Introduction
The tumor suppressor gene TP53 plays a
pivotal role in maintaining and regulating normal cellular
functions. The acquired TP53 mutations are the most common
genetic alterations in human cancer, which are mostly missense
mutations (1). Some p53 mutants
not only result in the loss of wild-type p53 activity, but also may
acquire new oncogenic properties known as gain-of-function. The
mutation usually leads to the formation of mutant p53 proteins,
which often accumulate at high levels in tumors (2–4). In
fact, immunohistochemical detection of p53 in tumors usually
indicates TP53 missense mutation and provides prognostic and
predictive information. As the notion that mutant p53 accumulation
augments their oncogenic potential, the field of molecular
modulators of mutant p53 level and activity is gaining interest. A
prominent notion underlying mutant p53 accumulation is that the
mutations in TP53 abrogate its ability to transactivate
MDM2, which typically regulates the ubiquitin-mediated
degradation of p53 (5,6). However, recent in vivo data
challenge this hypothesis by demonstrating that mutant Trp53
knock in mice (Trp53 encodes mouse p53) do not have mutant
p53 accumulation in normal tissues, whereas mutant p53 levels are
increased in most tumors (7). The
findings indicated that TP53 mutations alone are
insufficient for the accumulation of mutant p53 and that additional
events are required to prevent mutant p53 degradation. It has been
reported that molecules such as heat shock proteins (HSP90) or
phosphatase and tensin homolog (PTEN) interact with mutant p53 and
lead to its accumulation in tumors (8,9).
S100A4 (also known as Mts1) belongs to the S100
family of Ca2+-binding proteins that are overexpressed
in a multitude of cancers and that are accompanied by increased
metastatic capacity (10–12). S100A4 is involved in cancer
progression and metastasis through interaction with target
proteins. Using in vitro or in vivo binding assays,
several groups have demonstrated the interaction between wild-type
p53 and S100A4, and the functional consequence of wild-type
p53-S100A4 interaction has been reported. Mutant p53 and S100A4
interaction in mouse mammary cancer CSML-100 cells has also been
reported (13). However, whether
mutant p53 and S100A4 can interact in human cancer cells and the
functional consequences have yet to be revealed. The human gastric
cancer cell line MKN1 harbors mutant p53V143A (14). We investigated whether S100A4 and
mutant p53 interact in MKN1 cells and whether the interaction
contributes to the accumulation of mutant p53. We also studied the
role of S100A4 in regulating mutant p53-related molecular and
cellular effects.
Materials and methods
Cell culture
The human gastric cancer cell line MKN1 was kindly
provided by professor Kazunari K. Yokoyama (Riken, The Institute of
Physical and Chemical Research, Saitama, Japan). Cells were
cultured in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA)
supplemented with 10% fetal calf serum in a 37ºC humidified
incubator with 5% CO2.
Double immunofluorescence
MKN1 cells were seeded on glass coverslips
(1×105 cells/ml). After 24 h, the cells were fixed with
methyl alcohol and acetone, and then blocked with 10% normal goat
serum. The cell monolayer was treated overnight at 4ºC with primary
antibodies: rabbit anti-S100A4 antibody (1:100 dilution; Lab
Vision, Fremont, CA USA) and mouse anti-p53 antibody (DO-1; Santa
Cruz Biotechnology, Santa Cruz, CA, USA; 1:100 dilution). The p53
antibody (DO-1) is a mouse monoclonal antibody raised against amino
acids 11–25 of p53 of human origin, which was recommended for
detection of wild and mutant p53 of human origin according to the
instruction book. In the present study, we used DO-1 to detect
mutant p53V143A expression in MKN-1 cells. After
incubating in primary antibodies, the cells were treated with
fluorescein isothiocyanate-conjugated secondary goat anti-rabbit
IgG and tetramethylrhodamine isothiocyanate-conjugated goat
anti-mouse IgG (Sigma, St. Louis, MO, USA). Lastly, the nuclei were
stained with diaminophenylindole (DAPI) in phosphate-buffered
saline (PBS) for 10 min. Specimens were examined under a Leica TCS
SP2 AOBS confocal laser microscope (Leica Microsystems, Wetzlar,
Germany).
Co-immunoprecipitation (Co-IP)
Cells were washed with PBS and lysed in Triton lysis
buffer. Cell extracts were precleared by incubating with 50 μl
Protein G PLUS-Agarose (Santa Cruz Biotechnology) for 30 min at 4ºC
and centrifuged. Precleared cell extracts were incubated with 4 μg
mouse anti-p53 antibody or rabbit anti-S100A4 antibody at 4ºC for 1
h. IgG was used as the control for the Co-IP. Thereafter, 70 μl
resuspended Protein G PLUS-Agarose was added to the lysate-antibody
mix and incubated overnight at 4ºC on a rotating wheel. After
incubation, the beads were centrifuged and the supernatant was
discarded. The beads were washed four times with PBS, resuspended
and boiled in 20 μl SDS sample buffer, and immunoblotted with
anti-S100A4 antibody or anti-p53 antibody (1:200 dilution).
Plasmid construction and cell transient
transfection
The pCMV-Neo-Bam p53V143A expression
vector was a gift from Professor Bert Vogelstein (Johns Hopkins
University, Baltimore, MD, USA). The S100A4-specific shRNA
expression vector was constructed in a previous study by our group
(15). The double-stranded shRNA
oligo was cloned into a pSilencer 4.1-CMV neo vector (Ambion,
Austin, TX, USA) to construct pS100A4-shRNA. A scrambled sequence
without significant homology to human gene sequences was used as
the control (pControl-shRNA). Cell transfections were carried out
using Lipofectamine 2000 (Invitrogen) according to the
manufacturer's instruction.
RNA extraction and semi-quantitative
RT-PCR
Total cellular RNA was extracted using TRIzol
reagent (Invitrogen); 1 μg RNA was reverse-transcribed using
First-Strand cDNA Synthesis kit (Promega, Madison, WI, USA). The
newly synthesized complementary DNA was amplified by PCR. Primers
specific for human TP53, S100A4 and β-actin
were designed as follows: (TP53) sense,
5′-CAGCCAAGTCTGTGACTTGCACGTAC-3′ and antisense,
5′-CTATGTCGAAAAGTGTTTCTGTCATC-3′; (S100A4) sense,
5′-GATGTGATGGTGTCCACCTT-3′ and antisense,
5′-ATTTCTTCCTGGGCTGCTTA-3′; (β-actin) sense,
5′-CTCTTCCAGCCTTCCTTCCT-3′ and antisense,
5′-CACCTTCACCGTTCCAGTTT-3′. Amplification cycles were: 95ºC for 5
min, and then 30 cycles at 95ºC for 30 sec, 58ºC for 30 sec, and
72ºC for 30 sec, followed by 72ºC for 10 min. Aliquots of the PCR
product were electrophoresed on 1.5% agarose gels, and PCR
fragments were visualized by ethidium bromide staining.
Quantitative real-time RT-PCR
The RNA extraction and reverse transcription
reaction were performed as described above. Quantitative real-time
PCR analysis was performed by using SYBR Premix Ex Taq II (Takara
Bio, Dalian, China). Reactions were processed and analyzed on an
ABI 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA,
USA). The sense and antisense primers used for Id2,
c-Myc, and glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) were 5′-TCAGCCTGCATCACCAGAGA-3′ and
5′-CTGCAAGGACAGGATGCTGATA-3′; 5′-ACCAGATCCCGGAGTTGGAA-3′ and
5′-CGTCGTTTCCGCAACAAGTC-3′; and 5′-ATCATCAGCAATGCCTCC-3′ and
5′-CATCACGCCACAGTTTC-3′, respectively.
Western blotting
Whole cell extracts were prepared from cells by
homogenizing cells in a lysis buffer (50 mM Tris, pH 7.2, 1% Triton
X-100, 0.5% sodium deoxycholate, 0.1% SDS, 500 mM NaCl, 10 mM
MgCl2 with 10 μg/ml leupeptin, 10 μg/ml aprotinin and 1
mM PMSF) and quantified by Bradford method. Protein (100 μg) was
separated on a 12% polyacrylamide gel by electrophoresis,
transferred onto polyvinylidene difluoride membranes (Millipore,
Bedford, MA, USA) and blocked with TBS-T supplemented with 5%
non-fat milk. Membranes were incubated with rabbit anti-S100A4
antibody (1:500 dilution) and mouse anti-p53 antibody (1:500
dilution), rabbit anti-β-actin antibody (1:1,000 dilution; Santa
Cruz Biotechnology). After washing, membranes were incubated with a
peroxidase-conjugated second antibody (goat anti-rabbit IgG or goat
anti-mouse IgG) (1:2,000 dilution; Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd., Beijing, China). The reagent for enhanced
chemiluminescence (Amersham Biosciences, Freiburg, Germany) was
used for detection and developed by X-ray film. The experiments
were performed three times.
Chromatin immunoprecipitation assays
(ChIP)
ChIP assays were performed according to the
manufacturer's instructions (Active Motif, Carlsbad, CA, USA). The
target protein p53 was immunoprecipitated with either 3 μg anti-p53
antibody or 3 μg IgG as the negative control. DNA was extracted as
recommended by the protocol. To amplify the mutant p53 binding site
from nucleotides −163 to +22 in the Id2 gene promoter
region, PCR was performed using the forward and reverse primers
5′-GCACTTACTGTACTGTACTCTAT-3′ and 5′-GCTGGAGCTTCCCTTCGTC-3′,
respectively (16).
Induction and quantification of
autophagy
To analyze autophagosomes, we constructed
pEGFP-C1-LC3 expression vectors and transfected them into
MKN1 cells five days after pS100A4-shRNA or pControl-shRNA
transfection in the cells. After 36 h, cells were cultured for 12 h
in serum-free Hank's balanced salt solution medium, referred to as
nutrient-free medium, for serum and amino acid starvation (17). Autophagy was quantified by
determining the percentage of cells with GFP-LC3 accumulation in
vacuoles (GFP-LC3vac, of a minimum 100 cells per preparation in
three independent experiments). Under fluorescence microscopy,
cells presenting mostly diffuse distribution of GFP-LC3 in the
cytoplasm and nucleus were considered non-autophagic, whereas cells
with several intense punctate GFP-LC3 aggregates with no nuclear
GFP-LC3 were classified as autophagic (18). Each GFP-LC3 staining was read by
two independent investigators.
Measurement of ALP activity
MKN1 cells were harvested by trypsinization and
rinsed twice with PBS five days after pS100A4-shRNA or
pControl-shRNA transfection. Then, cells were incubated in 0.2%
Triton X-100 for 24 h at 4ºC, and sonicated. ALP activities in the
cell lysates were measured by determining the formation of
p-nitrophenol from p-nitro-phenol phosphate using a
commercially available kit (Nanjing Jiancheng Biotech Co., Ltd.,
Nanjing, China) according to the manufacturer's instructions. ALP
activity was expressed as ALP units per mg protein. All results
were normalized by protein quantitation. The results were repeated
in at least three independent experiments.
Statistical analysis
The data are presented as mean ± SD (standard
deviation). Each experiment was repeated at least three times.
Statistical analyses were performed using the Student's t-test or
analysis of variance (ANOVA) according to the number of groups
compared. Student-Newman-Keuls (SNK) test was used after the ANOVA
for pairwise comparison. All analyses used SPSS version 16.0
software. A P-value of <0.05 was considered statistically
significant.
Results
S100A4 and mutant p53V143A
colocalization in MKN1 cells
The colocalization of S100A4 and mutant
p53V143A in the nucleus and cytoplasm of MKN1 cells was
identified by double immunofluorescence staining (Fig. 1).
S100A4 interaction with mutant
p53V143A in MKN1 cells
As S100A4 and mutant p53V143A were
colocalized in MKN1 cells, we hypothesized that S100A4 could
interact with the mutant p53 in the cells. To investigate the
interaction, whole cell lysates of MKN1 cells were prepared and
precipitated with anti-S100A4 antibody, and the proteins from the
immunoprecipitates were detected by western blotting using anti-p53
antibody. We detected mutant p53V143A, which indicated
that it coprecipitated with S100A4. Similarly, when whole cell
lysates were precipitated with anti-p53 antibody, S100A4 was
detected from the immunoprecipitates (Fig. 2), indicating that S100A4 can
coprecipitate with mutant p53V143A. Normal IgG was used
as the negative control. These results demonstrate that S100A4
interacts with mutant p53V143A in MKN1 cells.
Effect of S100A4 on mutant
p53V143A levels
After demonstrating that S100A4 could interact with
mutant p53V143A, we used western blotting and RNA
interference to investigate whether inhibiting S100A4 would affect
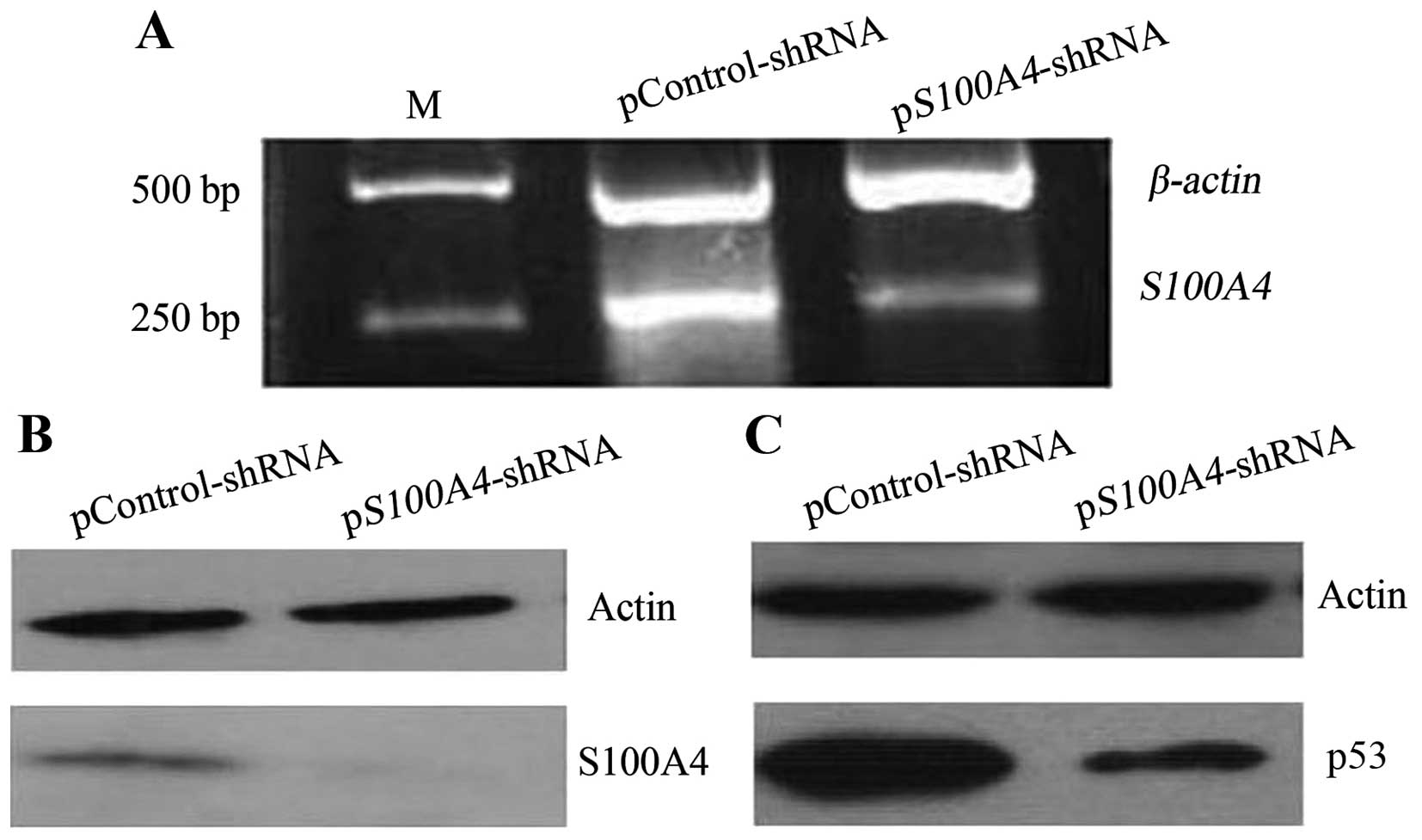
mutant p53 levels. S100A4 inhibition (Fig. 3A and B) decreased mutant
p53V143A levels (Fig.
3C), indicating that S100A4 could contribute to the
accumulation of mutant p53V143A in MKN1 cells.
Inhibitor of DNA binding 2 (Id2) was a
target gene of mutant TP53V143A
Id2 is a target gene of some p53 mutants,
such as p53R273H, p53P309S and
p53R248W (16), but it
is not clear whether it is the target gene of mutant
TP53V143A. To investigate this, we first
performed chromatin immunoprecipitation (ChIP), and the results
showed that mutant TP53V143A bound to the
promoter region of the Id2 gene (Fig. 4A). We further investigated the
effect of TP53V143A on Id2
expression via pCMV-Neo-Bam p53V143A transfection.
RT-PCR and western blotting showed that the levels of TP53 mRNA and
protein expression were significantly increased at 48 h after
pCMV-Neo-Bam p53V143A transfection compared to the
control cells (Fig. 4B and C),
indicating that the pCMV-Neo-Bam p53V143A transfection
led to mutant TP53V143A overexpression in
the cells. Mutant TP53V143A overexpression
decreased Id2 mRNA expression in the cells (Fig. 4D). The results indicate that
Id2 is a target gene of mutant
TP53V143A and that its expression is
directly repressed by mutant
TP53V143A.
S100A4 affected the expression of mutant
TP53V143A target genes
It has been reported that c-Myc is a target
gene of mutant p53V143A (19); our results above indicate that
Id2 is a target gene of mutant
TP53V143A. Therefore, we performed
real-time RT-PCR to investigate the effect of S100A4 on
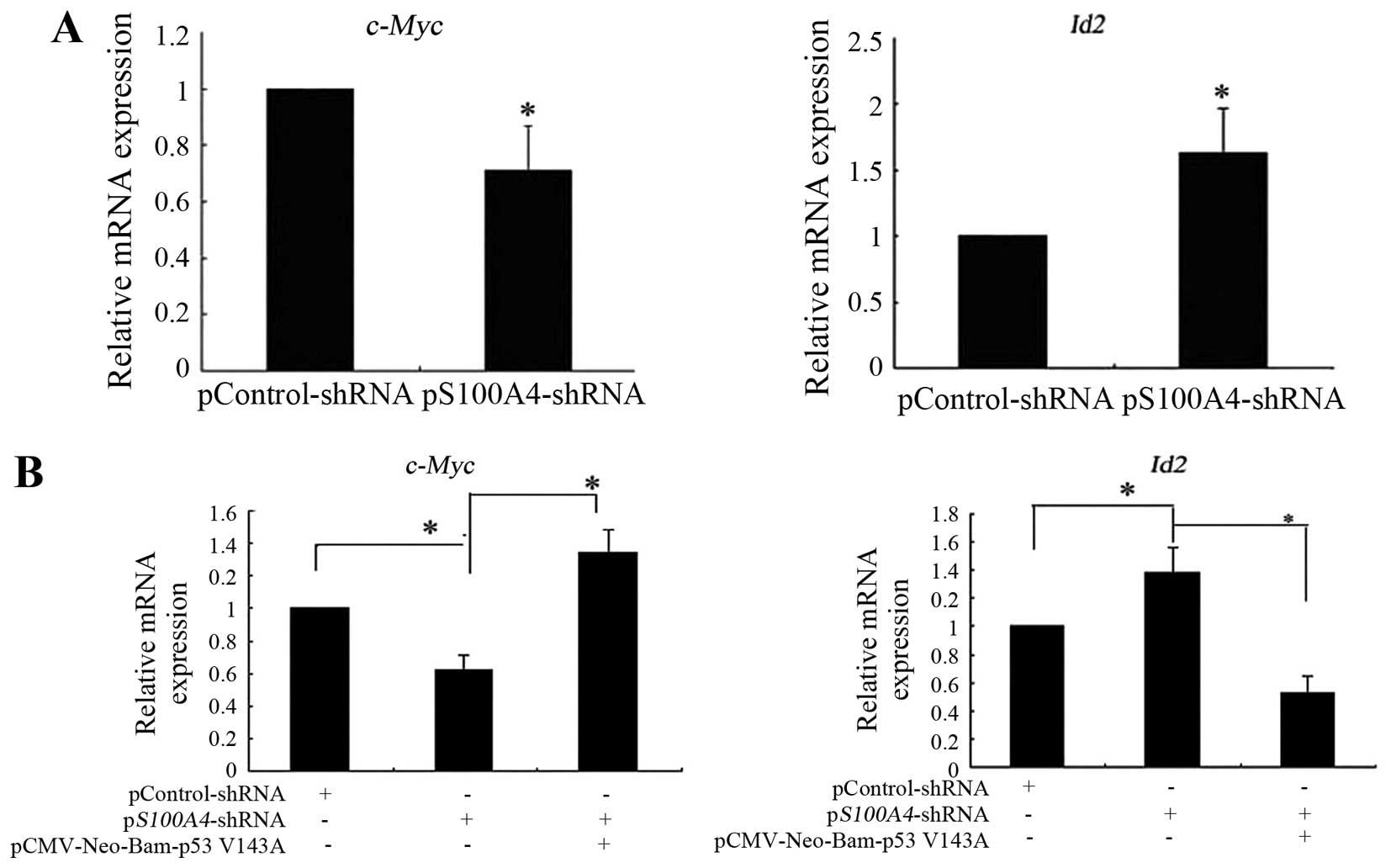
c-Myc and Id2 mRNA expression. S100A4 inhibition
decreased the expression of c-Myc mRNA and increased the
expression of Id2 mRNA (Fig.
5A). Rescue experiments showed that pCMV-Neo-Bam
p53V143A transfection into pS100A4-short hairpin
RNA (shRNA) cells attenuated the decreased expression of
c-Myc mRNA and the increased expression of Id2 mRNA
induced by S100A4 suppression (Fig.
5B). Collectively, the results suggest that S100A4 affects the
expression of mutant TP53V143A target
genes such as c-Myc and Id2, therefore, it affects
the molecular function of mutant
TP53V143A.
S100A4 suppression promoted MKN1 cell
autophagy and differentiation
We demonstrated that S100A4 interacts with mutant
p53 and affects mutant p53 levels. It has been reported that there
is a good correlation between many p53 variants (including
p53V143A) and their ability to inhibit autophagy
(20). It presents the possibility
that S100A4 may affect MKN1 cell autophagy through mutant
p53V143A. Therefore, we examined the potential effect of
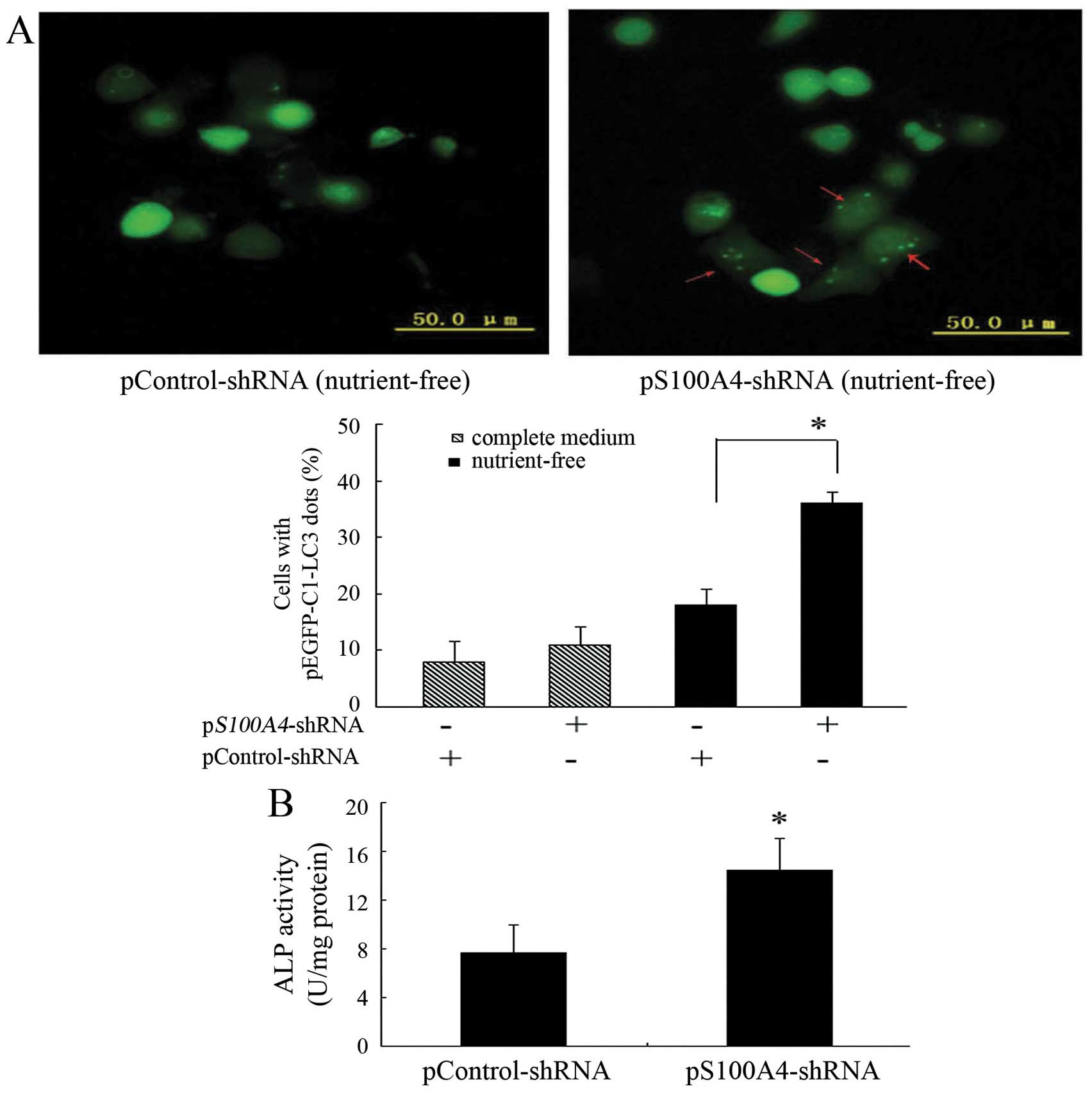
S100A4 in inducing autophagy. Knockdown of S100A4 significantly
increased green fluorescent protein-light chain 3 (GFP-LC3)
aggregation in the cytoplasmic dots induced by nutrient deprivation
(Fig. 6A), indicating that S100A4
suppression could increase MKN1 cell autophagy.
We determined that S100A4 affected the expression of
the mutant TP53V143A target genes
Id2 and c-Myc. The Id2 and c-Myc genes
are implicated in the regulation of cell differentiation (16,21).
We hypothesized that S100A4 may affect cell differentiation.
Alkaline phosphatase (ALP) activity is a marker of differentiation.
The expression of an ALP isoenzyme was stronger in
well-differentiated gastric carcinoma than in poorly differentiated
carcinoma (22). In the present
study, ALP activity was significantly increased in pS100A4-shRNA
cells compared to in pControl-shRNA cells (Fig. 6B), indicating that S100A4
inhibition may promote MKN1 cell differentiation.
Discussion
Using in vitro binding studies, several
different groups have demonstrated direct interaction between
recombinant p53 and S100A4 using different methods (23–25).
A report has also been published on the interaction between S100A4
and p53 in a complex sample involving coimmunoprecipitation (Co-IP)
of mouse mammary cancer cells harboring mutant p53 (13). Recent research has shown that
S100A4 interacted with wild-type p53 in human cancer cells
(26). However, whether S100A4 and
mutant p53 interact in human cancer cells, and whether S100A4
affects mutant p53 accumulation is not clear. The present study
marks the first report of the nuclear and cytoplasmic
colocalization of S100A4 and mutant p53, as well as the in
vivo interaction between S100A4 and mutant p53, which was
detected using double immunofluorescence and Co-IP in MKN1 cells,
which harbor mutant p53V143A. Importantly, our results
show that S100A4 inhibition decreased mutant p53V143A
expression, which suggests that S100A4 might promote mutant
p53V143A accumulation. The mechanism may depend on the
interaction between S100A4 and mutant p53V143A. A recent
report demonstrated the frequent combination of mutated
TP53-positive and S100A4-positive status in colorectal
carcinoma samples (27), which
supports our findings from a clinicopathological perspective. As
described above, it was reported recently that endogenous S100A4
and wild-type p53 interact in complex samples, and S100A4 knockdown
resulted in p53 stabilization in two wild-type p53 cell lines,
indicating that S100A4 promotes wild-type p53 degradation (26). These reports suggested that S100A4
has the opposite effect on wild-type p53 compared to its effect on
mutant p53, as reported in this study. Similar to the effect of
S100A4 on p53, the molecular chaperone HSP90, another partner of
p53, also has the opposite effect on wild-type p53 compared to its
effect on mutant p53. Wild-type p53 accumulated following HSP90
inhibition, whereas mutant p53 protein levels were reduced
(9). These findings suggest that
mutant and wild-type p53 might demonstrate different dependence on
the same partner.
It is well known that p53 exerts its functions by
regulating its target genes. Gain of function is dependent on the
ability of mutant p53 to transactivate or repress specific target
genes, such as c-Myc, Fas, and nuclear factor-κB
(NF-κB) (19,28,29).
The genes affected by the different p53 mutants vary. Id2 is a
member of the inhibitor of differentiation (Id) family, which plays
a role in tumor suppression in multiple tumor types. A previous
study showed that Id2 is a target gene of multiple p53
mutants, such as p53R273H, p53P309S and
p53R248W (16). The
present study is the first to demonstrate that mutant
p53V143A inhibits Id2 expression by binding to
the Id2 promoter, which suggests that Id2 is a target
gene of mutant p53V143A.
As S100A4 is frequently overexpressed in gastric
cancer (11), and we found that
S100A4 was responsible for mutant p53 accumulation by interacting
with it in gastric cancer cells, we speculated that S100A4 could
augment the oncogenic ability of mutant p53 in cells. How S100A4
affects mutant p53 activity requires further clarification. It has
been reported that S100A4 binding to wild-type p53 interferes with
the DNA binding activity of p53 in vitro and with reporter
gene transactivation in vivo (in two mouse cancer cell
lines: CSML-0 and VMR-Liv). In Tet-inducible cell lines expressing
wild-type p53, differential modulation of the wild-type p53 target
gene transcription [P21/WAF, BAX, thrombospondin-1
(THBS1), MDM2] was observed upon S100A4 induction
(13). We hypothesized that S100A4
may affect the target gene expression of mutant
p53V143A. We detected the effect of S100A4 on the
expression of c-Myc and Id2, two target genes of
mutant p53V143A. S100A4 knockdown decreased c-Myc
mRNA expression and increased Id2 mRNA expression. Rescue
assays showed that ectopic expression of mutant p53V143A
reversed the expression altered by S100A4 suppression. The above
findings suggest that interaction with S100A4 altered the
gene-specific regulation by mutant p53V143A. Thus, our
data add a new facet to the oncogenic properties of S100A4, where
it affects mutant p53 target gene expression.
We further investigated the cellular consequences
associated with mutant p53. In line with the gain-of-function
hypothesis, mutant p53 destabilization upon HSP90 inhibition is
accompanied by cell death (9). The
authors also demonstrated that S100A4 knockdown led to wild-type
p53-dependent cell cycle arrest and increased cisplatin-induced
apoptosis (26). Autophagy is a
catabolic process in which portions of the cytoplasmic organelles
are sequestered within autophagosomes, and then targeted for bulk
degradation. Disabled autophagy can accelerate tumor progression.
It was reported that p53 mutants, including p53V143A,
effectively repress autophagy (20). We therefore hypothesized that, as a
partner of mutant p53V143A, S100A4 could affect MKN1
cell autophagy, where S100A4 suppression increased it. These
results mark the first time the effect of S100A4 on gastric cancer
cell autophagy has been reported, and we hypothesize that mutant
p53V143A mediates this effect, although it remains to be
proven.
We found that S100A4 affected the expression of the
mutant TP53V143A target genes Id2
and c-Myc, which regulate cell differentiation (16,21).
Clinical reports have shown that S100A4 overexpression is
associated with poor differentiation in carcinoma (30). However, the role of S100A4 in
gastric cancer cell differentiation remains unclear. In the present
study, ALP activity showed that S100A4 inhibition might promote
differentiation in gastric cancer MKN1 cells. We speculate that
S100A4 may inhibit cell differentiation through mutant
p53V143A and its target genes such as Id2 and
c-Myc.
In conclusion, S100A4 interacts with mutant
p53V143A, and is responsible for the accumulation of
mutant p53V143A in human gastric cancer MKN1 cells.
Subsequently, S100A4 affects the expression of mutant
p53V143A target genes in the cells and further affects
cellular characteristics such as autophagy and cell
differentiation. S100A4 may be a powerful therapeutic target for
inhibiting gain-of-function p53 mutants in gastric cancer.
Acknowledgements
We would like to thank Professor Bert Vogelstein for
kindly providing the mutant p53V143A constructs
available to us. The gastric cancer cell line MKN1 was kindly
provided by Professor Kazunari K. Yokoyama. This project was
supported by grants from the National Natural Science Foundation of
China (nos. 81272717 and 30570848) and the Liaoning Natural Science
Foundation (no. 20102289).
References
|
1
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kastan MB and Berkovich E: p53: a
two-faced cancer gene. Nat Cell Biol. 9:489–491. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bossi G, Marampon F, Maor-Aloni R, Zani B,
Rotter V, Oren M, Strano S, Blandino G and Sacchi A: Conditional
RNA interference in vivo to study mutant p53 oncogenic gain of
function on tumor malignancy. Cell Cycle. 7:1870–1879. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Adorno M, Cordenonsi M, Montagner M,
Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo
V, et al: A Mutant-p53/Smad complex opposes p63 to empower
TGFbeta-induced metastasis. Cell. 137:87–98. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lukashchuk N and Vousden KH:
Ubiquitination and degradation of mutant p53. Mol Cell Biol.
27:8284–8295. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Muller P, Hrstka R, Coomber D, Lane DP and
Vojtesek B: Chaperone-dependent stabilization and degradation of
p53 mutants. Oncogene. 27:3371–3383. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Olive KP, Tuveson DA, Ruhe ZC, Yin B,
Willis NA, Bronson RT, Crowley D and Jacks T: Mutant p53 gain of
function in two mouse models of Li-Fraumeni syndrome. Cell.
119:847–860. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li Y, Guessous F, Kwon S, Kumar M, Ibidapo
O, Fuller L, Johnson E, Lal B, Hussaini I, Bao Y, et al: PTEN has
tumor-promoting properties in the setting of gain-of-function p53
mutations. Cancer Res. 68:1723–1731. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin K, Rockliffe N, Johnson GG,
Sherrington PD and Pettitt AR: Hsp90 inhibition has opposing
effects on wild-type and mutant p53 and induces p21 expression and
cytotoxicity irrespective of p53/ATM status in chronic lymphocytic
leukaemia cells. Oncogene. 27:2445–2455. 2008. View Article : Google Scholar
|
|
10
|
Zou M, Al-Baradie RS, Al-Hindi H, Farid NR
and Shi Y: S100A4 (Mts1) gene overexpression is associated with
invasion and metastasis of papillary thyroid carcinoma. Br J
Cancer. 93:1277–1284. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cho YG, Nam SW, Kim TY, Kim YS, Kim CJ,
Park JY, Lee JH, Kim HS, Lee JW, Park CH, et al: Overexpression of
S100A4 is closely related to the aggressiveness of gastric cancer.
APMIS. 111:539–545. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Matsumoto K, Irie A, Satoh T, Ishii J,
Iwabuchi K, Iwamura M, Egawa S and Baba S: Expression of S100A2 and
S100A4 predicts for disease progression and patient survival in
bladder cancer. Urology. 70:602–607. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Grigorian M, Andresen S, Tulchinsky E,
Kriajevska M, Carlberg C, Kruse C, Cohn M, Ambartsumian N,
Christensen A, Selivanova G, et al: Tumor suppressor p53 protein is
a new target for the metastasis-associated Mts1/S100A4 protein:
Functional consequences of their interaction. J Biol Chem.
276:22699–22708. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yamada Y, Yoshida T, Hayashi K, Sekiya T,
Yokota J, Hirohashi S, Nakatani K, Nakano H, Sugimura T and Terada
M: p53 gene mutations in gastric cancer metastases and in gastric
cancer cell lines derived from metastases. Cancer Res.
51:5800–5805. 1991.PubMed/NCBI
|
|
15
|
Hua J, Chen D, Fu H, Zhang R, Shen W, Liu
S, Sun K and Sun X: Short hairpin RNA-mediated inhibition of S100A4
promotes apoptosis and suppresses proliferation of BGC823 gastric
cancer cells in vitro and in vivo. Cancer Lett. 292:41–47. 2010.
View Article : Google Scholar
|
|
16
|
Yan W, Liu G, Scoumanne A and Chen X:
Suppression of inhibitor of differentiation 2, a target of mutant
p53, is required for gain-of-function mutations. Cancer Res.
68:6789–6796. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mizushima N, Yamamoto A, Matsui M,
Yoshimori T and Ohsumi Y: In vivo analysis of autophagy in response
to nutrient starvation using transgenic mice expressing a
fluorescent autophagosome marker. Mol Biol Cell. 15:1101–1111.
2004. View Article : Google Scholar :
|
|
18
|
Kanzawa T, Zhang L, Xiao L, Germano IM,
Kondo Y and Kondo S: Arsenic trioxide induces autophagic cell death
in malignant glioma cells by upregulation of mitochondrial cell
death protein BNIP3. Oncogene. 24:980–991. 2005. View Article : Google Scholar
|
|
19
|
Frazier MW, He X, Wang J, Gu Z, Cleveland
JL and Zambetti GP: Activation of c-myc gene expression by
tumor-derived p53 mutants requires a discrete C-terminal domain.
Mol Cell Biol. 18:3735–3743. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Morselli E, Tasdemir E, Maiuri MC,
Galluzzi L, Kepp O, Criollo A, Vicencio JM, Soussi T and Kroemer G:
Mutant p53 protein localized in the cytoplasm inhibits autophagy.
Cell Cycle. 7:3056–3061. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vaqué JP, Fernández-García B, García-Sanz
P, Ferrandiz N, Bretones G, Calvo F, Crespo P, Marín MC and León J:
c-Myc inhibits Ras-mediated differentiation of pheochromocytoma
cells by blocking c-Jun up-regulation. Mol Cancer Res. 6:325–339.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Watanabe H, Tokuyama H, Ohta H, Satomura
Y, Okai T, Ooi A, Mai M and Sawabu N: Expression of placental
alkaline phosphatase in gastric and colorectal cancers. An
immunohistochemical study using the prepared monoclonal antibody.
Cancer. 66:2575–2582. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
van Dieck J, Teufel DP, Jaulent AM,
Fernandez-Fernandez MR, Rutherford TJ, Wyslouch-Cieszynska A and
Fersht AR: Posttranslational modifications affect the interaction
of S100 proteins with tumor suppressor p53. J Mol Biol.
394:922–930. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
van Dieck J, Fernandez-Fernandez MR,
Veprintsev DB and Fersht AR: Modulation of the oligomerization
state of p53 by differential binding of proteins of the S100 family
to p53 monomers and tetramers. J Biol Chem. 284:13804–13811. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fernandez-Fernandez MR, Veprintsev DB and
Fersht AR: Proteins of the S100 family regulate the oligomerization
of p53 tumor suppressor. Proc Natl Acad Sci USA. 102:4735–4740.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Orre LM, Panizza E, Kaminskyy VO, Vernet
E, Gräslund T, Zhivotovsky B and Lehtiö J: S100A4 interacts with
p53 in the nucleus and promotes p53 degradation. Oncogene.
32:5531–5540. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Berge G, Costea DE, Berg M, Rasmussen H,
Grotterød I, Lothe RA, Mælandsmo GM and Flatmark K: Coexpression
and nuclear colocalization of metastasis-promoting protein S100A4
and p53 without mutual regulation in colorectal carcinoma. Amino
Acids. 41:875–884. 2011. View Article : Google Scholar
|
|
28
|
Scian MJ, Stagliano KE, Anderson MA,
Hassan S, Bowman M, Miles MF, Deb SP and Deb S: Tumor-derived p53
mutants induce NF-kappaB2 gene expression. Mol Cell Biol.
25:10097–10110. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zalcenstein A, Stambolsky P, Weisz L,
Müller M, Wallach D, Goncharov TM, Krammer PH, Rotter V and Oren M:
Mutant p53 gain of function: Repression of CD95 (Fas/APO-1) gene
expression by tumor-associated p53 mutants. Oncogene. 22:5667–5676.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rosty C, Ueki T, Argani P, Jansen M, Yeo
CJ, Cameron JL, Hruban RH and Goggins M: Overexpression of S100A4
in pancreatic ductal adenocarcinomas is associated with poor
differentiation and DNA hypomethylation. Am J Pathol. 160:45–50.
2002. View Article : Google Scholar : PubMed/NCBI
|