Introduction
Hypoxic conditions are associated with increased
tumor growth and metastasis, as well as poor survival in cancer
patients (1). Hypoxia inducible
factor-1α (HIF-1α) is a key protein that is expressed under hypoxic
conditions and promotes vascular remodeling by vascular endothelial
growth factor (VEGF). VEGF is one of the most critical factors that
stimulate angiogenesis. Therefore, inhibition of HIF-1α and VEGF
has demonstrated therapeutic efficacy in the treatment of several
types of cancer. Expression of VEGF is regulated by hypoxia, growth
factors, and oncogenes (2). HIF-1
is a heterodimeric transcription factor composed of HIF-1α and aryl
hydrocarbon receptor nuclear translocator (ARNT, HIF-1β), which
translocates to the nucleus and binds to hypoxia response element
(HRE) binding sites. Under normoxic conditions, HIF-1α protein is
efficiently degraded by the ubiquitin protein ligase Von
Hippel-Lindeau (VHL) and thus does not exert its effects (3). Several compounds have been tested as
inhibitors of hypoxia-induced HIF-1α expression in cancer cells
(4–6).
Various natural products and their analogues are
currently being researched as chemopreventive agents (7–9).
Sulforaphane is a potent isothiocyanate derivative found in
broccoli and other vegetables, such as Brussels sprouts and
cabbage, and has various health benefits, including anticancer and
antioxidant properties (8,10). Many studies have revealed that
sulforaphane activates phase 2 antioxidant enzymes via nuclear
factor E2-related factor 2 (Nrf2) (11–14).
In the last decade, many studies revealed that sulforaphane acts as
a chemopreventive agent by inducing apoptosis and cell cycle arrest
and inhibiting proliferation. In colon cancer cells, sulforaphane
induced apoptosis and G2/M phase cell cycle arrest (15–19).
In vivo studies showed that sulforaphane suppressed
azoxymethane-induced colonic aberrant crypt foci (ACF) (20) and prevented polyps in Apc/Min mice
(21).
Previous studies using hypoxic conditions revealed
that sulforaphane inhibited expression of HIF-1α in human tongue
squamous cancer cells and prostate cancer cells (7). However, the mechanisms by which
sulforaphane inhibits HIF-1α in colon cancer cells under hypoxic
conditions are not well understood.
In this study, we investigated the effects of
sulforaphane on expression of HIF-1α and VEGF, as well as migration
under hypoxic conditions, in human colon cancer cells.
Materials and methods
Chemicals
Sulforaphane was purchased from Sigma-Aldrich Co.
(St. Louis, MO, USA) and dissolved at a concentration of 100 mM in
dimethyl sulfoxide (DMSO) as a stock solution, which was stored
−20°C. The stock solution was diluted with cell culture medium to
the desired concentration prior to use. The maximum concentration
of DMSO did not exceed 0.1% (v/v), a concentration at which DMSO
did not influence cell growth (data not shown). The selective
proteasome inhibitor MG132 and protein synthesis inhibitor
cycloheximide (CHX) were purchased from Sigma-Aldrich.
Cell culture
HCT116 human colon cancer cells and AGS human
gastric cancer cells were obtained from American Type Culture
Collection (ATCC, Manassas, VA, USA). Cells were maintained in
RPMI-1640 medium (Hyclone, Logan, UT, USA) in a humidified
atmosphere of 5% CO2 at 37°C. The RPMI-1640 medium was
supplemented with 10% heat-inactivated fetal bovine serum (FBS,
Hyclone), 2 mM glutamine (Sigma-Aldrich), 100 U/ml penicillin
(Hyclone), and 100 μg/ml streptomycin (Hyclone).
Hypoxia experiments
Experiments to investigate the effects of hypoxia
were carried out in the hypoxia chamber of an anaerobic system
(Thormo, Marietta, OH, USA). The hypoxic condition was 1%
O2 and 5% CO2. The temperature was maintained
at 37°C. The normoxic condition was 21% O2 and 5%
CO2 (in a standard CO2 incubator). For
hypoxia experiments, HCT116 and AGS cells were grown to 50%
confluence in a standard CO2 incubator at 37°C.
Twenty-four hours prior to the experiment, cell culture media were
placed in normoxic and hypoxic chambers to allow equilibration.
Immediately before each experiment, cell culture media were
withdrawn from HCT116 and AGS cells and replaced with fresh media
that were equilibrated to normoxic and hypoxic conditions for 24
h.
MTT assay
Cell survival was quantified by an MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrasolium bromide;
Sigma-Aldrich) assay to measure mitochondrial activity in viable
cells. Cells seeded at a density of 1×105 per well were
allowed to adhere overnight, after which the culture media were
replaced with fresh media. Cells were exposed to sulforaphane at
concentrations of 12.5, 25 and 50 μM for 6 h under normoxic and
hypoxic conditions. The control groups were treated with DMSO equal
to the highest percentage (<0.1%) used in the experimental
conditions for the MTT assay. After 6 h, the medium was replaced.
MTT was freshly prepared at a concentration of 5 mg/ml in PBS and
passed through a filter (pore size, 0.2 μm). An aliquot of 2 ml of
MTT stock solution was added to each well and the plate was
incubated at 37°C for 4 h in a humidified 5% CO2
atmosphere. After 2 h, media were removed. To each well, 2 ml of
DMSO was added in order to solubilize the formazan crystals, which
were measured after 10 min. The optical density of each well was
measured with a spectrophotometer equipped with a 540-nm
filter.
Protein preparation and western blot
analysis
Cells were harvested and washed twice in PBS at 4°C.
Total cell lysates were lysed in lysis buffer [40 mM Tris (pH 8.0),
120 mM NaCl, 0.5% NP-40, 0.1 mM sodium orthovanadate, 2 μg/ml
aprotinin, 2 μg/ml leupeptin and 100 μg/ml phenymethylsulfonyl
fluoride (PMSF)]. The supernatants were collected and protein
concentrations were measured with protein assay reagents (Pierce,
Rockford, IL, USA). Equal amounts of protein were denatured by
boiling at 100°C for 5 min in sample buffer (0.5 M Tris-HCl, pH
6.8, 4% sodium dodecyl sulfate (SDS), 20% glycerol, 0.1%
bromophenol blue, 10% β-mercaptoethanol) at a 1:1 ratio. Equal
amount of the total proteins were subjected to 6–15%
SDS-polyacrylamide gel electrophoresis and transferred to
polyvinylidene difluoride membranes. The membranes were blocked
with 5% non-fat dry milk in Tris-buffered saline with Tween-20
buffer (TBS-T) (20 mM Tris, 100 mM NaCl, pH 7.5, and 0.1% Tween-20)
for 1 h at room temperature, after which the membranes were
incubated overnight at 4°C with the primary antibodies. The
membranes were washed thrice for 10 min with TBS-T buffer and
incubated for 1 h with horseradish peroxidase-conjugated
anti-rabbit or anti-mouse immunoglobin (Santa Cruz Biotechnology
Inc., Santa Cruz, CA, USA). The membranes were washed 4 times for
10 min with TBS-T buffer. Antigen-antibody complexes were detected
using the enhanced chemiluminescence (ECL) detection system (GE
Healthcare Biosciences, Pittsburgh, PA, USA).
ELISA assay
To analyze VEGF secretion, HCT116 cells were seeded
in 12-well plates, cultured to 50% confluence, pretreated with
sulforaphane or DMSO (control treatment) for 30 min, and switched
to fresh media that were pre-conditioned in normoxic or hypoxic
conditions. Cells were incubated with or without sulforaphane at
the corresponding conditions for 24 h. The supernatants in the
wells were collected, cleared by centrifugation, and stored at
−20°C. ELISA was performed using the human VEGF Quantikine kit
(R&D Systems, Minneapolis, MN, USA) according to the
manufacturer's protocol. Recombinant human VEGF was used for
calibration. Experiments were carried out at least 3 times in
triplicate.
In vitro migration assay
ell migration assays were performed using 24-well
modified Boyden chambers (Corning Life Science, Corning, NY, USA).
A cell migration kit was used for the cell migration assay
according to the manufacturer's protocol. Confluent cells were
added to the inner chamber of the insert in 100 μl of serum-free
medium. Medium (600 μl) with 10% FBS was added to the lower
chamber. To determine the effect of sulforaphane on cell migration,
5 or 10 μM sulforaphane was added to the lower chamber (DMSO was
used as a control). Cells were fixed and stained with the
Diff-Quick Stain kit (Baxter, McGaw Park, IL, USA) following the
procedure described by the manufacturer. The number of migrating
cells was counted under a microscope (×200 magnification) and the
results were expressed as the percentage of invaded cells per field
for each condition.
Statistical analysis
Results are expressed as the mean ± SD of 3 separate
experiments. Data were analyzed by Student's t-test. Means were
considered significantly different at p<0.05 or p<0.01.
Results
Sulforaphane inhibits hypoxia-induced
HIF-1a in HCT116 and AGS cells
To investigate the effects of hypoxia-induced
HIF-1α, HCT116 and AGS cells were grown to 70% confluence in a
standard CO2 incubator at 37°C (normoxic conditions of
21% O2 and 5% CO2) and transferred to a
hypoxia chamber (1% O2 and 5% CO2). Cell
cultures were exposed to hypoxia and harvested at various
time-points. Hypoxic conditions dramatically induced HIF-1α protein
expression in HCT116 and AGS cells (Fig. 1A). HIF-1α protein induction was
observed in cells 2 h after they were transferred to the hypoxic
environment and become pronounced from the 2 h time-point through
the 8 h time-point, reaching a maximum level at the 4 h and 6 h
time-points; therefore, the 6 h time-point was selected for further
experiments in HCT116 cells, while the 8 h time-point was selected
for AGS cells.
The next study was performed to assess whether
sulforaphane suppressed the observed responses to hypoxic
conditions in HCT116 and AGS cells. Cells were pretreated with
medium containing 12.5–100 μM sulforaphane for 1 h in normoxic
conditions and transferred to a hypoxia chamber. Sulforaphane
significantly inhibited HIF-1α expression in HCT116 and AGS cells
(Fig. 1B). Interestingly, low
concentration of sulforaphane slightly induced hypoxia-induced
HIF-1α in HCT116 cells. Therefore, to investigate the effects of
sulforaphane-induced cytotoxicity under normoxic and hypoxic
conditions and determine whether cytotoxicity is responsible for
suppression of HIF-1α accumulation, cell viability was determined
by MTT assay. No significant concentration-dependent reduction of
viability was observed when HCT116 and AGS cells were treated with
various concentrations of sulforaphane for 6 and 8 h under normoxic
and hypoxic conditions (data not shown). These data indicated that
the decrease in HIF-1α abundance under normoxic and hypoxic
conditions was not due to cell death.
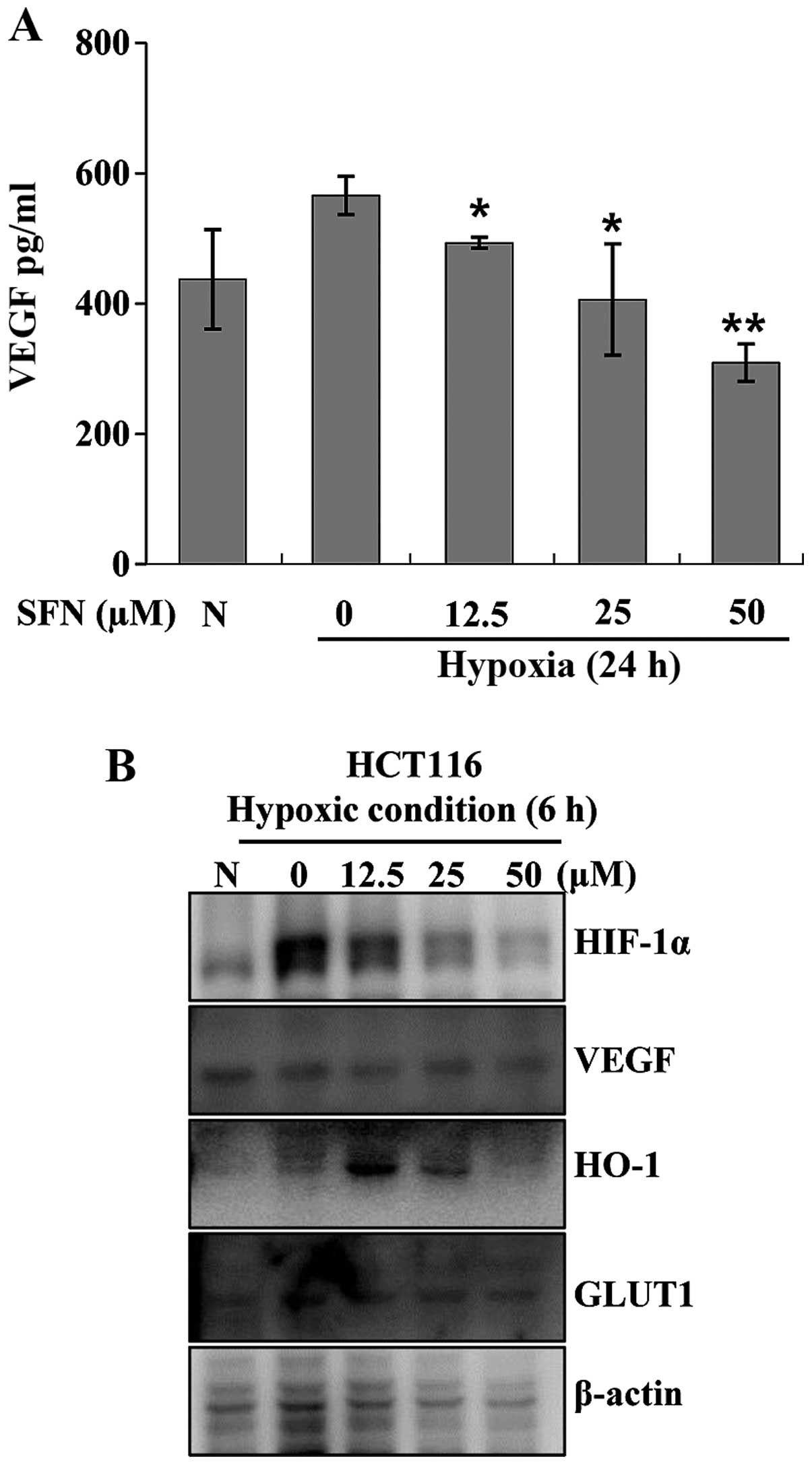
Sulforaphane suppresses hypoxia-induced
VEGF expression in HCT116 cells
VEGF is a target gene of HIF-1 that plays a crucial
role in tumor angiogenesis. HIF-1 regulates VEGF expression at the
transcriptional level (22). To
determine whether sulforaphane inhibits VEGF expressions in HCT116
cells, VEGF transcript abundance was measured using an ELISA kit.
Cells were incubated under hypoxic conditions with or without
12.5–50 μM sulforaphane. After 24 h of treatment, cell culture
media were collected and VEGF transcript abundance was measured.
VEGF transcript was increased under hypoxic conditions. However,
VEGF induction was decreased in a concentration-dependent manner by
sulforaphane (Fig. 2A).
Sulforaphane also inhibited hypoxia-related target protein
expressions such as VEGF, heme oxygenase (HO)-1 and glucose
transporter 1 (GLUT1) (Fig.
2B).
Sulforaphane affects the stability of
HIF-1α protein in HCT116 cells
To evaluate the mechanism by which sulforaphane
inhibits HIF-1α expression, HCT116 cells were exposed to hypoxic
conditions to induce HIF-1α protein expression. The exposure to
hypoxic conditions was necessary because very little HIF-1α is
detectable under normoxic conditions due to rapid protein
degradation. Cyclohexamide (CHX), a protein synthesis inhibitor, is
widely used in protein stability studies. HCT116 cells were
incubated for 6 h under hypoxic conditions and treated with CHX for
30 min in the presence or absence of 50 μM sulforaphane. Cells were
harvested at various time-points and cell lysates were subjected to
western blot analysis using anti-HIF-1α antibodies. Under hypoxic
conditions, the half-life of HIF-1α was not longer than 1 h when
the cells were treated with CHX alone (Fig. 3A). However, the half-life of HIF-1α
was ~15 min when the cells were treated with a combination of
sulforaphane and CHX (Fig. 3B).
These data revealed that sulforaphane reduced the half-life of
HIF-1α protein under hypoxic conditions, indicating that
sulforaphane treatment regulates HIF-1α expression by decreasing
protein stability. These results demonstrate that sulforaphane
affected hypoxia-induced HIF-1α protein stability in HCT116
cells.
Next, to examine whether sulforaphane-induced HIF-1α
protein degradation is mediated by the proteasome degradation
pathway, HCT116 cells were treated with proteasome inhibitor MG132
for 30 min, followed by treatment with medium containing
sulforaphane for 30 min, after which the cells were exposed to
hypoxic conditions for 6 h, washed with PBS, and lysed inside the
hypoxia chamber. Degradation of HIF-1α protein induced by
sulforaphane under hypoxic conditions was not completely prevented
by MG132 (Fig. 3C and D). These
data indicate that sulforaphane did not induce HIF-1α protein
degradation through the proteasome degradation pathway.
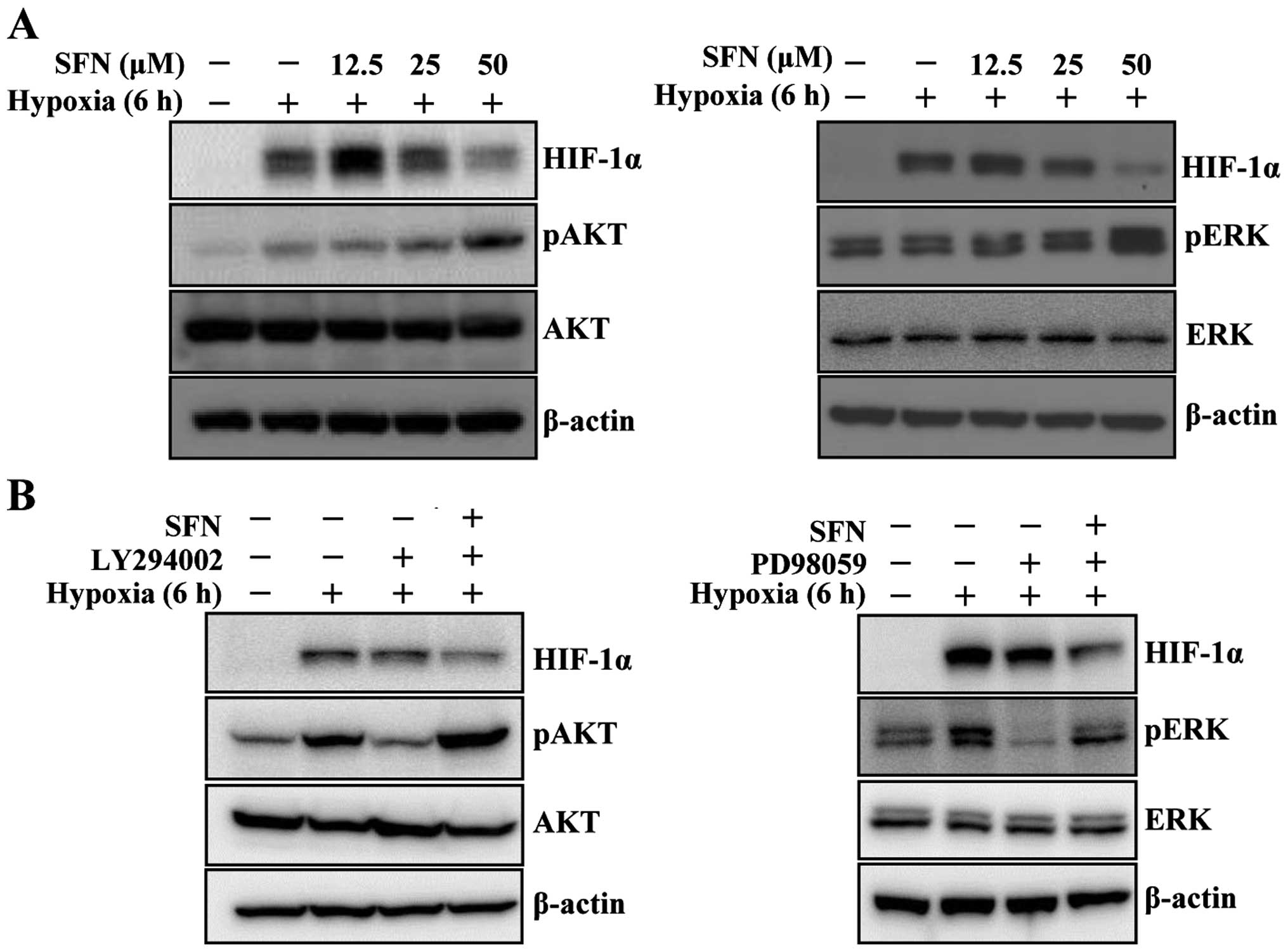
AKT and ERK signaling pathway is not
involved in down-regulation of HIF-1α protein by sulforaphane under
hypoxic conditions
Accumulating evidence has shown that multiple
signaling pathways, particularly phosphatidylinositol 3-kinase
(PI3K)/AKT and mitogen-activated protein kinase (MAPK)/ERK
pathways, are involved in hypoxia-induced HIF-1α protein
accumulation and downstream target gene expression (23). To determine whether sulforaphane
inhibits hypoxia-mediated activation of AKT, HCT116 cells were
pretreated with various concentrations of sulforaphane for 1 h
under normoxic conditions, followed by incubation for 6 h under
hypoxic conditions, after which the cells were washed with PBS and
lysed inside the hypoxia chamber. Sulforaphane did not inhibit
phosphorylation of AKT and ERK under hypoxic conditions (Fig. 4A). Interestingly, hypoxia-induced
HIF-1α was slightly increased by low concentration of sulforaphane.
To determine whether sulforaphane activates AKT and ERK, cells were
exposed to PI3K inhibitor LY294002 and ERK inhibitor PD98059.
LY294002 and PD98059 significantly decreased the elevated levels of
HIF-1α, pAKT, and pERK protein induced by hypoxic conditions
(Fig. 4B). However, combination of
the inhibitors with sulforaphane dramatically restored pAKT and
pERK expression under hypoxic conditions (Fig. 4B). These results show that the
decrease in HIF-1α expression produced by sulforaphane in HCT116
cells under hypoxic conditions is not mediated by regulation of
PI3K and ERK pathways.
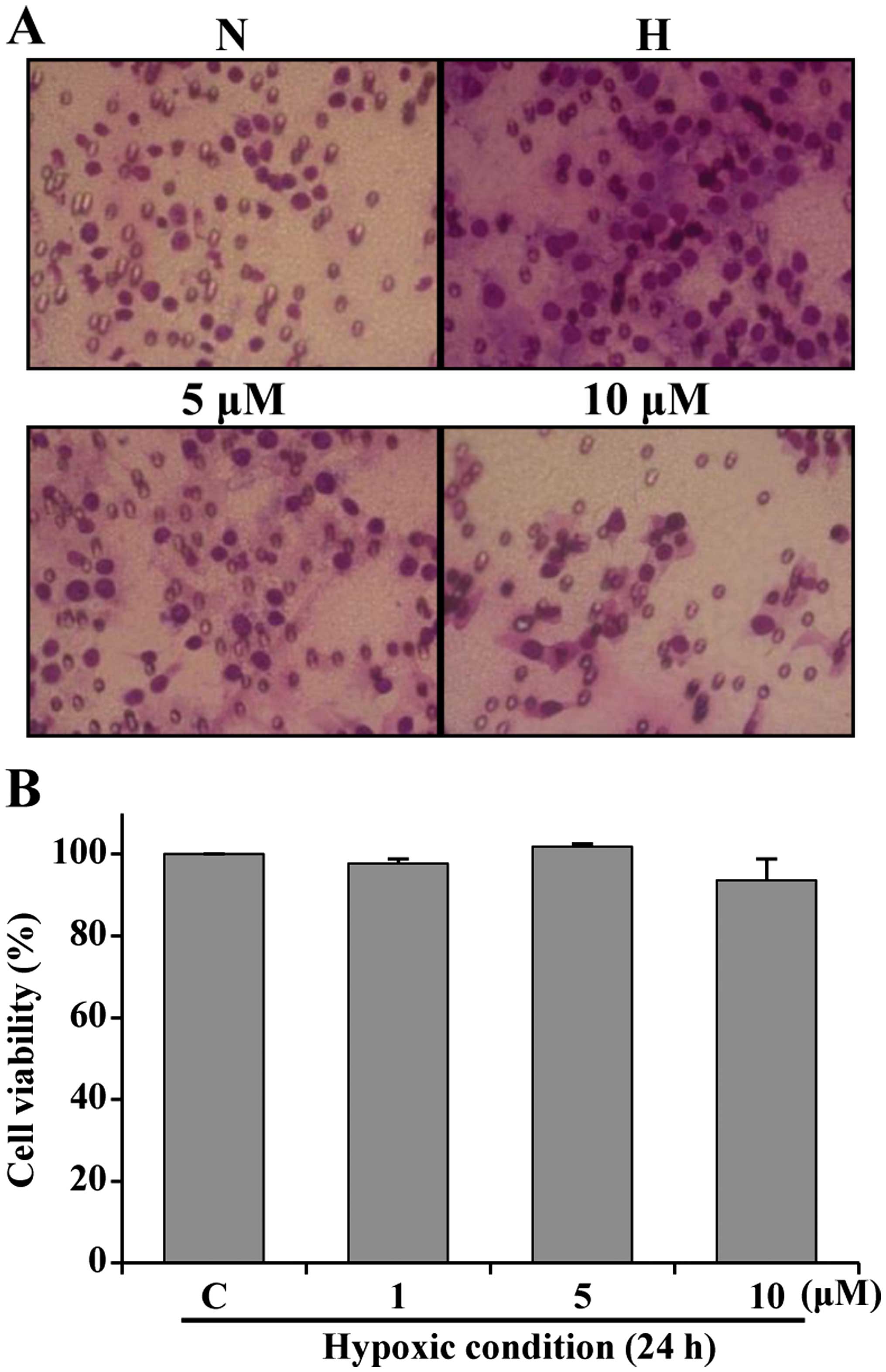
Sulforaphane inhibits hypoxia-induced
migration
Hypoxic conditions enhance metastasis of several
types of cancer cells, including colon and breast cancer cells.
In vitro migration experiments were performed to determine
whether sulforaphane inhibits HCT116 cell motility. HCT116 cells
were treated with sulforaphane under normoxic and hypoxic
conditions. As shown in Fig. 5A,
there was little cell migration under normoxic conditions. However,
cell migration increased under hypoxic conditions. Treatment with 5
and 10 μM sulforaphane suppressed migration activity. Sulforaphane
was not cytotoxic at concentrations of 5 and 10 μM under hypoxic
conditions (Fig. 5B). These
results indicate that treatment with sulforaphane suppressed
hypoxia-induced HCT116 cell migration.
Discussion
Phytochemicals are important cancer prevention
tools. The chemopreventive mechanisms of many phytochemicals,
including sulforaphane, are not well understood. Sulforaphane
induces phase 2 antioxidant enzymes such as glutathione
transferases, NAD(P)H:quinone reductase, epoxide hydrolase, heme
oxygenase, and UDP-glucuronosyltranferase, which play important
roles in detoxification of electrophiles and protect against
carcinogenesis and mutagenesis (11). During the last decade, many studies
on sulforaphane as a chemopreventive agent in various cancer cell
lines were released. However, the mechanism by which sulforaphane
inhibits HIF-1α expression under hypoxic conditions is
controversial and not well understood. In this study, we evaluated
inhibition of HIF-1α and VEGF expression under hypoxic conditions
by sulforaphane in human colon cancer cells.
Hypoxic conditions rapidly induced expression of
HIF-1α in HCT116 human colon cancer cells and AGS human gastric
cancer cells (Fig. 1A). However,
sulforaphane inhibited HIF-1α expression concentration-dependently
in both cell lines (Fig. 1B). Yao
et al showed similar results in human tongue and prostate
cancer cells (7). VEGF is a key
protein downstream of HIF-1 under hypoxic conditions. Induction of
VEGF expression under hypoxic conditions causes sprouting of new
blood vessels from existing endothelia, which is essential for
wound repair, organ regeneration, embryonic vascular system
development, and a variety of pathological conditions, including
tumor angiogenesis and metastasis of various solid tumors (24,25).
Treatment with sulforaphane under hypoxic conditions inhibited VEGF
activity in HCT116 cells (Fig.
2A). Sulforaphane also slightly suppressed HIF-1-regulated gene
expressions such as VEGF, HO-1 and GLUT1 (Fig. 2B).
Degradation of HIF-1α protein under normoxic
conditions is tightly regulated by ubiquitination and the 26S
proteasomal degradation system. Under hypoxic conditions,
ubiquitination and degradation of HIF-1α protein is suppressed,
leading to stabilization and accumulation of HIF-1α protein and
nuclear translocation (26). In
this study, expression of HIF-1α under hypoxic conditions was
inhibited by sulforaphane in HCT116 cells (Fig. 2B). Moreover, sulforaphane
significantly shortened the half-life of hypoxia-induced HIF-1α
protein (Fig. 3B). However,
inhibition of hypoxia-induced HIF-1α protein accumulation by
sulforaphane was not abolished in the presence of MG132, a potent
inhibitor of the 26S proteasome (Fig.
3C). These results were similar to those reported by others
previously (7). There are several
possible explanations for these results. One possible explanation
is that the lysosome pathway is involved in degradation of HIF-1α
following sulforaphane treatment under hypoxic conditions (27). Treatment with lysosomal inhibitors,
including bafilomycin A1 and chloroquine, induces HIF-1α expression
and activity, while hypoxic conditions induce chaperone-mediated
autophagy and lysosomal biogenesis in cancer cells (28). In addition, some studies have
reported that treatment with sulforaphane strongly enhanced the
proteasome pathway (29–31). Jung et al reported that
hypoxia-induced HIF-1α expression was inhibited by rhapontigenin
through interaction with von Hippel-Lindau in PC3 cells (32). Degradation of HIF-1α induced by
sulforaphane under hypoxic conditions did not involve the 26S
proteasome pathway in our culture system. Further studies are
required to determine the mechanisms by which HIF-1α degradation
induced by sulforaphane under hypoxic conditions exerts potent
anti-angiogenic activity.
Accumulating evidence indicates that hypoxic
conditions activate several signaling pathways, including PI3K/AKT,
glycogen synthase kinase 3 beta (GSK-3β), and ERK (4,5). In
this study, hypoxic conditions induced PI3K/AKT and ERK in HCT116
cells (Fig. 4A). Treatment of
HCT116 cells with PI3K/AKT inhibitor LY294002 and ERK inhibitor
PD98059 confirmed that both pathways are important for
hypoxia-mediated HIF-1α stabilization. However, treatment with
sulforaphane under hypoxic conditions induced pAKT and pERK, even
in the presence of LY294002 and PD98059 (Fig. 4B). Several studies in cancer cells
have shown that sulforaphane is an Nrf2 activator (8,33,34).
Treatment with sulforaphane strongly and concentration-dependently
induced expression of Nrf2 under hypoxic conditions in our culture
system (data not shown). Treatment with sulforaphane causes
autophagy in various human cancer cell lines, including colon,
pancreas, and prostate cancer cells (9,35,36).
Induction of PI3K and ERK pathway activity could be possible when
sulforaphane causes autophagy under hypoxic conditions. However,
more studies are necessary to determine the effects of sulforaphane
on PI3K and ERK pathways.
Accumulating evidence in solid tumors has shown that
HIF-1α overexpression, either as a result of intratumoral hypoxia
or genetic alterations, activates gene transcription, and the
protein products contribute to basement membrane migration and
invasion (25,37,38).
In this study, treatment with sulforaphane inhibited
hypoxia-induced migration by HCT116 cells (Fig. 5A). The sulforaphane concentrations
tested in the migration assay did not affect cell viability under
hypoxic conditions (Fig. 5B).
However, extensive studies are needed to identify the genes that
are directly or indirectly involved in sulforaphane-regulated
cancer cell migration and metastasis in response to hypoxic
conditions.
In this study, we showed that sulforaphane inhibited
HIF-1α protein expression in HCT116 and AGS cells under hypoxic
conditions. In addition, sulforaphane treatment inhibited
hypoxia-induced VEGF expression in HCT116 cells. Inhibition of
HIF-1α protein expression by sulforaphane was associated with
destabilization of HIF-1α protein and prevention of HIF-1α target
gene activation. Sulforaphane also inhibited hypoxia-induced cell
migration. These data suggest that sulforaphane may inhibit human
colon cancer angiogenesis and migration by inhibiting HIF-1α and
VEGF expression under hypoxic conditions. Taken together, these
results indicate that sulforaphane might be a new potent
chemopreventive agent against human cancer cells.
Acknowledgements
This study was financially supported by the 2015
Post-doc Development Program of Pusan National University.
References
|
1
|
Semenza GL: Hypoxia and human disease -
and the Journal of Molecular Medicine. J Mol Med Berl.
85:1293–1294. 2007. View Article : Google Scholar
|
|
2
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Semenza GL: Hypoxia-inducible factor 1
(HIF-1) pathway. Sci STKE. 2007:cm82007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mirzoeva S, Kim ND, Chiu K, Franzen CA,
Bergan RC and Pelling JC: Inhibition of HIF-1 alpha and VEGF
expression by the chemopreventive bioflavonoid apigenin is
accompanied by Akt inhibition in human prostate carcinoma PC3-M
cells. Mol Carcinog. 47:686–700. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim DH, Hossain MA, Kim MY, Kim JA, Yoon
JH, Suh HS, Kim GY, Choi YH, Chung HY and Kim ND: A novel
resveratrol analogue, HS-1793, inhibits hypoxia-induced HIF-1α and
VEGF expression, and migration in human prostate cancer cells. Int
J Oncol. 43:1915–1924. 2013.PubMed/NCBI
|
|
6
|
Chen MC, Lee CF, Huang WH and Chou TC:
Magnolol suppresses hypoxia-induced angiogenesis via inhibition of
HIF-1α/VEGF signaling pathway in human bladder cancer cells.
Biochem Pharmacol. 85:1278–1287. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yao H, Wang H, Zhang Z, Jiang BH, Luo J
and Shi X: Sulforaphane inhibited expression of hypoxia-inducible
factor-1alpha in human tongue squamous cancer cells and prostate
cancer cells. Int J Cancer. 123:1255–1261. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jo GH, Kim GY, Kim WJ, Park KY and Choi
YH: Sulforaphane induces apoptosis in T24 human urinary bladder
cancer cells through a reactive oxygen species-mediated
mitochondrial pathway: The involvement of endoplasmic reticulum
stress and the Nrf2 signaling pathway. Int J Oncol. 45:1497–1506.
2014.PubMed/NCBI
|
|
9
|
Wang M, Zhu JY, Chen S, Qing Y, Wu D, Lin
YM, Luo JZ, Han W and Li YQ: Effects of co-treatment with
sulforaphane and autophagy modulators on uridine
5′-diphospho-glucuronosyltransferase 1A isoforms and cytochrome
P450 3A4 expression in Caco-2 human colon cancer cells. Oncol Lett.
8:2407–2416. 2014.PubMed/NCBI
|
|
10
|
Juge N, Mithen RF and Traka M: Molecular
basis for chemoprevention by sulforaphane: A comprehensive review.
Cell Mol Life Sci. 64:1105–1127. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fahey JW and Talalay P: Antioxidant
functions of sulforaphane: A potent inducer of Phase II
detoxication enzymes. Food Chem Toxicol. 37:973–979. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kraft AD, Johnson DA and Johnson JA:
Nuclear factor E2-related factor 2-dependent antioxidant response
element activation by tert-butylhydroquinone and sulforaphane
occurring preferentially in astrocytes conditions neurons against
oxidative insult. J Neurosci. 24:1101–1112. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Boddupalli S, Mein JR, Lakkanna S and
James DR: Induction of phase 2 antioxidant enzymes by broccoli
sulforaphane: Perspectives in maintaining the antioxidant activity
of vitamins A, C, and E. Front Genet. 3:72012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen X, Liu J and Chen SY: Sulforaphane
protects against ethanol-induced oxidative stress and apoptosis in
neural crest cells by the induction of Nrf2-mediated antioxidant
response. Br J Pharmacol. 169:437–448. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gamet-Payrastre L, Li P, Lumeau S, Cassar
G, Dupont MA, Chevolleau S, Gasc N, Tulliez J and Tercé F:
Sulforaphane, a naturally occurring isothiocyanate, induces cell
cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer
Res. 60:1426–1433. 2000.PubMed/NCBI
|
|
16
|
Pledgie-Tracy A, Sobolewski MD and
Davidson NE: Sulforaphane induces cell type-specific apoptosis in
human breast cancer cell lines. Mol Cancer Ther. 6:1013–1021. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jeon YK, Yoo DR, Jang YH, Jang SY and Nam
MJ: Sulforaphane induces apoptosis in human hepatic cancer cells
through inhibition of
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase4, mediated by
hypoxia inducible factor-1-dependent pathway. Biochim Biophys Acta.
1814:1340–1348. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Parnaud G, Li P, Cassar G, Rouimi P,
Tulliez J, Combaret L and Gamet-Payrastre L: Mechanism of
sulforaphane-induced cell cycle arrest and apoptosis in human colon
cancer cells. Nutr Cancer. 48:198–206. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang M, Chen S, Wang S, Sun D, Chen J, Li
Y, Han W, Yang X and Gao HQ: Effects of phytochemicals sulforaphane
on uridine diphosphate-glucuronosyltransferase expression as well
as cell-cycle arrest and apoptosis in human colon cancer Caco-2
cells. Chin J Physiol. 55:134–144. 2012.PubMed/NCBI
|
|
20
|
Chung FL, Conaway CC, Rao CV and Reddy BS:
Chemo-prevention of colonic aberrant crypt foci in Fischer rats by
sulforaphane and phenethyl isothiocyanate. Carcinogenesis.
21:2287–2291. 2000. View Article : Google Scholar
|
|
21
|
Hu R, Khor TO, Shen G, Jeong WS, Hebbar V,
Chen C, Xu C, Reddy B, Chada K and Kong AN: Cancer chemoprevention
of intestinal polyposis in ApcMin/+ mice by sulforaphane, a natural
product derived from cruciferous vegetable. Carcinogenesis.
27:2038–2046. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Semenza GL: Regulation of hypoxia-induced
angiogenesis: A chaperone escorts VEGF to the dance. J Clin Invest.
108:39–40. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Semenza GL: Involvement of
hypoxia-inducible factor 1 in human cancer. Intern Med. 41:79–83.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Denes V, Lakk M, Makarovskiy A, Jakso P,
Szappanos S, Graf L, Mandel L, Karadi I and Geck P: Metastasis
blood test by flow cytometry: In vivo cancer spheroids and the role
of hypoxia. Int J Cancer. 136:1528–1536. 2015. View Article : Google Scholar
|
|
25
|
Liu ZJ, Semenza GL and Zhang HF:
Hypoxia-inducible factor 1 and breast cancer metastasis. J Zhejiang
Univ Sci B. 16:32–43. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pugh CW and Ratcliffe PJ: The von
Hippel-Lindau tumor suppressor, hypoxia-inducible factor-1 (HIF-1)
degradation, and cancer pathogenesis. Semin Cancer Biol. 13:83–89.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rudolf E and Cervinka M: Sulforaphane
induces cytotoxicity and lysosome- and mitochondria-dependent cell
death in colon cancer cells with deleted p53. Toxicol In Vitro.
25:1302–1309. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hubbi ME, Hu H, Kshitiz, Ahmed I,
Levchenko A and Semenza GL: Chaperone-mediated autophagy targets
hypoxia-inducible factor-1α (HIF-1α) for lysosomal degradation. J
Biol Chem. 288:10703–10714. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kwak MK, Cho JM, Huang B, Shin S and
Kensler TW: Role of increased expression of the proteasome in the
protective effects of sulforaphane against hydrogen
peroxide-mediated cytotoxicity in murine neuroblastoma cells. Free
Radic Biol Med. 43:809–817. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gan N, Wu YC, Brunet M, Garrido C, Chung
FL, Dai C and Mi L: Sulforaphane activates heat shock response and
enhances proteasome activity through up-regulation of Hsp27. J Biol
Chem. 285:35528–35536. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Balasubramanian S, Chew YC and Eckert RL:
Sulforaphane suppresses polycomb group protein level via a
proteasome-dependent mechanism in skin cancer cells. Mol Pharmacol.
80:870–878. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jung DB, Lee HJ, Jeong SJ, Lee HJ, Lee EO,
Kim YC, Ahn KS, Chen CY and Kim SH: Rhapontigenin inhibited hypoxia
inducible factor 1 alpha accumulation and angiogenesis in hypoxic
PC-3 prostate cancer cells. Biol Pharm Bull. 34:850–855. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kensler TW, Egner PA, Agyeman AS,
Visvanathan K, Groopman JD, Chen JG, Chen TY, Fahey JW and Talalay
P: Keap1-nrf2 signaling: A target for cancer prevention by
sulforaphane. Top Curr Chem. 329:163–177. 2013. View Article : Google Scholar :
|
|
34
|
Zhang C, Su ZY, Khor TO, Shu L and Kong
AN: Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP
C1 cells through epigenetic regulation. Biochem Pharmacol.
85:1398–1404. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Herman-Antosiewicz A, Johnson DE and Singh
SV: Sulforaphane causes autophagy to inhibit release of cytochrome
c and apoptosis in human prostate cancer cells. Cancer Res.
66:5828–5835. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Naumann P, Fortunato F, Zentgraf H,
Büchler MW, Herr I and Werner J: Autophagy and cell death signaling
following dietary sulforaphane act independently of each other and
require oxidative stress in pancreatic cancer. Int J Oncol.
39:101–109. 2011.PubMed/NCBI
|
|
37
|
Zhang J, Cao J, Ma S, Dong R, Meng W, Ying
M, Weng Q, Chen Z, Ma J, Fang Q, et al: Tumor hypoxia enhances
non-small cell lung cancer metastasis by selectively promoting
macrophage M2 polarization through the activation of ERK signaling.
Oncotarget. 5:9664–9677. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Matsuo Y, Ding Q, Desaki R, Maemura K,
Mataki Y, Shinchi H, Natsugoe S and Takao S: Hypoxia inducible
factor-1 alpha plays a pivotal role in hepatic metastasis of
pancreatic cancer: An immunohistochemical study. J Hepatobiliary
Pancreat Sci. 21:105–112. 2014. View
Article : Google Scholar
|