Introduction
Pancreatic ductal adenocarcinoma (PDAC) accounts for
>90% of all exocrine pancreatic cancers and is almost uniformly
lethal with an estimated annual number of 45,220 new cases ~38,460
annual deaths and a 5-year survival rate of <5% (1–3).
Late initial diagnosis, aggressive metastatic behavior and
resistance to chemo-radiotherapy render pancreatic cancer one of
the most difficult to treat of all malignancies. Systemic
chemotherapy with either gemcitabine or FOLFIRINOX (5-FU,
irinotecan and oxaliplatin) is the current standard of care for
advanced PDAC, providing short-term symptomatic improvement with
minor impact on survival (4–6).
Surgical resection is potentially curative; however, nearly 80% of
the patients are diagnosed with locally advanced disease,
precluding surgical intervention. Even in early stage PDAC patients
in whom cancerous tissue is surgically resected (Whipple
procedure), a majority of them relapse within two years after
surgery. Thus, there is a tremendous need for novel therapeutics
and treatment strategies for treating primary and residual PDAC for
preventing relapse after pancreatic cancer surgery.
Aberration of apoptosis has been implicated in
carcinogenesis and resistance to conventional anticancer therapies
(7). Thus, promotion of apoptosis
in cancer cells with novel agents could lead to tumor regression
and improved prognosis. Triterpenes or triterpenoids are members of
a large family of structurally related compounds known as
cyclosqualenoids that are widely distributed in nature (8). Oleanolic acid is naturally occurring
triterpenoid that has been used in traditional medicine as an
anticancer and anti-inflammatory agent (9–11).
In order to increase the anti-inflammatory and anticancer activity
of oleanolic acid several synthetic analogues have been
synthesized. Synthetic lealane triterpenoid
2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid (CDDO) and its C-28
methyl ester derivative
methyl-2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oate
(CDDO-Me) and C-28 imidazole derivative
1-(2-cyano-3,12-dioxooleana-1,9-dien-28-oyl) imidazole (CDDO-Im)
exhibit stronger anti-inflammatory activity than oleanolic acid
(12,13). CDDOs have also shown potent
antiproliferative and proapoptotic activity in diverse types of
tumor cell lines through the inhibition of MAPK (Erk1/2), NF-κB and
PPARγ signaling pathways and they induce cyto-protective phase 2
response through Nrf2 signaling (14–20).
In previous studies, we have demonstrated that the
antiproliferative and apoptosis inducing activity of CDDO-Me in
prostate cancer cell lines is mediated through the inhibition of
anti-apoptotic (prosurvival) Akt/NF-κB/mTOR signaling pathways
(21). Although the
antiproliferative activity of CDDOs in PDAC cell lines in
vitro has been reported their therapeutic efficacy for PDAC
in vivo has not been adequately studied. We have
investigated the therapeutic efficacy of CDDO-Me against
heterotopic and orthotopic tumor xenografts generated with human
PDAC cell lines with a focus on preventing or delaying
relapse/recurrence. Our data show therapeutic efficacy of CDDO-Me
in treating primary tumor growth as well as in preventing/delaying
recurrence of PDAC when administered after surgical removal of
tumors.
Materials and methods
Reagents
CDDO-Me was obtained from the National Cancer
Institute, Bethesda, MD, USA through the Rapid Access to
Intervention Development Program. Antibodies against p-Akt
(ser473), p-mTOR (Ser2448), PARP-1 and
β-actin were purchased from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA). CellTiter 96 AQueous One Solution Proliferation
assay system was from Promega (Madison, WI, USA) and Annexin V-FITC
apoptosis detection kit II was obtained from BD Pharmingen (San
Diego, CA, USA).
Cell culture
Human PDAC cell lines MiaPaCa-2 and BxPC-3 were
obtained from the American Type Culture Collection (ATCC,
Rockville, MD, USA). Cell lines were cultured at 37ºC in a
humidified atmosphere consisting of 5% CO2 and 95% air
and maintained by subculturing cells twice a week.
Mice
Six-week-old female Scid/Ncr mice were purchased
from the Charles River Laboratories, Inc., Frederic, MD, USA. Mice
were maintained in a temperature-controlled room (68–72ºF) with a
12-h light/dark cycle and provided semi-purified AIN-76A mouse chow
and water ad libitum. Mice were acclimated for one week
before starting the experiment. All animal treatments were
according to the protocol approved by the Institutional Animal Care
and Use Committee (IACUC).
Measurement of cell viability
Tumor cells (1×104) were seeded into each
well of a 96-well plate in 100 μl of tissue culture medium. After
incubation at 37ºC in a humidified atmosphere consisting of 5%
CO2 and 95% air for 24 h, cells were treated with
CDDO-Me at concentrations of 0.625–5 μM for 72 h. Cell viability
was then determined by the MTS assay using CellTiter 96®
AQueous One Solution Proliferation assay system.
Measurement of apoptosis
Apoptosis was assessed by the binding of Annexin
V-FITC to phosphotidylserine, which is externalized to the outer
leaflet of the plasma membrane early during induction of apoptosis.
Briefly, untreated cells and cells treated with CDDO-Me for 24 h
were resuspended in the binding buffer provided in the Annexin
V-FITC apoptosis detection kit and reacted with 5 μl of Annexin
V-FITC reagent and 5 μl of propidium iodide (PI) for 30 min at room
temperature in the dark. Stained cells were analyzed by flow
cytometry using Accuri C6 flow cytometer (Accuri Cytometers Inc.
Ann Arbor, MI, USA). The induction of apoptosis by CDDO-Me was
confirmed by the cleavage of PARP-1 by western blotting.
Western blotting
Cell lysates were prepared using NP 40 cell lysis
buffer. Lysates were clarified by centrifugation at 14,000 × g for
10 min at 4ºC and protein concentrations were determined. Samples
(50 μg) were boiled in an equal volume of sample buffer [20%
glycerol, 4% SDS, 0.2% bromophenol blue, 125 mM Tris-HCl (pH 7.5),
and 640 mM 2-ME] and separated on pre-casted Tris-glycine
polyacrylamide gels using the XCell Surelock™ Mini-Cell, in
Tris-glycine SDS running buffer, all from Novex (Invitrogen,
Carlsbad, CA, USA). Proteins resolved on the gels were transferred
to PVDF membranes. Membranes were blocked with 5% milk in 10 mM
Tris-HCl (pH 8.0), 150 mM NaCl with 0.05% Tween-20 (TPBS) and
probed using specific antibodies against proteins of interest or
β-actin (loading control) and HRP-conjugated secondary antibody.
Immune complexes were visualized with enhanced chemiluminescence.
Protein bands were imaged and band densities analyzed using the
NIH/Scion image analysis software. The protein band densities were
normalized to the corresponding β-actin band densities and percent
change in signal strength was calculated.
Generation of tumor xenografts
Heterotopic PDAC xenografts were generated by
implanting 1×106 BxPC-3 cells in 50 μl of PBS
subcutaneously in the right flank of 7-week-old Scid-Ncr female
mice. Cells were allowed to form tumors for 7 days before
initiating treatment with CDDO-Me. Orthotopic PDAC xenografts were
generated by implanting MiaPaCa-2 cells stably expressing
luciferase gene (MiaPaCa-2-Luc cells) in the pancreatic tail of
Scid/Ncr mice. Briefly, mice were anesthetized with
ketamine/xylazine (100/8 mg/kg i.p.) and a 2-cm long subcostal
laparotomy was performed after clipping hair. Pancreas were exposed
and 1×106 MiaPaCa-2-Luc cells in 20 μl of PBS were
injected in the pancreatic tail with a 30 G needle. Proper
deposition of cells was confirmed by bleb formation at the site of
injection. The fascia and skin incisions were sutured following
implantation of tumor cells.
Residual disease
To generate residual disease, 1×106
MiaPaCa-2-Luc cells in 20 μl of PBS were injected in the pancreatic
tail as described above. Cells were first allowed to form tumors
for 28 days and then mice were anesthetized with ketamine/xylazine
(100/8 mg/kg i.p.) and partial pancreatectomy was performed to
remove all visible tumor growth. After resection of tumors,
incisions were sutured and mice were either treated or not with
CDDO-Me.
Treatment with CDDO-Me
Two treatment paradigms were followed. To treat
primary tumor growth, treatment was started on day 7 after
implantation of BxPC-3 cells subcutaneously. To treat residual
disease, treatment was started one day after resection of tumors on
day 28 after implantation of MiaPaCa-2-Luc cells in the pancreas.
Mice in both treatment paradigms were administered CDDO-Me at a
dose of 15 μmol/kg (7.5 mg/kg), 5 days a week in 0.1 ml of vehicle
consisting of cremophor-EL:DMSO:PBS (1:1:8) by oral gavage until
day 40 (heterotopic xenografts) to treat primary tumor growth or
day 100 (orthotopic xenografts) to treat residual disease. Control
mice were treated with vehicle alone without CDDO-Me.
Bioluminecsent imaging (BLI)
The growth progression of orthotopic PDAC xenografts
generated by implanting MiaPaCa-2-Luc cells in the pancreas was
measured non-invasively by BLI imaging using Kodak Multispectral
Imaging system (Carestream Health, Woodbridge, CT, USA). Briefly,
mice were anesthetized with isofluorane and anesthesia was
maintained by inhalation of a mixture of isofluorane (2%) and
oxygen during the imaging procedure. First an X-ray image was taken
and then BLI was obtained for 2 min, 10 min after i.p. injection of
sodium luciferin (150 mg/kg). The X-ray and BLI images were
overlaid and peak photon flux (photon/sec/mm2) from the
tumors was plotted against time to obtain tumor growth curve.
Statistical analysis
Most data are presented as means ± SD and outcomes
for treated and untreated cells were compared by Student's t-test.
Differences were considered significant at p<0.05.
Results
CDDO-Me inhibits proliferation and
induces apoptosis in PDAC cells
To test the effect of CDDO-Me on the proliferation
of PDAC cells, 1×104 MiaPaCa-2 or BxPC-3 cells were
plated in 96-well microtiter plates for 24 h and then treated with
CDDO-Me for 72 h at concentrations ranging from 0 to 5 μM.
Viability of cultures was assessed by MTS assay and percent
reduction in viability of cultures was determined. As shown in
Fig. 1, significant decrease in
the viability of cell cultures was observed in both cell lines
treated with CDDO-Me at concentrations of 1.25–5 μM (MiaPaCa-2:
44–73%; BxPC-3: 57–79%).
To determine whether inhibition of proliferation of
PDAC cells by CDDO-Me was associated with induction of apoptosis,
binding of Annexin V-FITC to cells treated with CDDO-Me was
measured. For this, MiaPaCa-2 or BxPC-3 cells were treated or not
with CDDO-Me (0–5 μM) for 24 h and analyzed for Annexin V-FITC
binding by flow cytometry. As shown in Fig. 2A, the percentage of Annexin V-FITC
binding MiaPaCa-2 cells increased from <2% (untreated cells) to
17, 24, 37 and 62% following treatment with CDDO-Me at 0.625, 1.25,
2.5 and 5 μM, respectively. The percentage of Annexin V-FITC
binding BxPC-3 cells was 1, 9, 16, 56 and 67% at 0, 0.625, 1.25,
2.5 and 5 μM CDDO-Me.
The induction of apoptosis by CDDO-Me was confirmed
by the cleavage of native PARP-1. As shown in Fig. 2B, treatment with CDDO-Me caused the
emergence of 89 kDa cleaved PARP-1 fragment in both cell lines.
Together, increase in binding of Annexin V-FITC and cleavage of
PARP-1 demonstrated induction of apoptosis by CDDO-Me in PDAC
cells.
CDDO-Me inhibits p-Akt, NF-κB and p-mTOR
signaling proteins in PDAC cells
Akt, NF-κB and mTOR are anti-apoptotic (prosurvival)
signaling proteins that are constitutively active in a variety of
human cancers, providing survival advantage to cancer cells. We
investigated whether PDAC cells express constitutively active Akt
(p-Akt) and mTOR (p-mTOR) and the effect CDDO-Me has on the
expression of these signaling proteins. Cell lysates were prepared
from MiaPaCa-2 and BxPC-3 cells treated or not with CDDO-Me (0–5
μM) for 24 h and analyzed by western blotting for the levels of
these proteins. Fig. 3 shows that
both PDAC cell lines expressed p-Akt, NF-κB and p-mTOR and
treatment with CDDO-Me significantly or completely inhibited their
expression at concentrations of 1.25–5 μM. These data demonstrated
that induction of apoptosis by CDDO-Me is attributable at least in
part to the inhibition of anti-apoptotic Akt, NF-κB and mTOR.
CDDO-Me inhibits the growth of BxPC-3
xenografts
Next we determined the tumor inhibitory activity of
CDDO-Me in vivo against hetrotopic BxPC-3 xenografts. One
week after injection of 1×106 BxPC-3 PDAC cells in the
flanks of Scid-Ncr mice, mice were treated with CDDO-Me (15
μmol/kg) or vehicle only by oral gavage, 5 days/week for 5 weeks
(n=5) and experiment was terminated on day 40. To measure the
effect of CDDO-Me on progression of tumor growth, tumor dimentions
(length and width) were measured by a caliper and tumor volume was
calculated by the formula: (length × width2)/2. As shown
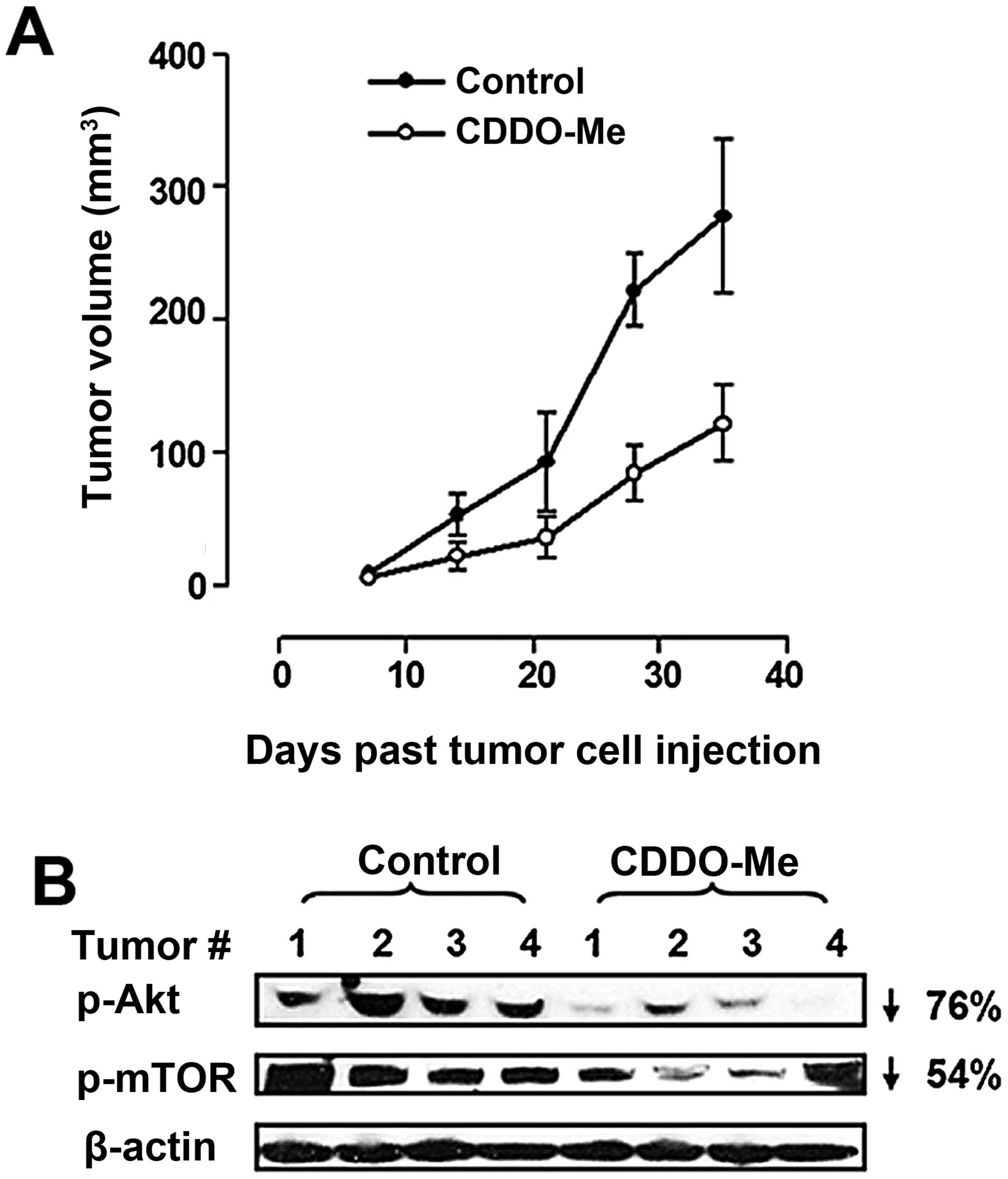
in Fig. 4A, treatment with CDDO-Me
significantly inhibited the growth of BxPC-3 xenografts compared to
vehicle contol animals [tumor volume (mm3 ± SD):
control, 53±10, 98±23, 226±21 and 284±39; CDDO-Me, 22±6, 37±10,
79±16 and 111±19 on days 14, 21, 28 and 35 after implantation of
tumor cells]. Furthermore, treatment with CDDO-Me also reduced
p-Akt and p-mTOR levels in the tumor tissue (76 and 54% reduction,
respectively) (Fig. 4B). These
data demonstrated therapeutic efficacy of CDDO-Me against PDAC
in vivo and provided evidence that activated Akt and mTOR
are therapeutic targets of CDDO-Me in PDAC tumors.
Generation of orthotopic PDAC xenografts
and efficacy of CDDO-Me for residual disease
To closely mimic physiological microenvironment of
PDAC in patients for treatment with CDDO-Me we generated orthotopic
xenografts by injecting MiaPaCa-2-Luc cells in the pancreatic tail
in Scid/Ncr mice. The development and progression of orthotopic
xenografts was monitored non-invasively by bioluminescence imaging
using Kodak Multispectral Imaging System (Carestream Health,
Woodbridge, CT, USA). Fig. 5 shows
BLI images of a xenograft on days 15 and 35 (Fig. 5A and B) and a metastatic lesion in
the liver (Fig. 5B, arrow). The
growth curve of xenografts was created by plotting photon flux
emission (photons/sec/mm2) from tumors at various time
points past tumor cell injection in the pancreas (Fig. 5C). The anatomical location of
tumors was visually confirmed upon opening the abdominal cavity and
tumors were harvested and identified as PDAC histologically
(Fig. 5D and E).
We used the orthotopic xenogaft model to generate
residual disease to investigate efficacy of CDDO-Me for preventing
relapse (recurrence) after surgical removal of tumors. In this
case, after injection of MiaPaCa-2-luc cells in the pancreatic
tail, tumor cells were first allowed to form tumors. On day 28,
tumors were resected to remove all of the visible tumor growth in
each animal. One day after removal of tumors, mice were treated
with vehicle alone (n=9) or CDDO-Me by oral gavage at a dose of 15
μmol/kg as described in Materials and methods (n=12). Separately,
mice were also treated with gemcitabine, the standard drug used for
treating pancreatic cancer, at a dose of 2 mg/kg, 2 times per week,
i.p. (n=9). Experiment was terminated on day 100 and survival data
were compared.
As shown in Fig.
5F, six of the nine mice in the vehicle control group died
during this period (33% survival; mean survival time =81.6±22
days). Five of the nine mice treated with gemcitabine survived
during the same period (56% survival; mean survival time =81±25
days, p>0.05). However, the group of mice treated with CDDO-Me
showed the best survival rate, i.e., eight of the 12 mice treated
with CDDO-Me revealed the presence of tumours in 2 of the 3 vehicle
control, 2 of the 5 gemcitabine-treated and 1 of the 8
CDDO-Me-treated surviving mice on day 100.
Discussion
Pancreatic ductal adenocarcinoma is an intractable
malignant disease with limited therapeutic options. Discovering
effective novel agents with defined therapeutic targets might
provide urgently needed therapeutics to treat this cancer. Natural
products have long been recognized as a rich source of chemical
diversity and compounds with a wide spectrum of anticancer
activities. From vincristine to paclitaxel, the effective antitumor
activity and novel mechanisms of action of many natural compounds
and their synthetic derivatives have been described (22,23).
CDDO and CDDO-Me are synthetic triterpenoids derived from oleanolic
acid with potent antiproliferative and pro-apoptotic activity.
Earlier we demonstrated efficacy of CDDO and CDDO-Me in
preventing/delaying the development and progression of prostate
cancer in the transgenic TRAMP mouse model of prostate
tumorigenesis (24,25). Others have shown efficacy of CDDOs
for breast, lung and colon cancers (26,27).
On the other hand, therapeutic efficacy of CDDOs for pancreatic
cancer has not been adequately investigated. Studies described in
this report were intended to determine therapeutic efficacy of
CDDO-Me for pancreatic ductal adenocarcinoma. Since Kras is
mutated in >90% of human PDAC and mutation of Kras at
codon G12 leads to constitutive activation of Kras
oncoprotein and more aggressive PDAC phenotype (28), we investigated the anticancer
activity of CDDO-Me in vitro and in vivo using two
PDAC cell lines with and without activating K-ras mutations
(MiaPaCa-2 and BxPC-3, respectively). CDDO-Me strongly inhibited
the proliferation of both cell lines in vitro independent of
K-ras mutations. Since inhibition of cell proliferation
induces apoptosis in cancer cells, we assessed induction of
apoptosis by CDDO-Me in PDAC cells. Induction of apoptosis was
measured by the binding of Annexin V-FITC to phosphotidylserine
which is externalized to the outer leaflet of the plasma membrane
early during induction of apoptosis. Treatment with CDDO-Me
significantly increased the percentage of Annexin V-FITC binding
cells in both cell lines. Induction of apoptosis was confirmed by
the cleavage of PARP-1, a hallmark of cells undergoing
apoptosis.
PI3K/Akt/mTOR and Akt/NF-κB are major
antiapoptotic/prosurvival signaling pathways that are
constitutively active in most cancers including pancreatic cancer.
Akt promotes cell growth and survival by inactivating downstream
substrates such as Bad, procaspase-9 and forkhead transcription
factors (28,29). Antiapoptotic NF-κB and progrowth
mTOR signaling pathways are downstream targets of activated
PI3K/AKT. The NF-κB family of transcription factors controls the
expression of genes involved in immune, inflammatory and oncogenic
responses and play a critical role in resistance of cancer cells to
anticancer therapies by protecting them from apoptosis (30). mTOR is a serine-threonine kinase,
which controls cell growth, survival and ribogenesis (31). CDDO-Me inhibited the expression of
p-AKT, NF-κB and p-mTOR in MiaPaCa-2 and BxPC-3 cells. The
inhibition of these anti-apoptotic and survival-promoting signaling
proteins suggested that they are involved in mediating the
antiproliferative and proapoptotic activity of CDDO-Me in PDAC
cells.
The results of in vitro studies showing
strong antiprolif-erative and proapoptotic activity of CDDO-Me for
PDAC cells prompted us to evaluate its therapeutic efficacy for
PDAC in vivo, since many times antitumor effects observed in
cell lines in vitro do not pan out in vivo. We first
examined CDDO-Me for therapeutic efficacy against heterotopic
xenografts generated with BxPC-3 cells with normal K-ras
expression. Treatment with CDDO-Me significantly inhibited the
growth of heterotopic xenografts (p<0.01). Western blot analysis
of the tumor tissue extract showed that the inhibition of tumor
growth was associated with the inhibition of
anti-apoptotic/prosurvival p-Akt, NF-κB and p-mTOR signaling
proteins. These data also demonstrated that in vitro effects
of CDDO-Me in PDAC cells does translate into therapeutic efficacy
in vivo.
To further analyze therapeutic efficacy of CDDO-Me
for PDAC, we tested it for preventing relapse/recurrence when
administered after surgical resection of tumors. This was
accomplished by creating a model of PDAC residual disease wherein
orthotopic PDAC xenografts generated by implanting MiaPaCa-2-Luc
cells in the pancreas were surgically removed after four weeks of
tumor growth. Treatment with CDDO-Me in this adjuvant setting
significantly prolonged survival of mice compared to untreated
animals. In fact, the survival advantage with CDDO-Me was even
better than that with gemcitabine, a standard drug for the
treatment of patients with advanced PDAC. Thus, therapeutic
outcomes in xenografts models generated with PDAC cells expressing
either normal K-ras (BxPC-3) or activated K-ras
(MiaPaCa-2) correlated with responses to CDDO-Me in vitro.
Although the exact mechanism of the antitumor activity of CDDO-Me
in vivo remain to be investigated, our data suggest that
downregulation of p-Akt, NF-κB and p-mTOR signaling proteins plays
a role in inhibition of cell proliferation and induction of
cellular apoptosis in tumor cells by CDDO-Me. Since treatment with
gemcitabine or CDDO-Me alone improved survival when administered
after removal of tumors suggests that combinatorial regimen of
gemcitabine and CDDO-Me might be even more beneficial in
preventing/delaying recurrence. This inference of course needs more
rigorous analysis in these preclinical models of PDAC.
In conclusion, our investigations show that CDDO-Me
exhibits therapeutic efficacy in xenograft models of PDAC
(heterotopic and orthotopic) for inhibiting primary tumor growth
and delaying/preventing relapse by destroying residual disease.
Because of its ability to inhibit p-AKT, NF-κB and p-mTOR in tumor
tissue, we speculate that CDDO-Me inhibits tumor growth by
inhibiting cell proliferation and promoting apoptosis through the
inhibition of anti-apoptotic/prosurvival AKT/NF-κB/mTOR signaling
pathways. Collectively, these findings unveiling antitumor
potential of CDDO-Me for PDAC warrant further studies for
development of CDDO-Me as an adjuvant to existing PDAC therapies to
obtain more durable responses and improved outcomes.
Acknowledgements
This study was supported by NIH grant 1R01 CA130948
and a grant from Elsa U. Pardee Foundation. Authors thank Dr Ali S.
Arbab for helping with BLI imaging.
References
|
1
|
Pancreatic Cancer-National Cancer
Institute. U.S. National Institutes of Health. Cancer Gov.
http:www.cancer.gov/cancer-topics/types/pancreatic.
Accessed April 6, 2010
|
|
2
|
Maitra A and Hruban RH: Pancreatic cancer.
Annu Rev Pathol. 3:157–188. 2008. View Article : Google Scholar
|
|
3
|
Li D, Xie K, Wolff R and Abbruzzese JL:
Pancreatic cancer. Lancet. 363:1049–1057. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mulcahy MF, Wahl AO and Small W Jr: The
current status of combined radiotherapy and chemotherapy for
locally advanced or resected pancreas cancer. J Natl Compr Canc
Netw. 3:637–642. 2005.PubMed/NCBI
|
|
5
|
Pino SM, Xiong HQ, McConkey D and
Abbruzzese JL: Novel therapies for pancreatic adenocarcinoma. Curr
Oncol Rep. 6:199–206. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vaccaro V, Sperduti I and Milella M:
FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N
Engl J Med. 365:768–769; author reply 769. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kerr JF, Winterford CM and Harmon BV:
Apoptosis. Its significance in cancer and cancer therapy. Cancer.
73:2013–2026. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dzubak P, Hajduch M, Vydra D, Hustova A,
Kvasnica M, Biedermann D, Markova L, Urban M and Sarek J:
Pharmacological activities of natural triterpenoids and their
therapeutic implications. Nat Prod Rep. 23:394–411. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang MT, Ho CT, Wang ZY, Ferraro T, Lou
YR, Stauber K, Ma W, Georgiadis C, Laskin JD and Conney AH:
Inhibition of skin tumorigenesis by rosemary and its constituents
carnosol and ursolic acid. Cancer Res. 54:701–708. 1994.PubMed/NCBI
|
|
10
|
Nishino H, Nishino A, Takayasu J, Hasegawa
T, Iwashima A, Hirabayashi K, Iwata S and Shibata S: Inhibition of
the tumor-promoting action of 12-O-tetradecanoylphorbol-13-acetate
by some oleanane-type triterpenoid compounds. Cancer Res.
48:5210–5215. 1988.PubMed/NCBI
|
|
11
|
Ryu SY, Oak MH, Yoon SK, Cho DI, Yoo GS,
Kim TS and Kim KM: Anti-allergic and anti-inflammatory triterpenes
from the herb of Prunella vulgaris. Planta Med. 66:358–360. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Honda T, Rounds BV, Gribble GW, Suh N,
Wang Y and Sporn MB: Design and synthesis of
2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, a novel and highly
active inhibitor of nitric oxide production in mouse macrophages.
Bioorg Med Chem Lett. 8:2711–2714. 1998. View Article : Google Scholar
|
|
13
|
Suh N, Honda T, Finlay HJ, Barchowsky A,
Williams C, Benoit NE, Xie QW, Nathan C, Gribble GW and Sporn MB:
Novel triterpenoids suppress inducible nitric oxide synthase (iNOS)
and inducible cyclooxygenase (COX-2) in mouse macrophages. Cancer
Res. 58:717–723. 1998.PubMed/NCBI
|
|
14
|
Ito Y, Pandey P, Sporn MB, Datta R,
Kharbanda S and Kufe D: The novel triterpenoid CDDO induces
apoptosis and differentiation of human osteosarcoma cells by a
caspase-8 dependent mechanism. Mol Pharmacol. 59:1094–1099.
2001.PubMed/NCBI
|
|
15
|
Konopleva M, Tsao T, Estrov Z, Lee RM,
Wang RY, Jackson CE, McQueen T, Monaco G, Munsell M, Belmont J, et
al: The synthetic triterpenoid
2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces
caspase-dependent and -independent apoptosis in acute myelogenous
leukemia. Cancer Res. 64:7927–7935. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao X, Deeb D, Jiang H, Liu Y, Dulchavsky
SA and Gautam SC: Synthetic triterpenoids inhibit growth and induce
apoptosis in human glioblastoma and neuroblastoma cells through
inhibition of prosurvival Akt, NF-kappaB and Notch1 signaling. J
Neurooncol. 84:147–157. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Konopleva M, Contractor R, Kurinna SM,
Chen W, Andreeff M and Ruvolo PP: The novel triterpenoid CDDO-Me
suppresses MAPK pathways and promotes p38 activation in acute
myeloid leukemia cells. Leukemia. 19:1350–1354. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shishodia S, Sethi G, Konopleva M,
Andreeff M and Aggarwal BB: A synthetic triterpenoid, CDDO-Me,
inhibits IkappaBalpha kinase and enhances apoptosis induced by TNF
and chemotherapeutic agents through down-regulation of expression
of nuclear factor kappaB-regulated gene products in human leukemic
cells. Clin Cancer Res. 12:1828–1838. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chintharlapalli S, Papineni S, Konopleva
M, Andreef M, Samudio I and Safe S:
2-Cyano-3,12-dioxoolean-1,9-dien-28-oic acid and related compounds
inhibit growth of colon cancer cells through peroxisome
proliferator-activated receptor gamma-dependent and -independent
pathways. Mol Pharmacol. 68:119–128. 2005.PubMed/NCBI
|
|
20
|
Liby K, Hock T, Yore MM, Suh N, Place AE,
Risingsong R, Williams CR, Royce DB, Honda T, Honda Y, et al: The
synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent
inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res.
65:4789–4798. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Deeb D, Gao X, Jiang H, Dulchavsky SA and
Gautam SC: Oleanane triterpenoid CDDO-Me inhibits growth and
induces apoptosis in prostate cancer cells by independently
targeting prosurvival Akt and mTOR. Prostate. 69:851–860. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chin YW, Balunas MJ, Chai HB and Kinghorn
AD: Drug discovery from natural sources. AAPS J. 8:E239–E253. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Itokawa H, Morris-Natschke SL, Akiyama T
and Lee KH: Plant-derived natural product research aimed at new
drug discovery. J Nat Med. 62:263–280. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gao X, Deeb D, Liu Y, Arbab AS, Divine GW,
Dulchavsky SA and Gautam SC: Prevention of prostate cancer with
oleanane synthetic triterpenoid CDDO-Me in the TRAMP mouse model of
prostate cancer. Cancers (Basel). 3:3353–3369. 2011. View Article : Google Scholar
|
|
25
|
Deeb D, Gao X, Liu Y, Jiang D, Divine GW,
Arbab AS, Dulchavsky SA and Gautam SC: Synthetic triterpenoid CDDO
prevents the progression and metastasis of prostate cancer in TRAMP
mice by inhibiting survival signaling. Carcinogenesis. 32:757–764.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ling X, Konopleva M, Zeng Z, Ruvolo V,
Stephens LC, Schober W, McQueen T, Dietrich M, Madden TL and
Andreeff M: The novel triterpenoid C-28 methyl ester of 2-cyano-3,
12-dioxoolen-1, 9-dien-28-oic acid inhibits metastatic murine
breast tumor growth through inactivation of STAT3 signaling. Cancer
Res. 67:4210–4218. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liby K, Royce DB, Williams CR, Risingsong
R, Yore MM, Honda T, Gribble GW, Dmitrovsky E, Sporn TA and Sporn
MB: The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl
amide prevent lung cancer induced by vinyl carbamate in A/J mice.
Cancer Res. 67:2414–2419. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Datta SR, Dudek H, Tao X, Masters S, Fu H,
Gotoh Y and Greenberg ME: Akt phosphorylation of BAD couples
survival signals to the cell-intrinsic death machinery. Cell.
91:231–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo
P, Hu LS, Anderson MJ, Arden KC, Blenis J and Greenberg ME: Akt
promotes cell survival by phosphorylating and inhibiting a Forkhead
transcription factor. Cell. 96:857–868. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Karin M, Cao Y, Greten FR and Li ZW:
NF-kappaB in cancer: From innocent bystander to major culprit. Nat
Rev Cancer. 2:301–310. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hay N and Sonenberg N: Upstream and
downstream of mTOR. Genes Dev. 18:1926–1945. 2004. View Article : Google Scholar : PubMed/NCBI
|