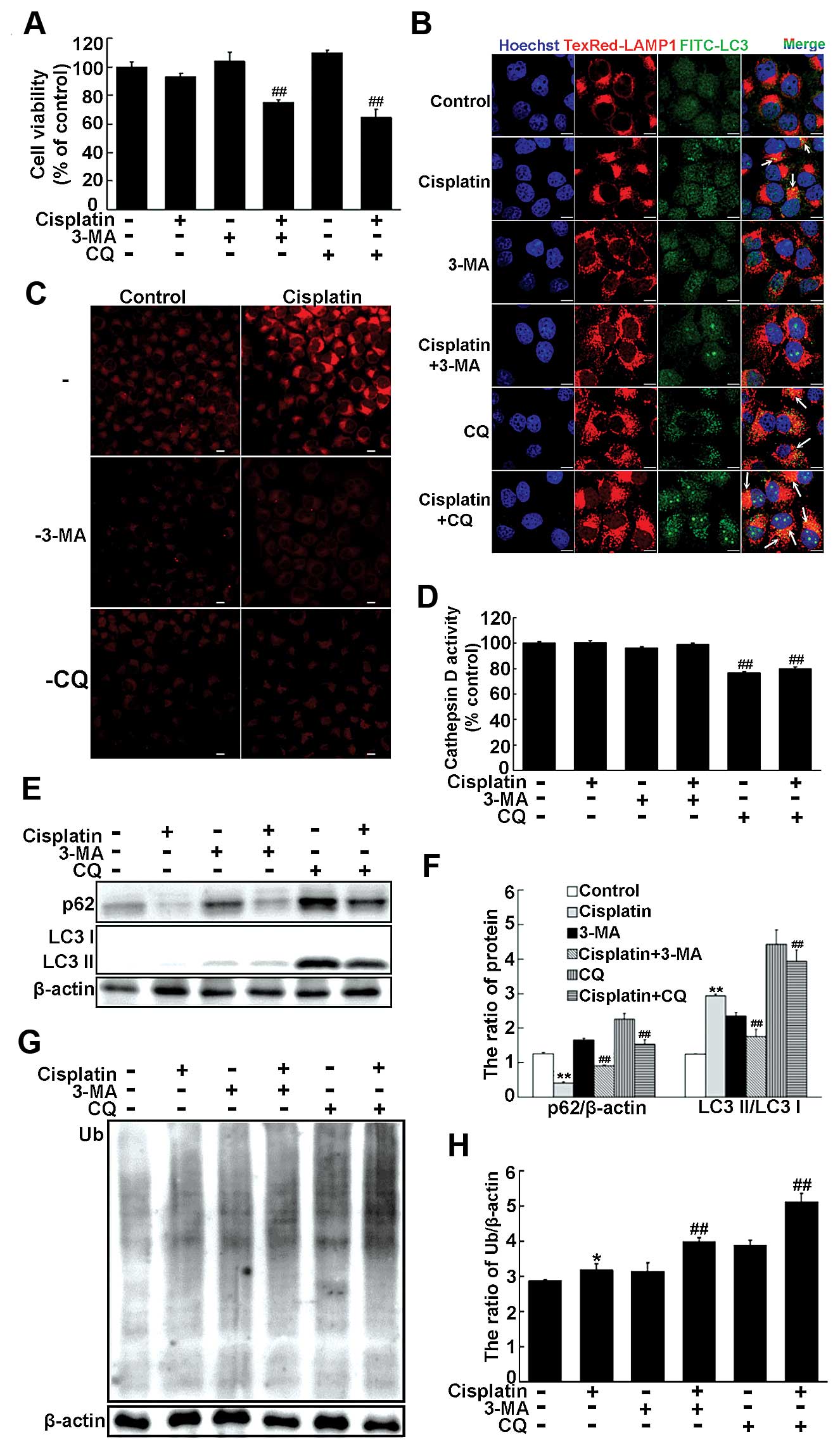 | Figure 3Inhibition of autophagy enhances
cisplatin cytotoxicity in cisplatin-resistant SKOV3/DDP cells, but
targeting the lysosome to disrupt autolysosome formation is more
effective (A) SKOV3/DDP cells were treated with cisplatin (6
μg/ml) in combination with 3-MA (10 mM) or CQ (50 μM)
for 24 h. Cell viability was determined by MTT assay. Data are
presented as mean ± SD, n=3. ##P<0.05 vs. cisplatin.
(B) Colocalization of LAMP1 and LC3 was observed by confocal
microscopy in SKOV3/DDP cells treated as indicated in panel A
(scale bar, 10 μm; arrows indicate colocalization of LAMP1
and LC3). (C) LysoTracker staining in SKOV3/DDP cells treated with
cisplatin (6 μg/ml) in combination with 3-MA (10 mM) or CQ
(50 μM) for 12 h was observed by confocal microscopy (scale
bar, 10 μm). (D) Following treatments indicated in panel C,
cells were lysed in CD cell lysis buffer and lysates analyzed using
a cathepsin D activity assay. Data are presented as mean ± SD, n=3.
##P<0.01 vs. cisplatin. (E and G) Western blot
analysis of LC3II/LC3I, p62, and ubiquitinated protein levels in
SKOV3/DDP cells treated as indicated in panel A. (F and H)
Quantitation of LC3II/LC3I, p62, and ubiquitinated protein levels.
Data are presented as mean ± SD, n=3. *P<0.05,
**P<0.01 vs. control; ##P<0.01 vs.
cisplatin. |
Compared with cells treated with cisplatin alone,
combination treatment of 3-MA and cisplatin in SKOV3/DDP cells
decreased the ratio of LC3II/LC3I and inhibited p62 degradation
(Fig. 3E and F). Additionally,
cisplatin-induced upregulation of LC3II was further augmented by
CQ-mediated inhibition of autolysosome formation. Cisplatin in
combination with CQ reduced degradation of p62 in SKOV3/DDP cells.
More ubiquitinated proteins were present in SKOV3/DDP cells treated
with a combination of cisplatin and 3-MA or CQ, with the greatest
amount found following cisplatin and CQ treatment (Fig. 3G and H). These results suggest that
inhibiting autophagy by targeting the lysosome enhances
cisplatin-induced cytotoxicity and disturbs autolysosome function
in SKOV3/DDP cells.
Inhibition of autophagy by targeting the
lysosome results in cell death through activation of
mitochondria-lysosome crosstalk in cisplatin-resistant SKOV3/DDP
cells
We next investigated apoptosis induction in
SKOV3/DDP cells treated with cisplatin in combination with
inhibition of autophagy.
Apoptotic populations of cells treated with
cisplatin and 3-MA or CQ were quantified using the Muse™ Annexin V
and Dead Cell assay. Compared with cisplatin alone, co-treatment
with cisplatin and 3-MA or CQ enhanced the rate of cell apoptosis,
with the CQ-cisplatin combination being more effective (Fig. 4A). We then examined whether the
mitochondrial apoptotic pathway was initiated by measuring
expression levels of Bax, Bcl-2 and cleaved caspase-3. Combined
treatment of SKOV3/DDP cells with cisplatin and 3-MA or CQ
increased the ratio of Bax/Bcl-2 and promoted activation of
caspase-3, with cisplatin-CQ being more effective (Fig. 4B and C). Because the lysosome
targeting effectively induced SKOV3/DDP cell apoptosis, we next
investigated whether lysosomal cell death occurred. The ratio of
tBid/Bid in SKOV3/DDP cells following treatment with CQ and
cisplatin was higher than that following treatment with 3-MA and
cisplatin (Fig. 4D and E). Cells
treated with combination CQ and cisplatin accumulated more
cathepsin D than cells treated with either cisplatin alone or a
combination of 3-MA and cisplatin (Fig. 4D and E). Therefore, while
inhibition of autophagy overcame cisplatin resistance in SKOV3/DDP
cells, targeting the lysosome could also enhance cisplatin-induced
cell death by triggering mitochondria-lysosome crosstalk.
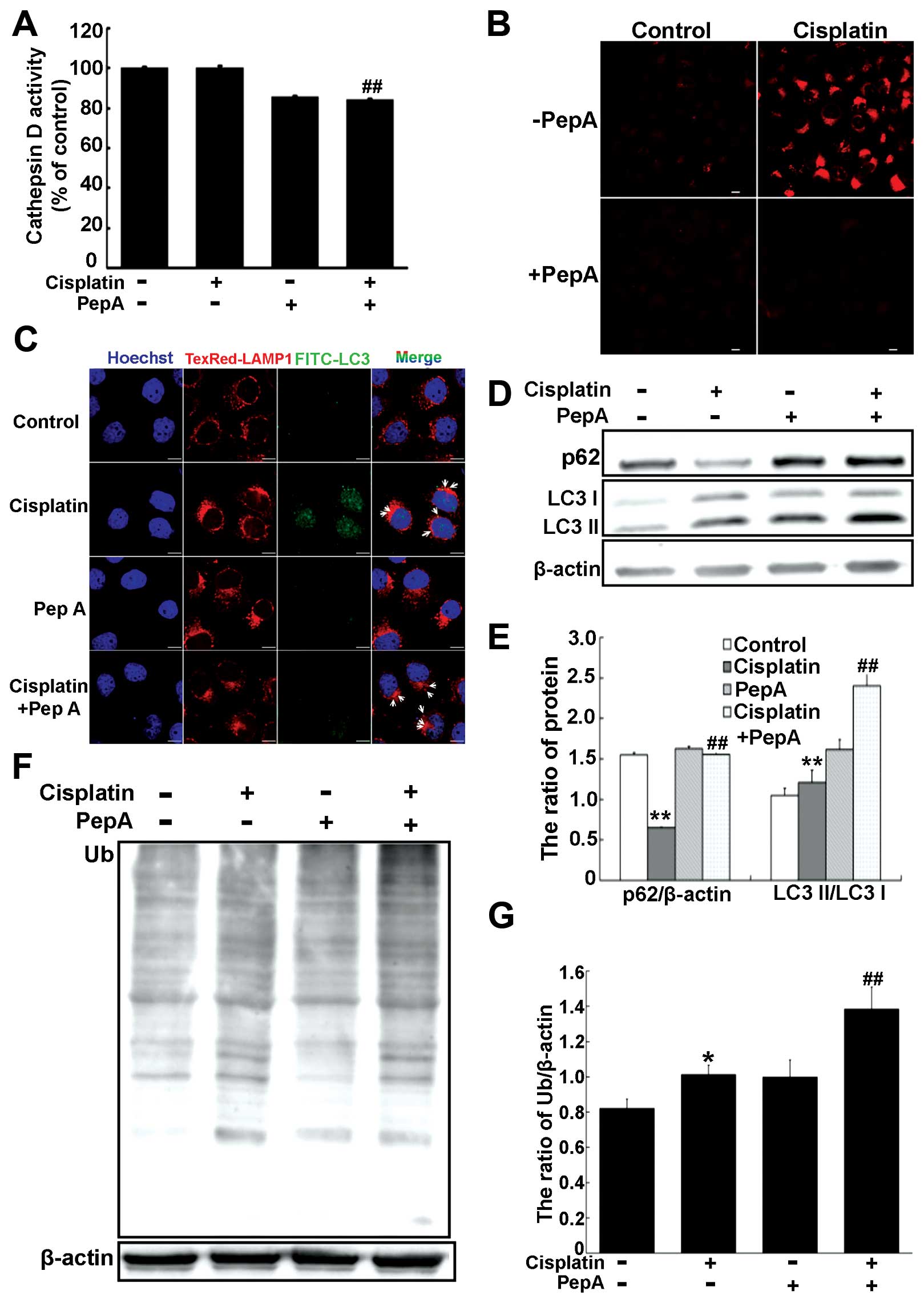
Inhibition of cathepsin D impairs
autophagic degradation in cisplatin-resistant SKOV3/DDP cells
We next investigated whether cathepsin D contributes
to protein degradation during autophagy in cisplatin-resistant
SKOV3/DDP cells by inhibiting cathepsin D and measuring autophagic
flux. Cells were treated with 80 μM pepstatin A, a potent
cathepsin D inhibitor. Cathepsin D activity was significantly
decreased in SKOV3/DDP cells treated with pepstatin A and/or
cisplatin (Fig. 5A). Consistently,
treatment with pepstatin A and/or cisplatin abolished LysoTracker
staining in SKOV3/DDP cells (Fig.
5B). These findings suggest that inhibition of autolysosomal
protein degradation by pepstatin A during cisplatin treatment could
impair lysosomal function.
Inhibition of cathepsin D activity also led to an
accumulation of enlarged autolysosomes, possibly because of
accumulated undegraded materials within autolysosomes (Fig. 5C). Western blot analysis identified
decreased p62 degradation and increased accumulation of LC3II
(Fig. 5D and E). Additionally,
inhibiting cathepsin D activity led to accumulation of
ubiquitinated proteins (Fig. 5F and
G). These data suggest that cathepsin D activity is required
for autophagic degradation and lysosomal function during autophagy
in cisplatin-resistant SKOV3/DDP cells.
Inhibition of lysosomal ATP accumulation
suppresses autophagic flux by regulating lysosomal function in
cisplatin-resistant cells
As accumulation of lysosomal ATP is critical for
normal lysosomal function (28),
we next measured the ATP content of lysosomes in
cisplatin-sensitive SKOV3 cells and cisplatin-resistant SKOV3/DDP
cells. Compared with un-treated SKOV3 cells, lysosomes of
un-treated SKOV3/DDP cells contained more ATP (Fig. 6A). Following treatment with
cisplatin for 12 h, the level of lysosomal ATP in SKOV3 cells
slightly increased. SKOV3/DDP cells always contained abundant ATP,
with cisplatin augmenting this high level. The anion transport
inhibitor DIDS inhibits lysosomal ATP accumulation by inhibiting
activity of SLC17A9, which is enriched in lysosomes (28). Therefore, we treated SKOV3/DDP cell
combinations of DIDS and cisplatin to investigate the effect of
inhibiting lysosomal ATP accumulation on cisplatin sensitivity.
Treatment with DIDS significantly decreased lysosomal ATP
accumulation, more so when combining DIDS with cisplatin (Fig. 6B).
We next investigated the effect of blocking
lysosomal ATP accumulation on lysosomal degradation of
autophagolysosomal materials in SKOV3/DDP cells. Compared with
cisplatin alone, co-treatment of SKOV3/DDP cells with DIDS and
cisplatin more effectively inhibited p62 degradation and increased
accumulation of LC3II (Fig. 6C and
D). Co-treatment with DIDS and cisplatin also yielded larger
and more numerous autolysosomes (Fig.
6E). We also evaluated lysosomal function following
co-treatment of SKOV3/DDP cells with DIDS and cisplatin by
LysoTracker staining and measuring cathepsin D activity.
Co-treatment with DIDS and cisplatin abolished LysoTracker
staining, and reduced cathepsin D enzyme activity (Fig. 6F and G). These results suggest that
lysosomes of cisplatin-resistant SKOV3/DDP cells have more ATP than
cisplatin-sensitive SKOV3 cells. Moreover, inhibition of lysosomal
ATP can impair lysosomal function to block autophagic flux.
Discussion
Platinum-based chemotherapy remains the primary
treatment strategy for ovarian cancer, with cisplatin commonly
employed (34). Non-small cell
lung cancer, cervical cancer, and ovarian cancer cells treated with
cisplatin commonly activate the autophagic process to mitigate
cisplatin-induced apoptosis (10,35).
We identified an increased ratio of LC3II/LC3I following treatment
of SKOV3 or SKOV3/DDP cells with cisplatin. This phenomenon was
more pronounced in cisplatin-resistant SKOV3/DDP cells, in which
levels of p62 were also decreased. These results suggest that
cisplatin induces activation of autophagy in both SKOV3 and
SKOV3/DDP cells, but a higher level of activation is induced in
SKOV3/DDP cells.
During tumor formation, autophagy activation
inhibits tumor growth. However, in solid tumors, autophagy promotes
tumor cell survival by mitigating the consequences of stress. In
the course of autophagy, the predominant role of lysosomes is
clearing autolysosomal material, and lysosomal proteases are
required for degradation of accumulated proteins within lysosomes
(36). We identified increased
expression of LAMP1 and cathepsin D in cisplatin-resistant
SKOV3/DDP cells following treatment with or without cisplatin.
These findings suggest that SKOV3/DDP cells possess more lysosomes
than SKOV3 cells. Moreover, lysosomal function is activated in
SKOV3/DDP cells during cisplatin-induced autophagy. Therefore,
lysosomes may be involved in mediating cisplatin resistance in
SKOV3/DDP cells. Yu et al have reported that many lysosomes
are consumed during autophagy, but autophagic lysosome reformation
(ALR) allows the cell to restore lysosomal homeostasis (33). We speculate that abundant formation
of autolysosomes may provide the basic conditions for ALR to
restore lysosomal homeostasis, though the precise mechanisms
require further elucidation.
Inhibiting autophagy can effectively enhance the
cytotoxicity of anticancer chemotherapeutics (35). For example, malignant glioma cells
are more responsive to imatinib following inhibition of late-stage
autophagy (37). We observed
enhanced cisplatin-induced cytotoxicity in SKOV3/DDP cells
following treatment with either 3-MA or CQ. Additionally,
combination treatment with cisplatin and CQ could more effectively
disturbs lysosomal function to lead to autolysosome accumulation.
Further, autophagic flux was blocked following cisplatin-CQ
combination treatment of SKOV3/DDP cells. Therefore, activation of
autophagy protects SKOV3/DDP cells from cisplatin-induced
apoptosis, and targeting the lysosome to inhibit autophagy could
more efficiently increase cisplatin-sensitivity of SKOV3/DDP
cells.
While lysosomes protect cancer cells from
chemotherapy-induced apoptosis, lysosome permeabilization leads to
a gradual leakage of cathepsins and other hydrolases from the
lysosomal lumen into the cytosol. These enzymes then initiate a
cell death pathway (38). During
induction of lysosomal-associated cell death, lysosomal cathepsins
B and D translocate to the cytosol where they activate Bid and
induce mitochondrial outer membrane permeabilization (38–40).
We found that combination treatment of SKOV3/DDP cells with
cisplatin and CQ could upregulate active tBID, increase the ratio
of Bax/Bcl-2, and stimulate cleavage of caspase-3. Thus, targeting
the lysosome could enhance cisplatin cytotoxicity in
cisplatin-resistant SKOV3/DDP cells by triggering
mitochondria-lysosome crosstalk. Our results further support a role
for lysosomes in chemotherapeutic resistance in certain
cancers.
Cathepsin D is normally localized within the
lysosome, where it degrades materials delivered to lysosomes
(39,41). Abnormal expression and/or function
of many lysosomal hydrolases can be found in various cancers, and
these abnormalities are frequently associated with tumor recurrence
and disease prognosis (42–44).
Cathepsin D is also associated with the activation of autophagy in
human malignant glioblastoma cells and cervical cancer cells
(45). Additionally, inhibition of
cathepsin D activity can impair degradation of autolysosome
contents (46). Reduced levels or
activity of cathepsin B and D impairs lysosomal function, leading
to accumulation of undegraded material in autolysosomes and delayed
ALR (47). We identified a
relatively high level of cathepsin D in SKOV3/DDP cells compared
with that in SKOV3 cells. Moreover, inhibition of cathepsin D
activity in SKOV3/DDP cells impaired lysosomal function to block
cisplatin-induced autophagic degradation. This led to an
enlargement of autolysosomes, and possibly disturbed ALR. These
findings reflect the importance of the lysosome and lysosomal
proteases such as cathepsin D in maintaining cell homeostasis by
removing accumulated proteins. Autophagy may play a critical role
in cisplatin resistance in ovarian cancer cells. However, the
precise mechanisms by which lysosomal function is regulated during
autophagy requires further study.
The lysosomes of astrocytes and other cell lines
contain a high level of ATP, which plays a crucial role in
maintaining lysosomal physiology and enables activation of
proteases such as cathepsin D (28,29,48,49).
ATP-sensitive sodium and potassium channels are required for
maintaining the low pH of the lysosomal lumen, and need ATP to
supply energy for their function (27). The lysosomes of untreated SKOV3/DDP
cells contained more ATP than the SKOV3 cells. Moreover, lysosomes
in both SKOV3 and SKOV3/DDP cells possessed slightly higher levels
of ATP following cisplatin treatment. This lysosomal ATP may be
needed to maintain lysosomal function and permit activation of
lysosomal proteases. Combination treatment with cisplatin and the
inhibitor of lysosomal ATP accumulation, DIDS, weakened LysoTracker
staining, inhibited cathepsin D activity, and increased
accumulation of autolysosomes and expression of p62. This suggests
a combination of inhibiting lysosomal ATP and cisplatin could
impair lysosomal function and autophagic degradation. Previous
studies showed changes to levels of lysosomal nutrients and energy
can activate mTORC1, facilitating the cycle of lysosomal
consumption and restoration by regulating ALR (28,33).
Therefore, we speculate that abundant lysosomal ATP induces
activation of mTORC1 and promotes ALR, thereby enabling lysosomal
function and the maintenance of lysosome homeostasis. These
processes then contribute to cisplatin resistance in cancer.
In summary, following cisplatin treatment, autophagy
was activated in SKOV3/DDP cells to enable cell survival. Targeting
the lysosome inhibited autophagy to effectively enhance
cisplatin-induced cytotoxicity and initiated lysosomal cell death
by triggering mitochondria-lysosome crosstalk in SKOV3/DDP cells.
The presence of sufficient cathepsin D enables maintenance of
lysosomal function and lysosome homeostasis in cisplatin-resistant
SKOV3/DDP cells. These effects may be dependent on lysosomal ATP
levels, because ATP-mediated lysosomal function is required for
cisplatin-induced autophagy. Our findings suggest that lysosomes
are involved in mediating cisplatin resistance in ovarian carcinoma
cells. Therefore, these observations indicate that lysosomes
represent a therapeutic target for re-sensitizing
cisplatin-resistant cancer cells to chemotherapy.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81202552,
20140520018JH, 81372793 and 81272876), and the Postdoctoral Science
Foundation of China (no. 2013M540256).
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shen W, Liang B, Yin J, Li X and Cheng J:
noscapine increases the sensitivity of drug-resistant ovarian
cancer cell line SKOV3/DDP to cisplatin by regulating cell cycle
and activating apoptotic pathways. Cell Biochem Biophys. Dec
16–2014.(Epub ahead of print). PubMed/NCBI
|
|
3
|
Yakirevich E, Sabo E, Naroditsky I, Sova
Y, Lavie O and Resnick MB: Multidrug resistance-related phenotype
and apoptosis-related protein expression in ovarian serous
carcinomas. Gynecol Oncol. 100:152–159. 2006. View Article : Google Scholar
|
|
4
|
Basu A and Krishnamurthy S: Cellular
responses to cisplatin-induced DNA damage. J Nucleic Acids.
2010:2013672010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang J and Wu GS: Role of autophagy in
cisplatin resistance in ovarian cancer cells. J Biol Chem.
289:17163–17173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sun Y, Liu JH, Jin L, Sui YX, Lai L and
Yang Y: Inhibition of Beclin 1 expression enhances
cisplatin-induced apoptosis through a mitochondrial-dependent
pathway in human ovarian cancer SKOV3/DDP cells. Oncol Res.
21:261–269. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang HQ, He B, Fang N, Lu S, Liao YQ and
Wan YY: Autophagy inhibition sensitizes cisplatin cytotoxicity in
human gastric cancer cell line SGC7901. Asian Pac J Cancer Prev.
14:4685–4688. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu N, Zhang J, Shen C, Luo Y, Xia L, Xue F
and Xia Q: Cisplatin-induced downregulation of miR-199a-5p
increases drug resistance by activating autophagy in HCC cell.
Biochem Biophys Res Commun. 423:826–831. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kang R, Wang ZH, Wang BQ, Zhang CM, Gao W,
Feng Y, Bai T, Zhang HL, Huang-Pu H and Wen SX: Inhibition of
autophagy-potentiated chemosensitivity to cisplatin in laryngeal
cancer Hep-2 cells. Am J Otolaryngol. 33:678–684. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Klionsky DJ: Autophagy: From phenomenology
to molecular understanding in less than a decade. Nat Rev Mol Cell
Biol. 8:931–937. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu WK, Coffelt SB, Cho CH, Wang XJ, Lee
CW, Chan FK, Yu J and Sung JJ: The autophagic paradox in cancer
therapy. Oncogene. 31:939–953. 2012. View Article : Google Scholar
|
|
12
|
Xu Y, Yu H, Qin H, Kang J, Yu C, Zhong J,
Su J, Li H and Sun L: Inhibition of autophagy enhances cisplatin
cytotoxicity through endoplasmic reticulum stress in human cervical
cancer cells. Cancer Lett. 314:232–243. 2012. View Article : Google Scholar
|
|
13
|
Yu C, Huang X, Xu Y, Li H, Su J, Zhong J,
Kang J, Liu Y and Sun L: Lysosome dysfunction enhances oxidative
stress-induced apoptosis through ubiquitinated protein accumulation
in Hela cells. Anat Rec (Hoboken). 296:31–39. 2013. View Article : Google Scholar
|
|
14
|
Yu H, Su J, Xu Y, Kang J, Li H, Zhang L,
Yi H, Xiang X, Liu F and Sun L: p62/SQSTM1 involved in cisplatin
resistance in human ovarian cancer cells by clearing ubiquitinated
proteins. Eur J Cancer. 47:1585–1594. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Korolchuk VI and Rubinsztein DC:
Regulation of autophagy by lysosomal positioning. Autophagy.
7:927–928. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kroemer G and Jäättelä M: Lysosomes and
autophagy in cell death control. Nat Rev Cancer. 5:886–897. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
de Duve C: The lysosome turns fifty. Nat
Cell Biol. 7:847–849. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rajapakshe AR, Podyma-Inoue KA, Terasawa
K, Hasegawa K, Namba T, Kumei Y, Yanagishita M and Hara-Yokoyama M:
Lysosome-associated membrane proteins (LAMPs) regulate
intracellular positioning of mitochondria in MC3T3-E1 cells. Exp
Cell Res. 331:211–222. 2015. View Article : Google Scholar
|
|
19
|
Carlsson SR, Roth J, Piller F and Fukuda
M: Isolation and characterization of human lysosomal membrane
glycoproteins, h-lamp-1 and h-lamp-2. Major sialoglycoproteins
carrying polylactosaminoglycan. J Biol Chem. 263:18911–18919.
1988.PubMed/NCBI
|
|
20
|
Chen JW, Pan W, D'Souza MP and August JT:
Lysosome-associated membrane proteins: Characterization of LAMP-1
of macrophage P388 and mouse embryo 3T3 cultured cells. Arch
Biochem Biophys. 239:574–586. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eskelinen EL, Schmidt CK, Neu S,
Willenborg M, Fuertes G, Salvador N, Tanaka Y, Lüllmann-Rauch R,
Hartmann D, Heeren J, et al: Disturbed cholesterol traffic but
normal proteolytic function in LAMP-1/LAMP-2 double-deficient
fibroblasts. Mol Biol Cell. 15:3132–3145. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kanai K: Biology of the mycobacterioses.
Some aspects of the lysosome-bacillus interaction in experimental
mouse tuberculosis. Ann N Y Acad Sci. 154(1 Biology of My):
177–193. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou J, Tan SH, Nicolas V, Bauvy C, Yang
ND, Zhang J, Xue Y, Codogno P and Shen HM: Activation of lysosomal
function in the course of autophagy via mTORC1 suppression and
autophagosome-lysosome fusion. Cell Res. 23:508–523. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhitomirsky B and Assaraf YG: Lysosomal
sequestration of hydrophobic weak base chemotherapeutics triggers
lysosomal biogenesis and lysosome-dependent cancer multidrug
resistance. Oncotarget. 6:1143–1156. 2015. View Article : Google Scholar :
|
|
25
|
Wang E, Lee MD and Dunn KW: Lysosomal
accumulation of drugs in drug-sensitive MES-SA but not
multidrug-resistant MES-SA/Dx5 uterine sarcoma cells. J Cell
Physiol. 184:263–274. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Braun M, Waheed A and von Figura K:
Lysosomal acid phosphatase is transported to lysosomes via the cell
surface. EMBO J. 8:3633–3640. 1989.PubMed/NCBI
|
|
27
|
Ishida Y, Nayak S, Mindell JA and Grabe M:
A model of lysosomal pH regulation. J Gen Physiol. 141:705–720.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cao Q, Zhao K, Zhong XZ, Zou Y, Yu H,
Huang P, Xu TL and Dong XP: SLC17A9 protein functions as a
lysosomal ATP transporter and regulates cell viability. J Biol
Chem. 289:23189–23199. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pillai S and Zull JE: Effects of ATP,
vanadate, and molybdate on cathepsin D-catalyzed proteolysis. J
Biol Chem. 260:8384–8389. 1985.PubMed/NCBI
|
|
30
|
Aits S and Jäättelä M: Lysosomal cell
death at a glance. J Cell Sci. 126:1905–1912. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Foghsgaard L, Wissing D, Mauch D, Lademann
U, Bastholm L, Boes M, Elling F, Leist M and Jäättelä M: Cathepsin
B acts as a dominant execution protease in tumor cell apoptosis
induced by tumor necrosis factor. J Cell Biol. 153:999–1010. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sathe MN, Woo K, Kresge C, Bugde A,
Luby-Phelps K, Lewis MA and Feranchak AP: Regulation of purinergic
signaling in biliary epithelial cells by exocytosis of
SLC17A9-dependent ATP-enriched vesicles. J Biol Chem.
286:25363–25376. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu L, McPhee CK, Zheng L, Mardones GA,
Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al: Termination of
autophagy and reformation of lysosomes regulated by mTOR. Nature.
465:942–946. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kelland L: The resurgence of
platinum-based cancer chemotherapy. Nat Rev Cancer. 7:573–584.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Amaravadi RK, Yu D, Lum JJ, Bui T,
Christophorou MA, Evan GI, Thomas-Tikhonenko A and Thompson CB:
Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest. 117:326–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sagulenko V, Muth D, Sagulenko E,
Paffhausen T, Schwab M and Westermann F: Cathepsin D protects human
neuroblastoma cells from doxorubicin-induced cell death.
Carcinogenesis. 29:1869–1877. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shingu T, Fujiwara K, Bögler O, Akiyama Y,
Moritake K, Shinojima N, Tamada Y, Yokoyama T and Kondo S:
Inhibition of autophagy at a late stage enhances imatinib-induced
cytotoxicity in human malignant glioma cells. Int J Cancer.
124:1060–1071. 2009. View Article : Google Scholar
|
|
38
|
Boya P and Kroemer G: Lysosomal membrane
permeabilization in cell death. Oncogene. 27:6434–6451. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Benes P, Vetvicka V and Fusek M: Cathepsin
D - many functions of one aspartic protease. Crit Rev Oncol
Hematol. 68:12–28. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yin L, Stearns R and González-Flecha B:
Lysosomal and mitochondrial pathways in
H2O2-induced apoptosis of alveolar type II
cells. J Cell Biochem. 94:433–445. 2005. View Article : Google Scholar
|
|
41
|
Diment S, Martin KJ and Stahl PD: Cleavage
of parathyroid hormone in macrophage endosomes illustrates a novel
pathway for intracellular processing of proteins. J Biol Chem.
264:13403–13406. 1989.PubMed/NCBI
|
|
42
|
Vasiljeva O, Reinheckel T, Peters C, Turk
D, Turk V and Turk B: Emerging roles of cysteine cathepsins in
disease and their potential as drug targets. Curr Pharm Des.
13:387–403. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kuester D, Lippert H, Roessner A and
Krueger S: The cathepsin family and their role in colorectal
cancer. Pathol Res Pract. 204:491–500. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Miao HQ, Liu H, Navarro E, Kussie P and
Zhu Z: Development of heparanase inhibitors for anti-cancer
therapy. Curr Med Chem. 13:2101–2111. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hah YS, Noh HS, Ha JH, Ahn JS, Hahm JR,
Cho HY and Kim DR: Cathepsin D inhibits oxidative stress-induced
cell death via activation of autophagy in cancer cells. Cancer
Lett. 323:208–214. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tatti M, Motta M, Di Bartolomeo S,
Cianfanelli V and Salvioli R: Cathepsin-mediated regulation of
autophagy in saposin C deficiency. Autophagy. 9:241–243. 2013.
View Article : Google Scholar :
|
|
47
|
Tatti M, Motta M, Di Bartolomeo S, Scarpa
S, Cianfanelli V, Cecconi F and Salvioli R: Reduced cathepsins B
and D cause impaired autophagic degradation that can be almost
completely restored by overexpression of these two proteases in Sap
C-deficient fibroblasts. Hum Mol Genet. 21:5159–5173. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang Z, Chen G, Zhou W, Song A, Xu T, Luo
Q, Wang W, Gu XS and Duan S: Regulated ATP release from astrocytes
through lysosome exocytosis. Nat Cell Biol. 9:945–953. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Huang P, Zou Y, Zhong XZ, Cao Q, Zhao K,
Zhu MX, Murrell-Lagnado R and Dong XP: P2X4 forms functional
ATP-activated cation channels on lysosomal membranes regulated by
luminal pH. J Biol Chem. 289:17658–17667. 2014. View Article : Google Scholar : PubMed/NCBI
|