Introduction
Cholangiocarcinoma (CCA) is a cancer arising from
bile duct epithelium. The highest prevalence of CCA in the world
has been reported in the northeastern regions of Thailand, where
liver fluke infection is highly endemic (1–3). The
etiology of the disease is related to chronic biliary inflammation
caused by the consumption of raw or undercooked fish that is
infected with the liver fluke Opisthorchis viverrini,
together with foods containing N-nitroso compounds (4,5).
Only CCA patients with early stage disease are curable by surgical
treatment (6). However, the early
stage disease is asymptomatic and difficult to diagnose. Treatment
for advance stage CCA is rather unsuccessful. Most CCA patients
have different chemotherapeutic responses even at the same stage,
leading to poor clinical outcomes with short survival times of ~5–7
months (7). 5-Fluorouracil (5-FU)
is recommended in low resource countries as the first line drug of
choice for the treatment of a variety of solid tumors including CCA
(8). However, 5-FU is rather
ineffective treatment for CCA, with a response rate <10%
(9). Several reports have noted
the effectiveness of other anticancer drugs in CCA such as
cisplatin (10,11), doxorubicin (10) and gemcitabine (12). Unfortunately, the survival time was
not significantly improved which was ~6–9 months (9). Therefore, the use of drug
combinations rather than single drug has been proposed for
obtaining better response in CCA. A response rate of ≤40% was
obtained with the triple combination of cisplatin, epirubicin and
5-FU (13). However, using more
anticancer drugs may produce additional adverse effects and
acquired drug resistance in patients.
5-FU is an inhibitor of thymidylate synthase leading
to inhibition of RNA and DNA synthesis of tumor cells. DNA damage
can trigger the induction of p53-dependent cell cycle arrest and
apoptosis, resulting in tumor cell death (14,15).
Although downregulation of p53 mRNA, a p73 family
member, has been reported in 5-FU-resistant CCA cell lines by
Namwat et al (16) no
information regarding p53 role in 5-FU-resistant CCA cells
has been reported. The p53 gene contains two alternative
promoters (P1 and P2). The P1 promoter generates the full-length
TAp53 and Δ40p53, a 40 amino acids deleted p53 variant via
alternative splicing and initiation of translation within intron 2
(17). The assembly of full-length
p53 molecules as a tetramer leads to normal function as a
transcription factor. The P2 promoter encodes larger amino-terminal
truncated proteins (133 and 160 deleted amino acids), Δ133p53
(17) and Δ160p53 (18), which exhibit anti-apoptotic
properties when oligomerized with TAp53 resulting in loss of the
transactivation function. The N-terminal deleted p53 protein
variant (ΔNp53) has been reported to disrupt wild-type p53 function
(19). Hence, the control of
promoter usage in p53 is proposed as an auto-regulation mechanism
for p53 functions (20). In
addition to the ΔNp53 isoforms, three alternate isoforms of p53; α,
β and γ at the carboxyl terminal are encoded by alternative
splicing. A full-length p53 or TAp53α is encoded from the normal
splice site, whereas TAp53β and TAp53γ are encoded from two
different alternative splicing sites of intron 9 at carboxyl end
(17). To date, any impact of β
and γ isoforms on tumor suppressor activities remains unclear. An
increase in Δ133p53 expression has also been reported in
renal cell (21), acute myeloid
leukemia (22), ovarian cancer
(23), breast (17), head and neck (24), melanoma (25), colon cancer (19). The correlation between Δ133p53 and
tumor progression has been found in colon carcinomas (19). Our previous study also found the
relationship between overexpression of defective p53 (mutant p53
and Δ133p53) with poor prognosis in CCA (26). Upregulation of Δ133p53 mRNA
level and ratio disruption of the p53 isoforms encoded from P2/P1
(Δ133p53/TAp53) was correlated with poor survival outcome of
CCA patients. The major factors affecting the patients survival
outcome may be the contribution from drug resistance. Reports of
Δ133p53 effects on drug resistance are few. Recently, upregulation
of Δ133p53α in response to a low dose of doxorubicin has been noted
in osteosarcoma and colon cancer cell lines (27). However, the role of Δ133p53
isoforms in drug resistance in CCA remains unclear.
This study attempts to demonstrate the association
between Δ133p53 overexpression and chemoresistance in CCA. 5-FU
sensitivity in clinical tissues of CCA was classified using an
ex vivo histoculture drug response assay (HDRA) and clinical
treatment outcome. Two 5-FU-resistant CCA cell lines were
established in this study and used as a model to evaluate the role
of Δ133p53 isoform in chemosensitivity.
Materials and methods
Clinical samples
A total of 22 tumor samples and 10 normal adjacent
tissues were collected from intrahepatic cholangiocarcinoma (ICC)
patients who were admitted to Srinagarind Hospital, Faculty of
Medicine, Khon Kaen University, Thailand. The project was approved
by the Khon Kaen University Ethics Committee in human research
(HE571044). All patients gave written informed consent. Fresh tumor
tissues were tested for Histoculture Drug Response Assay.
Paralleled tissues were kept under liquid nitrogen until used for
protein extraction.
Histoculture drug response assay
(HDRA)
Tumor tissues were classified as 5-FU sensitive and
5-FU resistant based on results obtained from an ex vivo
histoculture drug response assay (HDRA), using the median of
inhibition index (% II) as previously described (28). In brief, fresh tumor tissues were
washed and a 3-mm diameter punch was used aseptically to take
samples that were placed on collagen gel sponges. Each was cultured
at 37ºC in a 6-well plate containing RPMI medium supplemented with
2.5% v/v penicillin-streptomycin-fungizone (PSF; Invitrogen,
Carlsbad, CA, USA) and 5-FU at 200 μM. For a control sample, no
5-FU was added in the culture medium. After 4 days of culture, the
viability of tumor cells in the cultured tissues was examined using
TUNEL staining. TUNEL-positive cells were identified as dead cells.
The efficacy of 5-FU was calculated and expressed as the % II using
the following formula: % II = (1 − % viable tumor cells in
5-FU-treated tumor tissue/% living tumor cells in control tissue) ×
100.
CCA cell lines and cell culture
Two CCA cell lines, KKU-M139 and KKU-M214 were
established from primary tumors of human intrahepatic CCA at the
Liver Fluke and Cholangiocarcinoma Research Center, Khon Kaen
University Thailand (16,29,30).
Both were cultured at 37ºC in RPMI medium (Gibco BRL, Grand Island,
NY, USA) supplemented with 10% fetal bovine serum (FBS), 1% v/v
penicillin-streptomycin solution (Gibco, BRL) under 5%
CO2 atmosphere. These CCA cells were named as KKU-M139P
and KKU-M214P, to identify the parental cell lines at the beginning
of drug resistance induction.
Establishment of 5-FU-resistant CCA cell
lines
5-FU-resistant cell lines were generated from the
parental cell lines KKU-M139P and KKU-M214P by stepwise increases
of the concentration of 5-FU (Boryung Pharm, Korea) as described
previously (16). In brief,
1×105 cells were seeded in 25-cm2 flasks and
cultured without 5-FU for 24 h. Subsequently, cells were exposed
with 5-FU at 6 μM for KKU-M139 (1X IC50 value of
KKU-M139P) and 4 μM for KKU-M214 (1X IC50 value of
KKU-M214P) for 72 h. Cells were then cultured in a drug-free medium
until they reached 70% confluence. These cells were continuously
maintained in 1X IC50 for several passages until these
cells were stable before being subjected into 2X IC50.
The 5-FU-resistant clones were finally obtained after continuous
selection by several passages for 18 months. The IC50 of
the resistant clones was checked by the SRB assay (31). The resistant clones were then
passaged into a drug-free medium for 2 weeks before being stored as
a stock of the resistant cell lines (KKU-M139R and KKU-M214R) at
−80ºC. KKU-M139R and KKU-M214R were cultured in 5-FU-free medium
for ≥2 weeks to eliminate potential long-term effects of 5-FU
unrelated to drug resistance prior to being performed in all
experiments.
Transient silencing of Δ133p53 by
siRNA
Expression of Δ133p53 in KKU-M139R and KKU-M214R
cells was suppressed using a siRNA technique. The sequences of two
specific siRNA targeting human Δ133p53 (Δ133p53a and Δ133p53b) as
described previously (18), were
purchased from Ambion (Austin, TX, USA). The cells
(2×106 cells/well) were seeded in a 6-well plate and
cultured overnight before being transfected separately with 100 pM
of siΔ133p53a and siΔ133p53b, while siGFP (Green fluorescence
protein, Applied Biosystems/Ambion, Carlsbad, CA, USA) was used as
a siRNA control. Transfection was carried out using Lipofectamine
RNAiMAX (Invitrogen) according to the manufacturer's instructions.
After 24 h of transfection, culture medium was added and the plates
were incubated at 37ºC for a further 48 h. At 72 h-post
transfection, total proteins were extracted using TRIzol reagent
(Invitrogen). The level of Δ133p53 protein was determined by
western blot analysis using β-actin as a loading control.
Measurement of IC50 by
Sulforhodamine B (SRB) assays
The parental CCA cells (KKU-M139P and KKU-M214P) and
the 5-FU-resistant cells (KKU-M139R and KKU-M214R) were seeded at
1×104 cells/well in triplicate into a 96-well culture
plate and incubated at 37ºC for 24 h. All cell lines were then
treated with various concentrations of 5-FU ranging from 2–128 μM
in triplicate for 72 h, while 0.9% saline was used as a negative
control. The cytotoxicity was performed using a sulforhodamine B
(SRB) assay as previously described (31). The cells were fixed using a 10%
cold trichloroacetic acid (TCA), washed and air-dried at room
temperature. SRB (Sigma-Aldrich, MO, USA) solution (100 μl/well)
was added and followed by three quick rinses with 1% acetic acid to
remove unbound dye. SRB was solubilized in a 10 mM Tris base
solution and the absorbance at 490 nm was measured using a
microplate reader (Tecan Ltd., Reading, UK). Percentage of cell
viability was calculated [(mean ODsample − mean
ODday0/mean ODnegative control − mean
ODday0) × 100] and used to generate the curve by which
IC50 was calculated. Resistance index was defined as a
ratio of the IC50 value of drug resistant cells to
parental cells.
Population doubling time (PDT)
To assess cell growth, the population doubling time
(PDT) of the cells was assessed in triplicate. Cells
(2×105) were cultured in a drug free medium supplemented
with 10% FBS at 37ºC in a humidified 5% CO2 atmosphere.
When reaching 70% confluence, the cells were trypsinized, stained
with trypan blue and counted on a hemocytometer. PDT was calculated
using the following formula as described previously (32): (T-T0)
log2/logN-logN0, where N0 is the initial cell
number, N is the final cell number, T is the time interval between
N0 and N, and T0 is the initial time.
Colony forming assay
KKU-M139R and KKU-M214R cells were pre-treated with
either siΔ133p53 or siRNA control. The cells were seeded at 200
cells/well in a 6-well plate containing 2 ml RPMI culture medium
supplemented with 10% v/v FBS and cultured at 37ºC in a humidified
5% CO2 atmosphere for 5 days. The cells were washed
twice with PBS, stained with H&E and the colonies were
counted.
Analysis of apoptosis by Annexin V/PI
staining
The analysis of Annexin V binding was carried out
with the Annexin V-FITC Detection Kit I (eBioscience, San Diego,
CA, USA) according to the manufacturer's instructions. Briefly,
cells were incubated with or without siΔ133p53 or a control
scramble RNA for 48 h. Cells were collected, washed twice with cold
PBS, centrifuged at 1,800 rpm for 3 min, and resuspended in 1X
binding buffer at a concentration of 106 cells/ml. Then
100 μl of the solution (105 cells) was transferred to a
5-ml culture tube; 5 μl of Annexin V-FITC and 5 μl of PI were
added. Cells were incubated for 15 min at room temperature in the
dark. Furthermore, 200 μl of 1X binding buffer were added to each
tube, and samples were analyzed by FACScan flow cytometry (BD
FACSCanto II; BD, USA). For each sample, 10,000 ungated events were
acquired. Annexin V+/PI− cells represented
the early apoptotic populations, and Annexin
V+/PI+ cells the late apoptotic
populations.
Cell cycle analysis by flow
cytometry
KKU-M139R and KKU-M214R cells (105
cells/ml) were incubated with or without siΔ133p53 or a siRNA
control for 48 h. The cells were collected, washed with cold PBS,
fixed in cold 100% ethanol, treated with DNase-free RNase, and
stained with 40 μg/ml of propidium iodide (PI). The distribution of
the cells between phases of the cell cycle was deduced from the DNA
content on a FACScan flow cytometer (BD FACSCanto II; BD, USA). For
each sample, 10,000 gated events were acquired.
Western blot analyses
Protein was extracted from CCA tissues and cell
lines using TRIzol (Invitrogen) and 40 μg aliquots were
fractionated on 15% polyacrylamide gel electrophoresis. Primary
antibodies; CM-1 (1:100, Signet, Emeryville, CA, USA), p21 (1:400),
p27 (1:400), Bcl-2 (1:200), Bax (1:200), and p73 (ab-4) (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) as well as a loading control,
β-actin (1:4,000, Sigma Chemical Co.) were used followed by the
secondary antibody peroxidase-labeled anti-rabbit (1:10,000, Abcam,
UK) The proteins were detected by chemiluminescence using the ECL
Plus system (GE Healthcare, UK). Protein band intensity was
calculated by Scion image program, and normalized to β-actin.
Immunocytochemical staining of p53
The paraffin-embedded sections (5 μm) of CCA cell
pellets fixed with 10% formalin solution were deparaffinized.
Antigen retrieval was performed in boiling 0.01 M citrate buffer
(pH 6.0) as described previously (26). Endogenous peroxidase was
inactivated with 100 μl of 3% H2O2.
Non-specific binding was further treated with a blocking buffer
containing phosphate-buffered saline with Tween-20 (PBS-T), 30%
casein and 5% FBS. For p53 protein detection, mutant p53 was
detected with the primary antibody clone DO-7, 1:100 (Dako,
Glostrup, Denmark), which recognizes an epitope between amino acids
1–45 of human p53. The slides were incubated overnight at room
temperature with primary antibody. Proteins were detected using the
EnVision system (Dako) for 1 h at room temperature. Color was
developed with DAB solution (Dako) and nuclei were counterstained
with hematoxylin. Positive staining was observed as brown color in
blue/gray nuclei. Positive with DO-7 antibody indicates
overexpression of the mutated p53 due to its stability (26,33–35).
Statistical analyses
Statistical analyses were performed using SPSS for
Windows version 15 (SPSS, Inc., IL, USA). The Mann-Whitney U test
was used for comparison of two groups. Data are expressed as mean ±
SD from three independent experiments. Statistically significant
differences are indicated in the figures as *P<0.05,
**P<0.01, ***P<0.001.
Results
Assessment of 5-FU sensitivity in CCA
samples
5-FU sensitivity in 22 CCA patients was classified
based on the results obtained from histoculture drug response
assays (HDRA) and treatment outcome data. For the HDRA-based
classification, we used the median of the % inhibition index (% II)
which was 36.5% and this identified 11 patients as 5-FU-sensitive
and 11 as 5-FU-resistant. Only 12 of 22 CCA patients underwent
complete course of chemotherapy. The treatment outcome was further
followed up every 6 months for ≥12 months. Poor response (n=7) and
favorable response (n=5) were defined in terms of having tumor
progression before and after 6 months after treatment,
respectively. All clinical data are summarized in Table I.
 | Table IClinicopathological data of 22 CCA
patients. |
Table I
Clinicopathological data of 22 CCA
patients.
| | | | | |
Chemosensitivityb |
|---|
| | | | | |
|
|---|
| No. | Sex | Age | Survival
timea | Stage | Chemotherapy | HDRA | Clinical
outcome |
|---|
| 1 | M | 53 | Long | IVB | Treated | Sensitive | Poor response |
| 2 | M | 64 | Long | II | Treated | Sensitive | Favorable
response |
| 3 | M | 52 | Long | III | Treated | Sensitive | Favorable
response |
| 4 | M | 61 | Short | II | Treated | Sensitive | Poor response |
| 5 | F | 51 | Short | IVA | Untreated | Sensitive | NA |
| 6 | F | 65 | Short | III | Treated | Sensitive | Poor response |
| 7 | M | 58 | Short | IIIA | Untreated | Sensitive | NA |
| 8 | M | 70 | Short | IVA | Untreated | Sensitive | NA |
| 9 | M | 69 | Long | IVA | Treated | Sensitive | Poor response |
| 10 | F | 51 | Short | II | Treated | Sensitive | Favorable
response |
| 11 | F | 64 | Long | III | Treated | Sensitive | Favorable
response |
| 12 | M | 57 | Long | IVA | Treated | Resistant | Poor response |
| 13 | M | 53 | Short | IIIA | Untreated | Resistant | NA |
| 14 | F | 58 | Long | IVA | Treated | Resistant | Favorable
response |
| 15 | F | 51 | Long | IIIA | Treated | Resistant | Poor response |
| 16 | F | 64 | Short | IVA | Untreated | Resistant | NA |
| 17 | M | 63 | Short | III | Untreated | Resistant | NA |
| 18 | M | 59 | Long | III | Untreated | Resistant | NA |
| 19 | F | 50 | Short | IVA | Untreated | Resistant | NA |
| 20 | M | 69 | Short | IVB | Untreated | Resistant | NA |
| 21 | M | 62 | Long | IIIA | Treated | Resistant | Poor response |
| 22 | M | 69 | Long | IVA | Untreated | Resistant | NA |
Levels of the ΔNp53 isoform are increased
significantly in 5-FU-resistant CCA samples
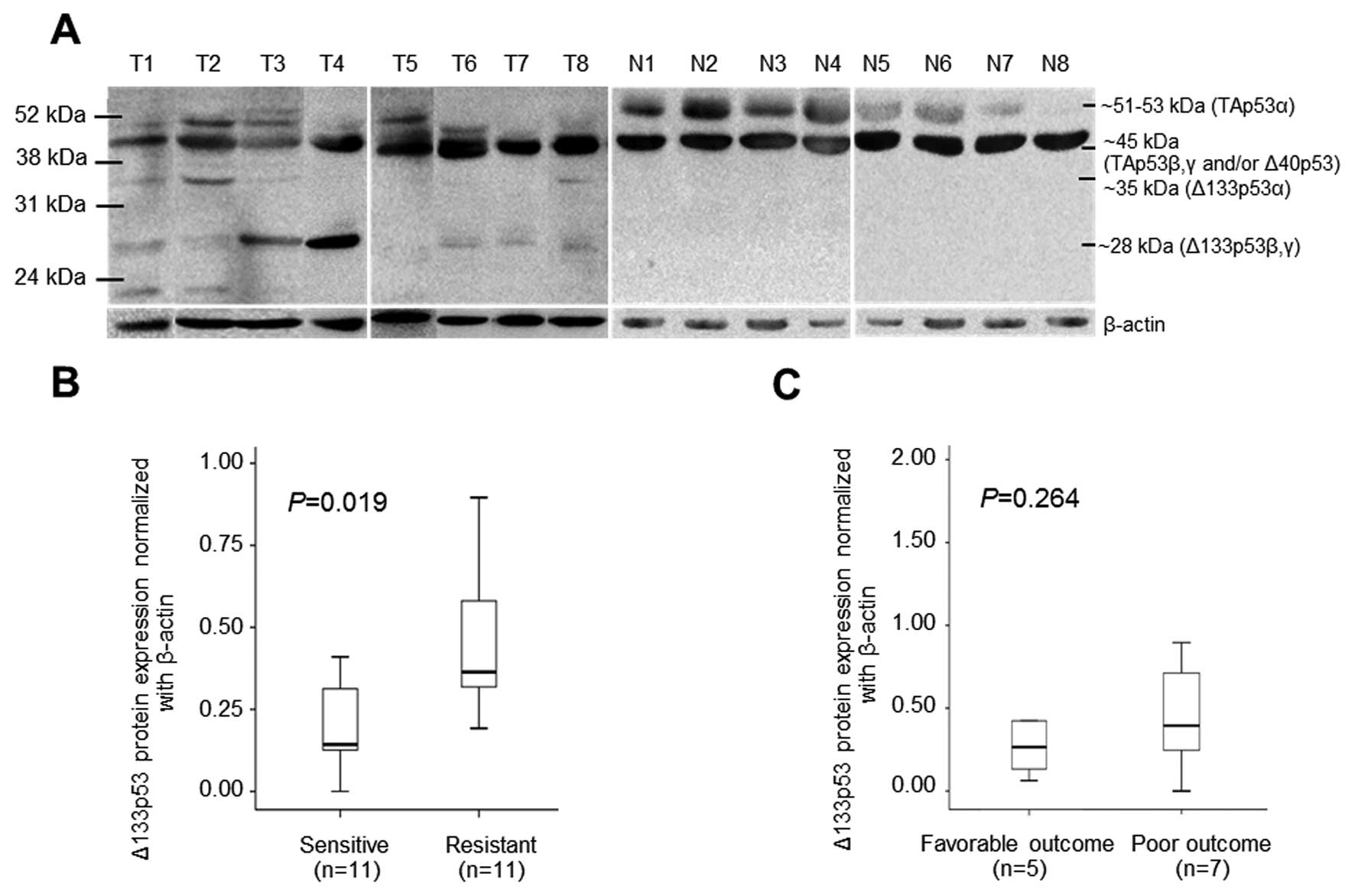
Western blot analysis of 22 tumors and 10 normal
paired tissues using CM-1 antibody revealed the presence of various
p53 isoforms (Fig. 1). At least 3
isoforms; TAp53, Δ40p53 and Δ133p53 were observed in tumor tissues,
in which two different sizes of Δ133p53 at 35 kDa corresponding to
Δ133p53α and 28 kDa to Δ133p53β, γ were defined (Fig. 1A). Interestingly, no Δ133p53
isoform was observed in the normal tissues (Fig. 1A). The box-plot analysis of Δ133p53
protein expression normalized to β-actin and 5-FU sensitivity
classified by HDRA showed significantly increased Δ133p53 in
5-FU-resistant cases compared to sensitive ones (P=0.019) (Fig. 1B). It seemed that Δ133p53 protein
was highly expressed in CCA patients with poor outcome compared to
CCA cases with favorable outcome but was not statistically
significant (P=0.264) (Fig.
1C).
Characteristics of 5-FU-resistant CCA
cell lines
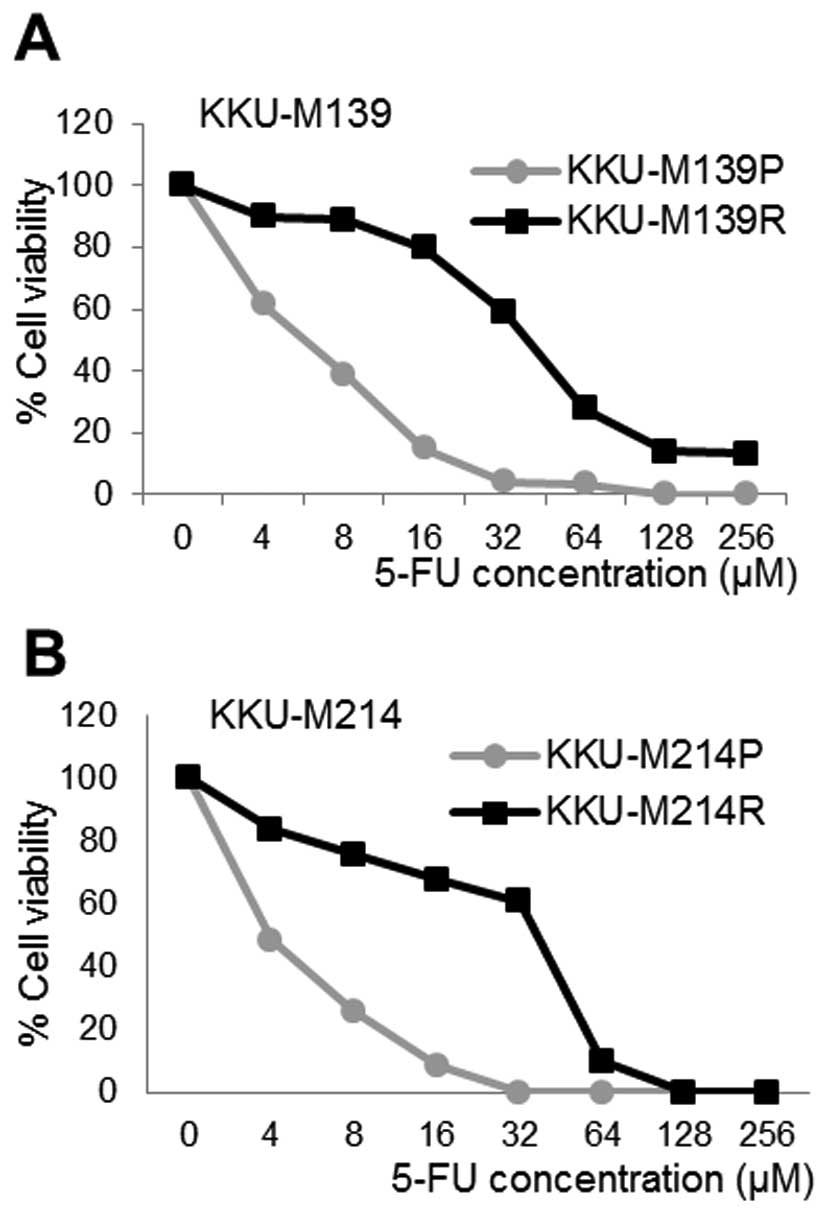
Two resistant CCA cell lines; KKU-M139R and
KKU-M214R were successfully induced from the parental cells;
KKU-M139P and KKU-M214P. Drug toxicity is presented in Fig. 2. The IC50 values of 5-FU
in KKU-M139P, KKU-M139R, KKU-M214P and KKU-M214R cells were
6.2±2.09, 38.8 ±7.11, 3.9±2.04 and 39.5±3.45 μM, respectively. The
resistance index calculated from the ratio of IC50 of
resistant to parental CCA cell lines was 6.26 and 10.12 for
KKU-M139 and KKU-M214, respectively. Furthermore, population
doubling times of both resistant cell lines were shorter than those
of parental cell lines as summarized in Table II.
| Table IIPopulation doubling time (PDT) and
IC50 of 5-FU at 72-h culture of KKU-M139 and KKU-M214
cell lines. |
Table II
Population doubling time (PDT) and
IC50 of 5-FU at 72-h culture of KKU-M139 and KKU-M214
cell lines.
| Cell lines | Population doubling
time (PDT) (h) (mean ± SD) | IC50 of
5-FU (μM) (mean ± SD) | Resistant index
(IC50 of resistant/parental cells) |
|---|
| KKU-M139P | 30.13±0.86 | 6.2±2.09 | 6.26 |
| KKU-M139R | 17.81±0.74 | 38.8±7.11 | |
| KKU-M214P | 40.16±1.12 | 3.9±2.04 | 10.12 |
| KKU-M214R | 22.92±1.03 | 39.5±3.45 | |
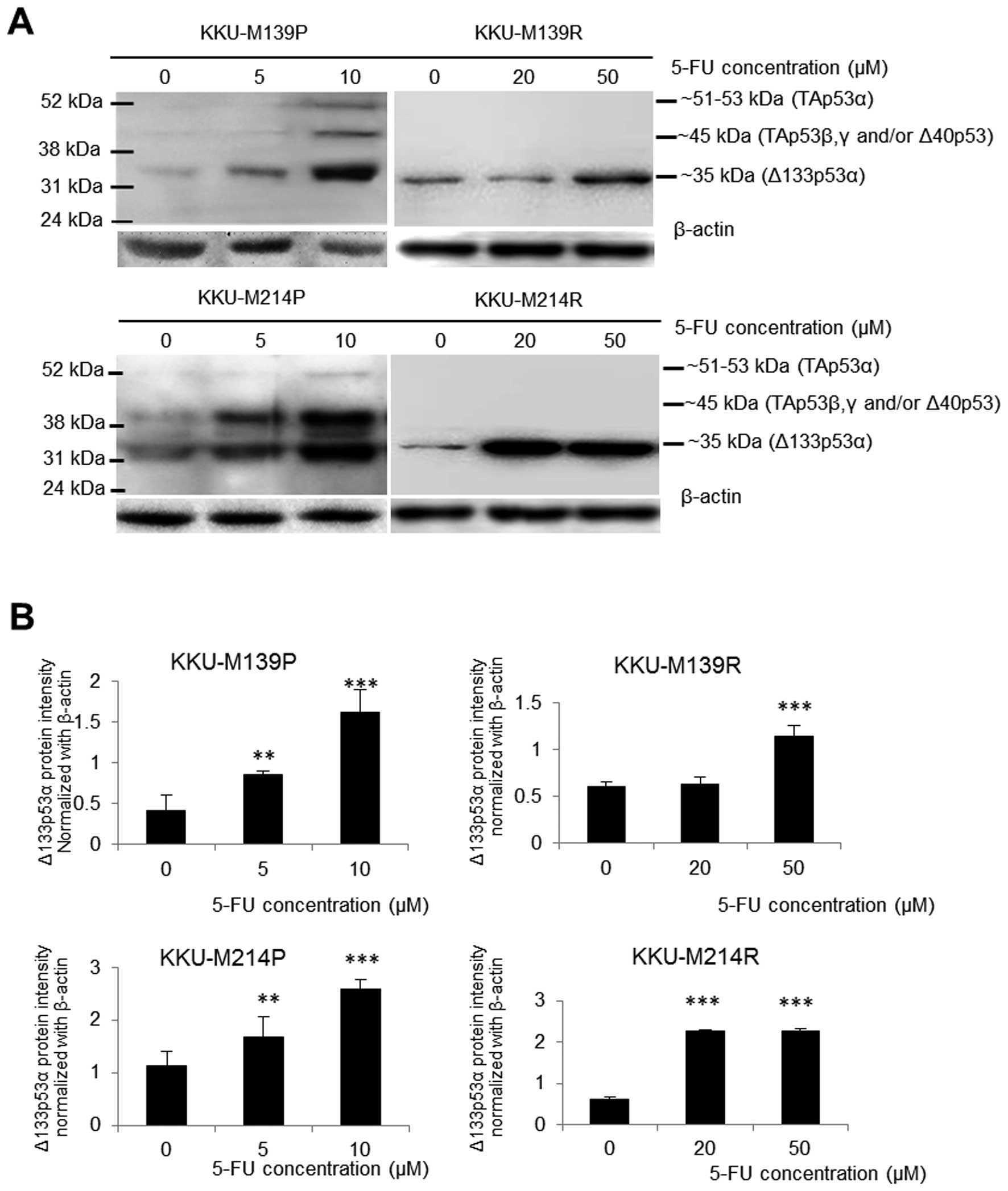
Upregulation of the Δ133p53 isoform
induced by 5-FU
The expression of p53 isoforms was assessed in both
parental and resistant CCA cell lines in response to 5-FU
concentration covering their IC50 values. When
challenged with 5-FU with 5 and 10 μM, TAp53, Δ40p53 and Δ133p53
protein isoforms were markedly increased in both parental cell
lines (KKU-M139P and KKU-M214P) in a dose-dependent manner
(Fig. 3). This finding indicates
the increased usage of both P1 and P2 promoters under 5-FU stress.
In contrast to parental cells, only Δ133p53 protein was upregulated
in both resistant cell lines (KKU-M139R and KKU-M214R) induced by
5-FU (Fig. 3) suggesting the
enhancement of P2 promoter usage. Upon 5-FU challenge with 20 and
50 μM, KKU-M214R showed rapid response to 5-FU in which the
upregulation of Δ133p53 was detected at a lower dose (20 μM of
5-FU) compared to KKU-M139R (50 μM of 5-FU).
Silencing of Δ133p53 promotes apoptosis
and cell cycle arrest in 5-FU resistant CCA cells
Silencing of Δ133p53 in both KKU-M214R and KKU-M139R
was used to investigate the role of Δ133p53 in 5-FU-resistant CCA
cells. The expression of Δ133p53 was successfully suppressed with
both siΔ133p53a and siΔ133p53b compared to the siRNA control
(Fig. 4A and B) with normal
apparent morphology (Fig. 4C). The
effect of silenced Δ133p53 on apoptotic and cell cycle markers in
KKU-M139R and KKU-M214R was investigated. The suppression of
Δ133p53 protein resulted in significant upregulation of Bax
expression in both KKU-M214R and KKU-M139R (P<0.01) as well as
downregulation of Bcl-2 in KKU-M214R (P<0.001) and KKU-M139R
(P<0.01) (Fig. 4D).
Accordingly, Annexin V/PI staining showed significantly increased
cell apoptosis in the silenced siΔ133p53 of both KKU-M139R and
KKU-M214R (siΔ133p53a at P<0.01 and siΔ133p53b at P<0.001,
respectively) compared with control (Fig. 5).
Additionally p21 and p27 proteins were significantly
upregulated with P<0.01 and P<0.001 in M139R and KKU-M214R
compared to the siRNA control (Fig. 4A
and 4D). Moreover, cells were significantly arrested at G2 in
both siΔ133p53a and siΔ133p53b treated KKU-M139R and KKU-M214R
compared to the siRNA controls (Fig.
6). Interestingly, significant upregulation of p73 was observed
in both types of silenced CCA cells (Fig. 4A and D). Moreover, no mutated p53
was revealed in siΔ133p53 treated (KKU-M139R and KKU-M214R) or
siRNA control compared to their parental cells (KKU-M139P and
KKU-M214P) using immunostaining with DO-7 (Fig. 7). These results imply that the
induced p73 protein might help mediating apoptosis upon the absence
of wild-type TAp53 (Fig. 3A) and
mutated p53 (Fig. 7). Moreover,
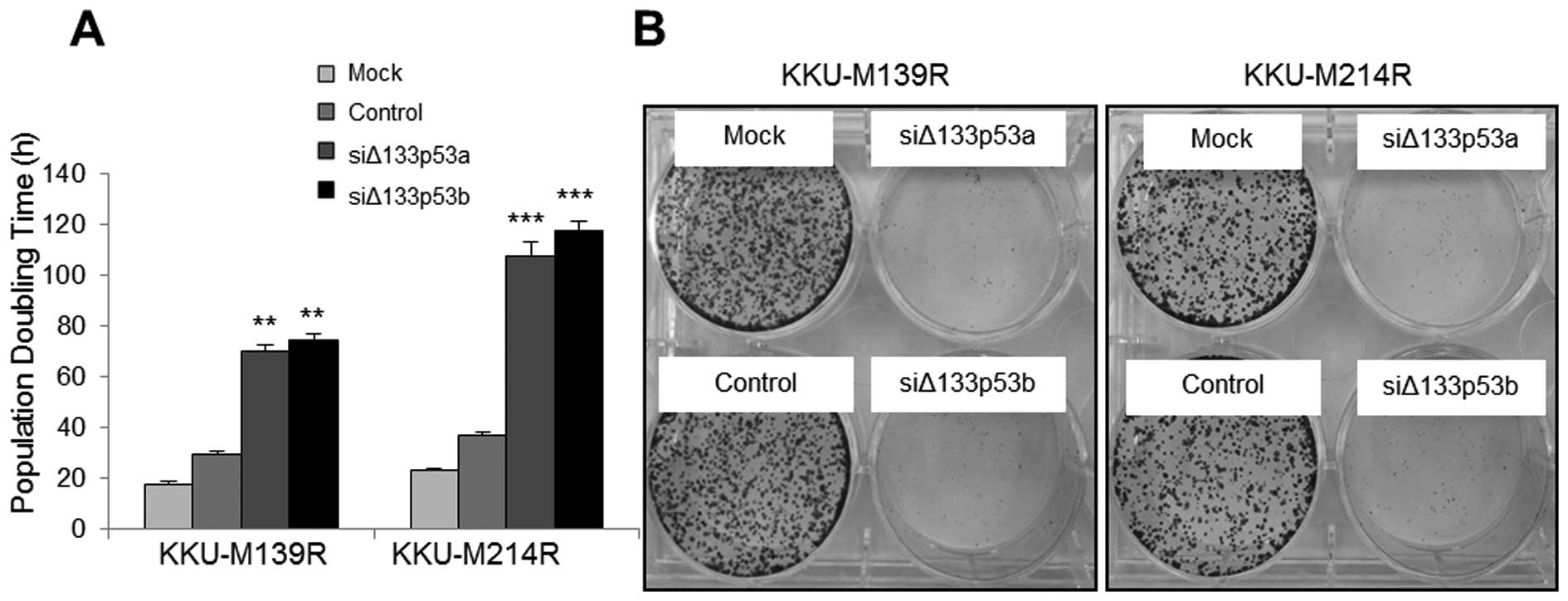
the induced cell cycle arrest by Δ133p53 silencing leads to growth
retardation, as shown by the prolonged PDT (Fig. 8A) and the inhibition of colony
forming capability (Fig. 8B).
Hence, suppression of Δ133p53 expression affects certain tumor
characteristics of these CCA resistant cells.
Attenuation of Δ133p53 levels enhances
the chemosensitivity of 5-FU-resistant CCA cells
The chemosensitivity of KKU-M139R and KKU-M214R
cells transfected with siΔ133p53a and siΔ133p53b was re-assessed
using an SRB assay of cell numbers (Table III). Strikingly, the
IC50 of KKU-M139R-siΔ133p53a and KKU-M139R-Δ133p53b
cells to 5-FU was decreased 12- and 45-fold, as compared with the
IC50 of the siRNA control cells. Similar effects were
also observed in KKU-M214R with 11- and 20-fold decreases,
respectively. The IC50 of silenced CCA cells was lower
than those of parental cells suggesting that silencing of Δ133p53
can re-sensitize 5-FU resistance in CCA cell lines.
| Table IIIThe IC50 of 5-FU at 72-h
culture of KKU-M139R and KKU-M214R cell lines after siRNA
treatment. |
Table III
The IC50 of 5-FU at 72-h
culture of KKU-M139R and KKU-M214R cell lines after siRNA
treatment.
| Cell lines | 5-FU
IC50 at 72 h (μM) (mean ± SD) | Fold reduction of
IC50 a |
|---|
| KKU-M139R |
| Mock | 38.83±7.11 | - |
| Control | 31.47±4.98 | - |
| siΔ133p53a | 2.7±1.18 | 11.66 |
| siΔ133p53b | 0.7±0.94 | 44.96 |
| KKU-M214R |
| Mock | 39.5±3.45 | - |
| Control | 35.24±3.91 | - |
| siΔ133p53a | 3.2±2.84 | 11.01 |
| siΔ133p53b | 1.8±1.48 | 19.58 |
Discussion
A variety of ΔNp53 isoforms generated from both P1
and P2 promoters were reported in clinical tumors, however, limited
evidence of chemoresistance has been noted. This study is the first
to demonstrate the correlation between high levels of Δ133p53
expression in clinical CCA tissues with 5-FU resistant based HDRA.
A limited sample sizing, resulting from an incomplete treatment,
affected the statistical testing for correlation in clinical
treatment. Even though Δ133p53 expression showed no significant
correlation with clinical treatment outcome, Δ133p53 level seemed
to increase in patients with poor response to treatment. This
result suggested that Δ133p53 might be involved on drug
responsiveness. Aoubala et al reported the upregulation of
Δ133p53α with response to a low dose of doxorubicin in osteosarcoma
and colon cancer (27). However,
p53 function can be inactivated by either mutation or Δ133p53
overexpression, thus, the mutant p53 should also be considered in
clinical CCA. The incidence of p53 mutation has been reported in
clinical CCA samples as up to 41–44% (26,36).
Defective p53 due to mutation has been reported with drug
resistance to 5-FU-based therapy in colorectal cancer (37–39).
A correlation between p53 mutation and platinum-based
chemotherapy resistance, early relapse, and shortened overall
survival was reported in ovarian cancer patients (40). Of note, the incidence of ΔNp53
overexpression without mutant p53 in CCA has been previously found
at 54% (26). Therefore, the
5-FU-resistant CCA cell lines KKU-M139R and KKU-M214R were used as
an in vitro model to address the impact of Δ133p53 on 5-FU
resistance without the influence of p53 mutation. These
5-FU-resistant CCA cell lines contain only Δ133p53 protein without
the full length p53 (TAp53) or the mutated p53 as shown by negative
western blotting with CM-1 and negative DO-7 staining. The
IC50 of 5-FU resistance remained stable even cultured in
drug free medium for ≥4 weeks, supporting the claim for stable
resistant clone.
For both parental CCA cell lines, 5-FU can enhance
the upregulation of both TAp53 and Δ133p53 in a dose-dependent
manner. The enhancement of both P1 and P2 promoter usage might
provide an advantage of apoptosis evasion via p53 inactivation
which enabling an acquired 5-FU resistance upon drug exposure.
Similar finding of Δ133p53 upregulation in response to 5-FU was
revealed in both resistant cells except TAp53 existence. The
enhancement of P2 promoter regardless of TAp53 may result from
continuous selective pressure upon induction of 5-FU resistance.
The rapid response to lower dose of 5-FU found in KKU-M214R, may
explain the higher resistance index of KKU-M214R (10.12-fold) than
that of KKU-M139R (6.26-fold). Targeting of Δ133p53 in 5-FU
resistant cells by siRNA is therefore verified the role of Δ133p53
on chemoresistance in these 5-FU-resistant CCA cell lines which was
successfully obtained by both siΔ133p53a and siΔ133p53b with 75–90%
suppression. Interestingly, the targeting of Δ133p53 helps
restoring the 5-FU sensitivity with markedly reduced
IC50 compared to that of parental cells. The molecular
underlying mechanism of 5-FU resensitization can be explained by an
increase of cell apoptosis via an upregulation of pro-apoptotic BAX
and downregulation of anti-apoptotic Bcl-2. Moreover, G2 arrest was
induced by upregulation of p21 and p27 in comparison with siRNA
control cells. This evidence is relevant to the antitumor activity
of 5-FU which is known to be involved in the induction of
p53-dependent cell cycle arrest and apoptosis (37,39,41).
Regardless of TAp53, the suppressed Δ133p53 can
explain only the withdrawal of p53 inactivation, but is unable to
provide clues for p53 activation. The increase of p73 expression
observed by western blotting in both types of silenced Δ133p53 CCA
cells might be responsible for p53 function restoration.
p73, as a p53 family member, has been shown to
possess the capability to restore p53 function via p21 activation
in a neuroblastoma cell line (42). The p73 protein can activate
upstream transcriptional regulation of p21 and p27, resulting in
cell cycle arrest in G2 (43–45).
In human lung adenocarcinoma, p73 overexpression can enhance
chemosensitivity by apoptosis induction (46,47).
Namwat et al (16),
reported the association between downregulation of TAp73
mRNA and 5-FU-resistant CCA cell lines. Evasion of apoptosis and
cell cycle arrest are evident as a common mechanism of 5-FU
resistance in various cancers such as colorectal cancer (48), breast cancer (49), and CCA (16,29).
Recently, evasion of both intrinsic and extrinsic apoptotic pathway
has been reported in gemcitabine-resistant CCA cell lines (32). Thus, Δ133p53 may exert a signature
of chemoresistant cells to evade p53-dependent cell apoptosis and
cell cycle arrest.
Collectively, the silencing of Δ133p53 and increased
p73 expression may modulate the chemosensitivity of 5-FU
resistance in CCA cell lines. Low incidence of p73 mutation
has been reported in CCA (50),
downregulation via p73 methylation has been frequently found
in various cancers (51–54) including CCA (55). Data on the alteration of p73
for chemo-resistance in CCA is still limited. The status of
p73 should be investigated in further study.
In conclusion, this study is the first to
demonstrate the important role of Δ133p53 in 5-FU resistance in
CCA. The attenuation of p53 by molecular targeting of Δ133p53 may
modulate the chemosensitivity in CCA, hence the potential for use
of Δ133p53 as a candidate for targeted therapy.
Acknowledgements
This study was supported by the Higher Education
Research Promotion and National Research University Project of
Thailand, Office of the Higher Education Commission, through the
Health Cluster (SHeP-GMS), Khon Kaen University (Grant no.
H-2553-Ph.D-06); the Centre for Research and Development of Medical
Diagnostic Laboratories, Faculty of Associated Medical Sciences,
Khon Kaen University.
References
|
1
|
Vatanasapt V, Sriamporn S and Vatanasapt
P: Cancer control in Thailand. Jpn J Clin Oncol. 32(Suppl):
S82–S91. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sriamporn S, Pisani P, Pipitgool V,
Suwanrungruang K, Kamsa-ard S and Parkin DM: Prevalence of
Opisthorchisviverrini infection and incidence of cholangiocarcinoma
in Khon Kaen, Northeast Thailand. Trop Med Int Health. 9:588–594.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Patel T: Worldwide trends in mortality
from biliary tract malignancies. BMC Cancer. 2:102002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thamavit W, Bhamarapravati N, Sahaphong S,
Vajrasthira S and Angsubhakorn S: Effects of dimethylnitrosamine on
induction of cholangiocarcinoma in Opisthorchis viverrini-infected
Syrian golden hamsters. Cancer Res. 38:4634–4639. 1978.PubMed/NCBI
|
|
5
|
Thamavit W, Kongkanuntn R, Tiwawech D and
Moore MA: Level of Opisthorchis infestation and carcinogen
dose-dependence of cholangiocarcinoma induction in Syrian golden
hamsters. Virchows Arch B Cell Pathol Incl Mol Pathol. 54:52–58.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Patel T: Cholangiocarcinoma. Nat Clin
Pract Gastroenterol Hepatol. 3:33–42. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shaib Y and El-Serag HB: The epidemiology
of cholangiocarcinoma. Semin Liver Dis. 24:115–125. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Patel T: Cholangiocarcinoma -
controversies and challenges. Nat Rev Gastroenterol Hepatol.
8:189–200. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thongprasert S: The role of chemotherapy
in cholangiocarcinoma. Ann Oncol. 16(Suppl 2): ii93–ii96. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Patt YZ, Hassan MM, Lozano RD, Waugh KA,
Hoque AM, Frome AI, Lahoti S, Ellis L, Vauthey JN, Curley SA, et
al: Phase II trial of cisplatin, interferon alpha-2b, doxorubicin,
and 5-fluorouracil for biliary tract cancer. Clin Cancer Res.
7:3375–3380. 2001.PubMed/NCBI
|
|
11
|
Lee MA, Woo IS, Kang JH, Hong YS and Lee
KS: Epirubicin, cisplatin, and protracted infusion of 5-FU (ECF) in
advanced intrahepatic cholangiocarcinoma. J Cancer Res Clin Oncol.
130:346–350. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sookprasert A, Chindaprasert J and
Wirasorn K: Systemic therapy for locally advanced and metastatic
cholangiocarcinoma. Asian Pac J Cancer Prev. 13(Suppl): S3–S6.
2012.
|
|
13
|
Choi CW, Choi IK, Seo JH, Kim BS, Kim JS,
Kim CD, Um SH, Kim JS and Kim YH: Effects of 5-fluorouracil and
leucovorin in the treatment of pancreatic-biliary tract
adenocarcinomas. Am J Clin Oncol. 23:425–428. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Longley DB, Harkin DP and Johnston PG:
5-fluorouracil: Mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Noordhuis P, Holwerda U, Van der Wilt CL,
Van Groeningen CJ, Smid K, Meijer S, Pinedo HM and Peters GJ:
5-Fluorouracil incorporation into RNA and DNA in relation to
thymidylate synthase inhibition of human colorectal cancers. Ann
Oncol. 15:1025–1032. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Namwat N, Amimanan P, Loilome W,
Jearanaikoon P, Sripa B, Bhudhisawasdi V and Tassaneeyakul W:
Characterization of 5-fluorouracil-resistant cholangiocarcinoma
cell lines. Chemotherapy. 54:343–351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bourdon JC, Fernandes K, Murray-Zmijewski
F, Liu G, Diot A, Xirodimas DP, Saville MK and Lane DP: p53
isoforms can regulate p53 transcriptional activity. Genes Dev.
19:2122–2137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Marcel V, Perrier S, Aoubala M, Ageorges
S, Groves MJ, Diot A, Fernandes K, Tauro S and Bourdon JC: Δ160p53
is a novel N-terminal p53 isoform encoded by Δ133p53 transcript.
FEBS Lett. 584:4463–4468. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fujita K, Mondal AM, Horikawa I, Nguyen
GH, Kumamoto K, Sohn JJ, Bowman ED, Mathe EA, Schetter AJ, Pine SR,
et al: p53 isoforms Delta133p53 and p53beta are endogenous
regulators of replicative cellular senescence. Nat Cell Biol.
11:1135–1142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu X: Tied up in loops: Positive and
negative autoregulation of p53. Cold Spring Harb Perspect Biol.
2:a0009842010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song W, Huo SW, Lü JJ, Liu Z, Fang XL, Jin
XB and Yuan MZ: Expression of p53 isoforms in renal cell carcinoma.
Chin Med J (Engl). 122:921–926. 2009.
|
|
22
|
Anensen N, Oyan AM, Bourdon JC, Kalland
KH, Bruserud O and Gjertsen BT: A distinct p53 protein isoform
signature reflects the onset of induction chemotherapy for acute
myeloid leukemia. Clin Cancer Res. 12:3985–3992. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hofstetter G, Berger A, Fiegl H, Slade N,
Zori A, Holzer B, Schuster E, Mobus VJ, Reimer D, Daxenbichler G,
et al: Alternative splicing of p53 and p73: The novel p53 splice
variant p53delta is an independent prognostic marker in ovarian
cancer. Oncogene. 29:1997–2004. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Boldrup L, Bourdon JC, Coates PJ, Sjöström
B and Nylander K: Expression of p53 isoforms in squamous cell
carcinoma of the head and neck. Eur J Cancer. 43:617–623. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Avery-Kiejda KA, Zhang XD, Adams LJ, Scott
RJ, Vojtesek B, Lane DP and Hersey P: Small molecular weight
variants of p53 are expressed in human melanoma cells and are
induced by the DNA-damaging agent cisplatin. Clin Cancer Res.
14:1659–1668. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nutthasirikul N, Limpaiboon T, Leelayuwat
C, Patrakitkomjorn S and Jearanaikoon P: Ratio disruption of the
133p53 and TAp53 isoform equilibrium correlates with poor clinical
outcome in intrahepatic cholangiocarcinoma. Int J Oncol.
42:1181–1188. 2013.PubMed/NCBI
|
|
27
|
Aoubala M, Murray-Zmijewski F, Khoury MP,
Fernandes K, Perrier S, Bernard H, Prats AC, Lane DP and Bourdon
JC: p53 directly transactivates Δ133p53α, regulating cell fate
outcome in response to DNA damage. Cell Death Differ. 18:248–258.
2011. View Article : Google Scholar :
|
|
28
|
Hahnvajanawong C, Chaiyagool J, Seubwai W,
Bhudhisawasdi V, Namwat N, Khuntikeo N, Sripa B, Pugk hem A and
Tassaneeyakul W: Orotate phosphoribosyl transferase mRNA expression
and the response of cholangiocarcinoma to 5-fluorouracil. World J
Gastroenterol. 18:3955–3961. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tepsiri N, Chaturat L, Sripa B, Namwat W,
Wongkham S, Bhudhisawasdi V and Tassaneeyakul W: Drug sensitivity
and drug resistance profiles of human intrahepatic
cholangiocarcinoma cell lines. World J Gastroenterol. 11:2748–2753.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Thanasai J, Limpaiboon T, Jearanaikoon P,
Sripa B, Pairojkul C, Tantimavanich S and Miwa M: Effects of
thymidine phosphorylase on tumor aggressiveness and 5-fluorouracil
sensitivity in cholangiocarcinoma. World J Gastroenterol.
16:1631–1638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Voigt W: Sulforhodamine B assay and
chemosensitivity. Methods Mol Med. 110:39–48. 2005.PubMed/NCBI
|
|
32
|
Wattanawongdon W, Hahnvajanawong C, Namwat
N, Kanchanawat S, Boonmars T, Jearanaikoon P, Leelayuwat C,
Techasen A and Seubwai W: Establishment and characterization of
gemcitabine-resistant human cholangiocarcinoma cell lines with
multidrug resistance and enhanced invasiveness. Int J Oncol.
47:398–410. 2015.PubMed/NCBI
|
|
33
|
Nakano Y, Naoe T, Kiyoi H, Kitamura K,
Minami S, Miyawaki S, Asou N, Kuriyama K, Kusumoto S, Shimazaki C,
et al: Prognostic value of p53 gene mutations and the product
expression in de novo acute myeloid leukemia. Eur J Haematol.
65:23–31. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Albaric O, Bret L, Amardeihl M and
Delverdier M: Immunohistochemical expression of p53 in animal
tumors: A methodological study using four anti-human p53
antibodies. Histol Histopathol. 16:113–121. 2001.PubMed/NCBI
|
|
35
|
Limpaiboon T, Sripa B, Wongkham S,
Bhudhisawasdi V, Chau-in S and Teerajetgul Y: Anti-p53 antibodies
and p53 protein expression in cholangiocarcinoma.
Hepatogastroenterology. 51:25–28. 2004.PubMed/NCBI
|
|
36
|
Ong CK, Subimerb C, Pairojkul C, Wongkham
S, Cutcutache I, Yu W, McPherson JR, Allen GE, Ng CC, Wong BH, et
al: Exome sequencing of liver fluke-associated cholangiocarcinoma.
Nat Genet. 44:690–693. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liang JT, Huang KC, Cheng YM, Hsu HC,
Cheng AL, Hsu CH, Yeh KH, Wang SM and Chang KJ: P53 overexpression
predicts poor chemosensitivity to high-dose 5-fluorouracil plus
leucovorin chemotherapy for stage IV colorectal cancers after
palliative bowel resection. Int J Cancer. 97:451–457. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Elsaleh H, Powell B, McCaul K, Grieu F,
Grant R, Joseph D and Iacopetta B: P53 alteration and
microsatellite instability have predictive value for survival
benefit from chemotherapy in stage III colorectal carcinoma. Clin
Cancer Res. 7:1343–1349. 2001.PubMed/NCBI
|
|
39
|
Kaeser MD, Pebernard S and Iggo RD:
Regulation of p53 stability and function in HCT116 colon cancer
cells. J Biol Chem. 279:7598–7605. 2004. View Article : Google Scholar
|
|
40
|
Reles A, Wen WH, Schmider A, Gee C,
Runnebaum IB, Kilian U, Jones LA, El-Naggar A, Minguillon C,
Schönborn I, et al: Correlation of p53 mutations with resistance to
platinum-based chemotherapy and shortened survival in ovarian
cancer. Clin Cancer Res. 7:2984–2997. 2001.PubMed/NCBI
|
|
41
|
Longley DB, Boyer J, Allen WL, Latif T,
Ferguson PR, Maxwell PJ, McDermott U, Lynch M, Harkin DP and
Johnston PG: The role of thymidylate synthase induction in
modulating p53-regulated gene expression in response to 5-
fluorouracil and antifolates. Cancer Res. 62:2644–2649.
2002.PubMed/NCBI
|
|
42
|
Goldschneider D, Blanc E, Raguénez G,
Barrois M, Legrand A, Le Roux G, Haddada H, Bénard J and Douc-Rasy
S: Differential response of p53 target genes to p73 overexpression
in SH-SY5Y neuroblastoma cell line. J Cell Sci. 117:293–301. 2004.
View Article : Google Scholar
|
|
43
|
Fang L, Lee SW and Aaronson SA:
Comparative analysis of p73 and p53 regulation and effector
functions. J Cell Biol. 147:823–830. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fulco M, Costanzo A, Merlo P, Mangiacasale
R, Strano S, Blandino G, Balsano C, Lavia P and Levrero M: p73 is
regulated by phosphorylation at the G2/M transition. J Biol Chem.
278:49196–49202. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhu J, Jiang J, Zhou W and Chen X: The
potential tumor suppressor p73 differentially regulates cellular
p53 target genes. Cancer Res. 58:5061–5065. 1998.PubMed/NCBI
|
|
46
|
He Y, Fan S, Jiang Y, Chen J, Li Z, Zhou P
and Zhou Y: Effect of exogenous p73 gene on chemosensitivity of
wild-type p53 human lung adenocarcinoma cell A549. Zhongguo Fei Ai
Za Zhi. 7:331–335. 2004.(In Chinese). PubMed/NCBI
|
|
47
|
He Y, Fan SZ, Jiang YG, Chen JM, Li ZP,
Zhou P and Zhou YG: Effect of p73 gene on chemosensitivity of human
lung adenocarcinoma cells H1299. Ai Zheng. 23:645–649. 2004.(In
Chinese). PubMed/NCBI
|
|
48
|
Adamsen BL, Kravik KL, Clausen OP and De
Angelis PM: Apoptosis, cell cycle progression and gene expression
in TP53-depleted HCT116 colon cancer cells in response to
short-term 5- fluorouracil treatment. Int J Oncol. 31:1491–1500.
2007.PubMed/NCBI
|
|
49
|
Guo X, Goessl E, Jin G, Collie-Duguid ES,
Cassidy J, Wang W and O'Brien V: Cell cycle perturbation and
acquired 5-fluorouracil chemoresistance. Anticancer Res. 28A:9–14.
2008.
|
|
50
|
Levrero M, De Laurenzi V, Costanzo A, Gong
J, Wang JY and Melino G: The p53/p63/p73 family of transcription
factors: Overlapping and distinct functions. J Cell Sci.
113:1661–1670. 2000.PubMed/NCBI
|
|
51
|
Jha AK, Nikbakht M, Jain V, Sehgal A,
Capalash N and Kaur J: Promoter hypermethylation of p73 and p53
genes in cervical cancer patients among north Indian population.
Mol Biol Rep. 39:9145–9157. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang YL, Guo XR, Shen DH, Cheng YX, Liang
XD, Chen YX and Wang Y: Expression and promotor methylation of p73
gene in ovarian epithelial tumors. Zhonghua Bing Li Xue Za Zhi.
41:33–38. 2012.PubMed/NCBI
|
|
53
|
House MG, Wistuba II, Argani P, Guo M,
Schulick RD, Hruban RH, Herman JG and Maitra A: Progression of gene
hypermethylation in gallstone disease leading to gallbladder
cancer. Ann Surg Oncol. 10:882–889. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kawano S, Miller CW, Gombart AF, Bartram
CR, Matsuo Y, Asou H, Sakashita A, Said J, Tatsumi E and Koeffler
HP: Loss of p73 gene expression in leukemias/lymphomas due to
hypermethylation. Blood. 94:1113–1120. 1999.PubMed/NCBI
|
|
55
|
Yang B, House MG, Guo M, Herman JG and
Clark DP: Promoter methylation profiles of tumor suppressor genes
in intrahepatic and extrahepatic cholangiocarcinoma. Mod Pathol.
18:412–420. 2005. View Article : Google Scholar
|