Introduction
Notch signaling controls various biological events
and is vital for many types of cell fate determination in embryonic
development. Four Notch receptors (Notch1-4) and two groups of
ligands, Jagged and Delta-like, have been identified in mammals
(1). The activation of Notch
signaling is initialized by the binding of Notch receptors and
ligands, which leads to the release of intracellular region of
Notch (ICN) from Notch transmembrane subunit (NTM) and then
translocation to the nucleus to activate transcription of a group
of downstream genes (2). Recent
studies have revealed the deregulation of expression and function
of Notch receptors and ligands in a series of malignant tumors. The
expression of Notch1 and Jagged1 are upregulated in human colon
adenocarcinoma (3). Notch1,
Notch2, and hairy and enhancer of split 1 (HES1) show high
expression in biliary tract cancer (4). Notch3 has a stronger positive degree
of expression in lung squamous cell carcinoma and adenocarcinoma
compared with the corresponding non-tumor tissue (5). The expression of Notch1 and Jagged1
in both metastatic prostate cancer and high grade prostate cancer
is significantly higher than those in low grade prostate cancer and
in benign prostatic tissues (6).
Besides, Notch2 and Jagged1 expression levels correlate with
survival of colorectal cancer patients (7), and nuclear expression of Notch1 and
Notch3 is also found to be associated with tumor recurrence in
colorectal cancer (8).
Overexpression of Notch1 is also identified as a significant and
independent prognostic factor for esophageal squamous cell cancer
(9).
HBV is a major cause of HCC, and HBx has been
reported as an important viral protein in the carcinogenesis and
progression of HBV-associated HCC (10). Studies show that HBx interacts with
transcription factors in the nucleus and activates signal
transduction cascades in the cytoplasm (11,12),
which in the end induces foci in cells and leads to liver cancer in
transgenic mice (13,14). There have been some studies on the
relationship between HBx and Notch. Researchers from a Chinese
laboratory reported that HBx promotes the growth of human hepatic
L02 and HCC HepG2 cells via the activated Notch pathway, and Notch1
is a potential therapeutic target for the treatment of
HBx-associated HCC (15–17). However, this same team also
published a study reporting that HBx downregulates NF-κB signaling
through decreasing Notch signaling pathway in L02 cells (18), which seems the opposite to their
previous results and is therefore puzzling. Besides, there was
still another study showing that HBx decreases ICN1 in HCC Huh7
cells by reducing Notch1 cleavage, which results in enhanced cell
proliferation (19). These results
are inconsistent, and we noted that they are all based on cell
research and data from tissue samples is lacking.
Therefore, it is very necessary to investigate the
overall expression of Notch receptors and their relationship with
HBx in HBV-associated HCC tissues. In the present study, we
examined the expression and activity of Notch in both
HBV-associated HCC tissues and HBx expressed HCC cells and found
that HBx activated Notch signaling by its effects on Notch1 and
Notch4 in HBV-associated HCC.
Materials and methods
Human tissue samples, cell lines and
drugs
HCC tissue samples were obtained from 44 patients
with HBV infection who received surgical resection at Xijing
Hospital of the Fourth Military Medical University from 2002 to
2004 with informed consents of the the patients and with the
approval of the Human Research Committee of the University and
Wuhan General Hospital. pcDNA3-HBx was constructed by inserting a
wild-type HBx cDNA fragment into pcDNA3 (Invitrogen, Carlsbad, CA,
USA). pcDNA3 stable transfected HepG2 cells (HepG2-pc) and HepG2X
cells were established in our perious study (20). HeLa cells were purchased from the
American Type Culture Collection (ATCC; Manassas, VA, USA). For
drug treatment, HepG2-pc and HepG2X cells were treated with the
following drugs for 24 h at different final concentrations (5, 10,
20 and 40 μM) to inhibit the activity of MEK, PI-3K and p38 MAPK:
PD98059 (Calbiochem, Darmstadt, Germany), LY294002 (Calbiochem),
SB203580 (Alexis Biochemicals, San Diego, CA, USA). DMSO (20 μM)
was used as drug control. For suppression of Notch signaling,
HepG2-pc and HepG2X cells were treated with
N-[N-(3,5-difluorophenacetyl-L-alanyl)]-S-phenyl glycine t-Butyl
Ester (DAPT; Calbiochem) at different final concentrations (2.5, 5
and 10 μM). DMSO (5 μM) was used as drug control.
Luciferase assay
pGa981-6, a reporter gene plasmid which contained a
hexamerized 50 bp Epstein-Barr virus nuclear antigen 2 response
element (EBNA2RE) of the TP1 promotor in front of the luciferase
gene, was strictly dependent on RBP-J (21). pRL-TK (Promega, Madison, WI, USA)
was co-transfected as an internal control for transfection
efficiency. A negative control plasmid (neg-pGa981-6) was
constructed by replacing EBNA2RE with an irrelevant DNA segment.
For luciferase assay, pcDNA3-HBx, pGa981-6 and pRL-TK were
co-transfected into HepG2 cells with Lipofectamine™ 2000
(Invitrogen). DAPT at different final concentrations (2.5, 5 and 10
μM) was added to the transactivation system to suppress Notch
signaling. pcDNA3 and neg-pGa981-6 replacing pcDNA3-HBx and
pGa981-6, respectively, were used as negative controls. Forty-eight
hours later, co-transfected cells were lysed and luciferase assays
were performed in Luminometer TD-20/20 (Turner Designs, Inc.,
Sunnyvale, CA, USA).
Immunohistochemical staining
Horseradish peroxidase staining was used to
visualize antigens on paraffin-embedded 5-μm sections. Sections
were incubated with anti-Notch1 primary goat polyclonal antibody
(pAb), anti-Notch2, 3 and 4 primary rabbit pAb (1:100; Santa Cruz
Biotechnology, Santa Cruz, CA, USA), or anti-HBx mouse monoclonal
antibody (mAb) (1:50; Chemicon International, Temecula, CA, USA),
and then detected with Histostain™-SP (Zymed
Laboratories, San Francisco, CA, USA). In negative controls,
non-immune IgGs were used to substitute for the primary antibody.
All stained sections were evaluated by two independent
investigators who were blinded to the groups. The scoring was based
on intensity and the extent of the staining (22). The immunoreactive score of each
section was calculated by sum of these two parameters and graded as
negative (I), weak (II), moderate (III) and strong (IV).
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total cellular RNA was extracted using the One Step
RT-PCR kit (MBI, Vilnius, Lithuania). Semi-qualitative PCR was
performed using human primer sequences for Notch1-4 and for HBx as
in our previous reports (20,22).
β-actin was used as a housekeeping gene. Gene expression was
presented by the relative yield of the PCR product from target
sequences to that from β-actin gene.
Western blot analysis
Immunoblotting was performed using the anti-Notch1-4
pAbs (1:300), the anti-HBx mAb (1:100), anti-phospho-MAPKAPK-2
rabbit mAb (1:500; Cell Signaling Technology, Boston, MA, USA),
anti-phospho-p44/42 MAPK (Erk1/2) rabbit mAb (1:1,000; Cell
Signaling Technology) and anti-phospho-Akt rabbit mAb (1:500; Cell
Signaling Technology) followed by incubation with peroxidase
coupled anti-goat, anti-rabbit or anti-mouse IgG, respectively.
HeLa cells were used as positive control for the expression of
Notch receptors. Autoradiograms were quantified by densitometry
using Bio Image Intelligent Quantifier (IQ) software. Relative
protein levels were calculated by referring them to the amount of
β-actin protein.
Double immunofluorescence staining
HBV-associated HCC tissues and HepG2X cells were
stained with anti-Notch1 goat pAb and anti-HBx mouse mAb, or
anti-Notch4 rabbit pAb and anti-HBx mouse mAb, respectively. After
incubated with tetraethyl rhodamine isothiocyanate-labeled rabbit
anti-goat IgG (1:100; Chemicon) and fluorescein isothiocyanate
(FITC)-conjugated goat anti-mouse IgG (1:80; Chemicon) or
tetraethyl rhodamine isothiocyanate-labeled goat anti-rabbit IgG
(1:100; Chemicon) and FITC-conjugated goat anti-mouse IgG, all
sections were examined by a laser scanning confocal microscope
(Bio-Rad Laboratories, Hercules, CA, USA). Non-immune IgGs in
substitution for the primary antibodies were considered negative
controls.
Co-immunoprecipitation assay
HepG2X and HepG2-pc cells were lysed and the lysate
was pretreated with Protein G Plus/Protein A Agarose (Calbiochem)
to remove non-specifically bound proteins. Immunoprecipitation (IP)
was carried out with anti-HBx mouse mAb or non-immune mouse IgG and
the agarose beads. After incubated at 4°C for 4 h, the beads were
washed and subjected to western blot analysis with the anti-Notch1
pAb, anti-Notch4 pAb or anti-HBx mAb.
Cell proliferation and colony-forming
assay
For
3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT)
assay, HepG2-pc and HepG2X cells were seeded and cultured
overnight, then DAPT at different final concentrations was added
and 5 μM DMSO was used as drug control. After 1, 2, 3, 4, 5, 6 and
7 days of drug treatment, the cell survival rates were assessed
using MTT assay. The growth inhibitory rate was calculated as:
(control A value - treated A value)/control A value × 100%. For
colony-forming assay, HepG2-pc and HepG2X cells were treated as
above. After 2 weeks of incubation, cell colonies were fixed and
stained with Giemsa dye and colonies containing >50 cells were
counted. For anchorage-independent cell proliferation, 24-well
plates were coated with 0.5% of bottom agar solution, and
2×103 HepG2-pc and HepG2X cells were embedded into 0.3%
top agar gel containing DAPT of 2.5, 5 and 10 μM, respectively. The
dishes were examined with vertical microscope for colony formation
after a 2-week incubation period. Colonies of >75 mm was
counted.
Propidium iodide (PI) staining and flow
cytometry (FCM)
HepG2-pc (1×106) and HepG2X cells were
seeded and incubated overnight, then 5 μM DAPT was added. After 24
h of incubation, cells were harvested, fixed and resuspended in PI
solution. Samples were then analyzed for their DNA content by flow
cytometer (Beckman Coulter, Inc., Brea, CA, USA).
Apoptosis assay
For Annexin V binding assay, HepG2-pc and HepG2X
cells were treated with 5 μM DAPT, respectively. PI and Annexin V
were added to those cells successively, and the cells were analyzed
by FCM. Mean values from three independent experiments were
accepted as results. For transmission electron microscopy (TEM),
HepG2-pc and HepG2X cells treated with DAPT were fixed, dehydrated
and embedded. Cells were then sectioned and stained. The cell
ultrastructure was assessed by TEM at 4,000-fold magnification.
Statistical analysis
Statistical analysis was performed using the SPSS
software (version 13.0; SPSS, Inc., Chicago, IL, USA). Correlations
between Notch receptors and HBx were analyzed using Spearman's rank
correlation. Two-sided Student's t-test was used for comparisons.
P<0.05 was considered as statistically significant.
Results
HBx activates Notch signaling in HepG2
cells
To investigate whether Notch signaling could be
activated by HBx, luciferase assay was carried out with
co-transfection of pGa981-6 and pcDNA3-HBx into HepG2 cells. The
data showed that HBx activated Notch signaling in a dose-dependent
manner (P<0.05), and the activation was significantly suppressed
by 5 or 10 μM DAPT (Fig. 1). The
control sample with pcDNA3 replacing pcDNA3-HBx showed luciferase
activity to some extent, indicating the endogenous activity of
Notch signaling in HepG2 cells.
HBx upregulates the expression of Notch1
through p38 MAPK pathway and activates Notch4 in HBV-associated HCC
tissues and HepG2X cells
Since Notch signaling could be activated by HBx, we
wonder which Notch receptor is involved in the activation. For
this, we examined the expression of Notch1-4 in 44 HBV-associated
HCC tissues by immunohistochemical staining. The result showed that
Notch1-4 were all expressed in the neoplastic cells of HCCs with
different intensity and extensity. Cytoplasmic Notch1 expression
was strong and could be detected in 93.2% (41/44) of HCCs. Nuclear
Notch1 was weak and the positive rate was 11.4% (5/44). Notch2 and
Notch3, detected only in the cytoplasm, were expressed in 31.8%
(14/44) and 61.4% (27/44) of HCCs, respectively. Cytoplasmic and
nuclear expression of Notch4 was detected in 75.0% (33/44) and
63.6% (28/44) of HCCs, respectively. HBx immunoreactivity was
observed in 86.4% (38/44) of HCCs, with positive signal in the
cytoplasm (Fig. 2A). Spearman
analysis showed that there was a significant correlation between
HBx and cytoplasmic Notch1 or nuclear Notch4 immunoreactivity, with
rs=0.396, P<0.01 (HBx and Notch1), and rs=0.407, P<0.01 (HBx
and Notch4), whereas other Notch molecules and HBx did not show
such a relationship (Table I).
 | Table IExpression of Notch receptors and HBx
in HBV-associated HCC tissues. |
Table I
Expression of Notch receptors and HBx
in HBV-associated HCC tissues.
| Positive cases | Score | rs |
|---|
|
|---|
| I | II | III | IV |
|---|
| Notch1
(cytoplasmic) | 41 | 3 | 10 | 26 | 5 | 0.396a |
| Notch1
(nuclear) | 5 | 39 | 4 | 1 | 0 | |
| Notch2 | 14 | 30 | 12 | 2 | 0 | |
| Notch3 | 27 | 17 | 18 | 9 | 0 | |
| Notch4
(cytoplasmic) | 33 | 11 | 15 | 17 | 1 | |
| Notch4
(nuclear) | 28 | 16 | 11 | 16 | 1 | 0.407a |
| HBx | 38 | 6 | 26 | 12 | 0 | |
The expression of Notch molecules was further
examined in HepG2X cells. RT-PCR showed that the mRNA level of
Notch1 was much higher in HepG2X than that in HepG2-pc cells
(P<0.05). Notch2, Notch3 and Notch4 mRNA showed no difference
between the two cell lines (Fig.
2B). For the protein level, the result of the western blot
analysis showed that the proteins of Notch1-4 were all expressed in
HepG2-pc and HepG2X cells, with specific bands at ~70–120 kDa.
Notch1, Notch2 and Notch3 showed only one band, which was the Notch
transmembrane subunit (NTM) form (23). The expression of Notch1 was higher
in HepG2X than in HepG2-pc cells (P<0.05). Notch2 and Notch3
were equally expressed between the two cell lines. Notch4 showed
two bands, one of which with a higher molecular mass was its NTM
form, and the lower one was its intracellular region of Notch (ICN)
form (24). Although the NTM form
of Notch4 was equally expressed between HepG2X and HepG2-pc cells,
the ICN form appeared only in HepG2X (Fig. 2C).
Since HBx activates Ras/Raf/MEK, PI-3K/Akt and p38
MAPK pathways (25–27), and MAPK and p38 signaling can
upregulate the expression of various Notch molecules in certain
context (28,29), we wonder whether the regulation of
Notch1 by HBx is also mediated by these pathways. For this,
inhibitors of p38, MEK and PI-3K were used to treat HepG2X and
HepG2-pc cells, and the expression of their target as well as
Notch1 was examined. The result showed that 10 μM SB203580, 5 μM
PD98059 and 20 μM LY294002 began to inhibit phospho-MAPKAPK-2,
phospho-Erk1/2 and phospho-Akt in HepG2X cells, respectively, and
only SB203580 decreased the expression of Notch1 (NTM) (P<0.05).
In HepG2-pc cells, although 5 μM SB203580, 5 μM PD98059 and 10 μM
LY294002 began to suppress their targets respectively, none of them
could influence the expression of Notch1 (Fig. 2D). These results demonstrated that
HBx upregulated Notch1 and activated Notch4 in HBV-associated HCC,
and the upregulation of Notch1 by HBx was through p38 MAPK
pathway.
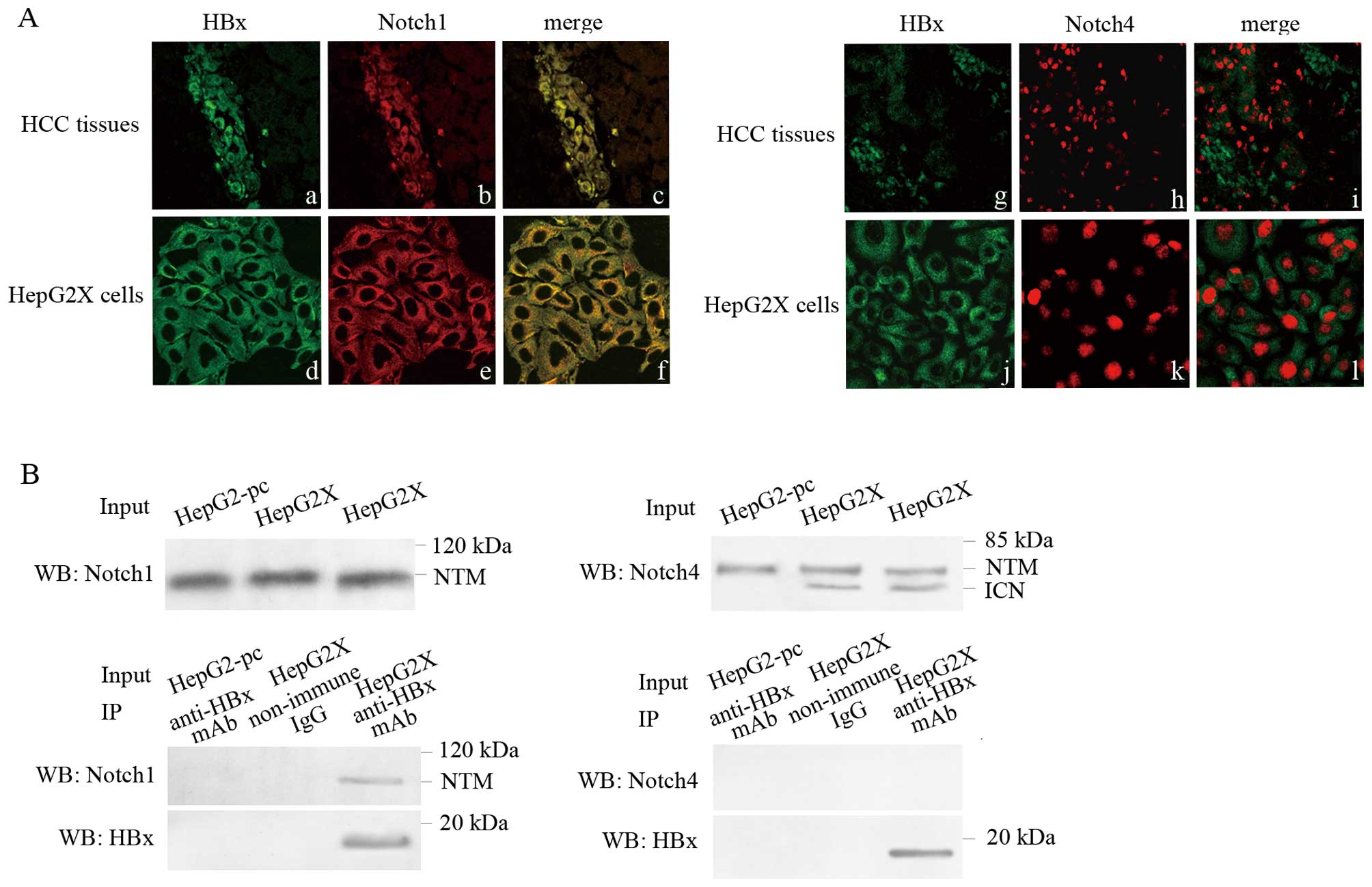
Notch1 morphologically co-localizes and
physically interacts with HBx
The relationship of Notch1, Notch4 with HBx
suggested a possible co-localization, and we explored this
possibility by confocal microscopy. As the result showed, HBx was
stained green and Notch1 was stained red. The yellow staining in
dual-labeling experiments indicated overlapping areas of red and
green fluorescent labels and suggested co-localization of Notch1
with HBx in the cytoplasm of neoplastic cells of HBV-associated HCC
tissues and HepG2X cells. For HBx and Notch4, no apparent yellow
fluorescent was observed in dual-labeling experiments (Fig. 3A). No fluorescent showed up in
negative control sections. Confocal analysis demonstrated that
Notch1, but not Notch4, morphologically co-localized with HBx.
We further examined the possible physical
interaction of HBx with these two Notch receptors in HepG2X cells
by co-immunoprecipitation assay. The result showed that Notch1
(NTM) was co-immunoprecipitated with HBx in HepG2X cells. No
interaction was observed between Notch4 and HBx. No interaction was
observed in HepG2-pc cells. In addition, no non-specific
interaction was observed from control non-immune IgG, indicating
the specificity of Notch1-HBx interaction (Fig. 3B).
Suppression of Notch signaling in HepG2X
inhibits cell growth and blocks cell cycle progression
To investigate the effects of the activation of
Notch signaling by HBx on HCC, the activity of Notch signaling in
HepG2X and HepG2-pc cells was suppressed by a highly specific GSI,
DAPT and cell growth was examined. The result from MTT showed that
the inhibition of DAPT on the growth of HepG2X cells were observed
at the final concentrations (5 and 10 μM) for 1 day (P<0.05),
and the degree of inhibition was positively correlated with
exposure time. The inhibition effect of 10 μM was similar with that
of 5 μM DAPT, which indicated 5 μM a suitable concentration
(Fig. 4A). The result of
colony-formation assay and soft agar assay also showed that 5 or 10
μM DAPT while not DMSO significantly reduced the capacity of single
cell colony formation and anchorage-independent cell proliferation
in HepG2X cells (P<0.05). Cell growth and colony formation in
HepG2-pc was not influenced significantly by DAPT (Fig. 4B and C). For cell cycle, PI
staining and FCM analysis demonstrated a significant increase in
the number of cells in G1 phase after HepG2X cells was treated with
5 μM DAPT while not 5 μM DMSO (P<0.05). Neither DAPT nor DMSO
influenced HepG2-pc cell cycle distribution (Fig. 4D). The above results showed that
suppression of Notch signaling in HepG2X inhibited cell growth and
proliferation, and blocked cell cycle progression.
Suppression of Notch signaling in HepG2X
induces cell apoptosis
Cell apoptosis was further examined. Apoptosis
histograms from PI staining showed that the percentage of apoptotic
cells in HepG2X dramatically increased to 23.4% after treated with
DAPT, compared to 0.98% when treated with DMSO (P<0.01)
(Fig. 5A). Apoptosis was also
assessed by Annexin V and PI double-staining assay. The percentage
of apoptotic cells (Annexin V+/PI−, early
stage; Annexin V+/PI+, late stage) of HepG2X
treated with DMSO was 2.3%. After treatment with 5 μM DAPT for 24
h, the percentages of apoptotic cells significantly increased to
19.1% (P<0.01) (Fig. 5B). We
also examined ultrastructure of cell apoptosis by TEM. DAPT (5 μM)
led to a characteristic pattern of apoptosis in HepG2X cells. No
such characteristic apoptosis pattern was observed in the DMSO
control (Fig. 5C). In all the
above assays, DAPT did not induce apparent cell apoptosis in
HepG2-pc. These results demonstrated that suppression of activity
of Notch signaling could lead to cell apoptosis in HepG2X
cells.
Discussion
The role of Notch in HCC has been studied
comprehensively. Our previous research demonstrated that Notch1
activation contributes to HCC cell growth and proliferation
(30). We also found that Jagged1
is highly expressed in HCC tissues and its expression is closely
related with HBx (20).
Afterwards, studies from other research teams showed that
downregulation of Notch1 inhibits the invasion and migration of HCC
cells by inactivating the cyclooxygenase-2/Snail/E-cadherin
pathway, and also by regulation of phosphatase and tensin homolog
(PTEN) and focal adhesion kinase (FAK) (31,32).
There are also some studies investigating the role of Notch in the
prognosis of HCC patients. Researchers found that patients with
high Notch1 or Notch3 expression are at a significantly increased
risk for shortened survival time in HCC patients (33). Both Notch1 expression and Notch4
overexpression are independent predictors of shorter
disease-specific survival in HCC (34). These reports indicate an oncogenic
role of Notch in HCC.
HBx has been reported to have an important effect on
the carcinogenesis and progression of HBV-associated HCC by its
regulation of a series of signal pathways and molecules and thus
also affects signal transduction and biological behavior of the
host cells. Although HBx has been inferred as an important
carcinogenic factor, more vital downstream effectors remain to be
identified. Since Notch signaling has been demonstrated to play an
oncogenic role in HCC, we studied its relationship with HBx and
investigated its role in HBV-associated HCC. Our results show that
HBx activates Notch signaling in a dose-dependent manner. To
explore which Notch receptor is involved in the activation, we
examined the expression of Notch1-4 in HBV-associated HCC tissues
and HepG2X cells. The results show that the level of Notch1 (NTM)
is upregulated by HBx. The inhibitor of p38 MAPK blocks the
increase of Notch1 by HBx, which indicates that HBx regulates
Notch1 through the p38 MAPK pathway. Moreover, ICN4 is found to
accumulate in HBV-associated HCC tissues and HepG2X cells. Since
ICN is rapidly degraded after it activates transcription in the
nucleus, the accumulation of ICN4 in our case is obviously due to
the activation of Notch4 by HBx. Therefore, we conclude that HBx
activates Notch signaling by its activation of Notch4.
Notch1 was found in a very small number of nuclear
positive samples in HBV-associated HCC tissues and did not detect
its activated form in HepG2X cells, which seems to indicate that it
is the expression, not the activity, of Notch1 which is regulated
by HBx. However, there is still another possible reason for the
undetectable ICN1, that is, the extremely poor stability of ICN1 in
our case. It has been found that ICN1 is degraded in the nucleus by
Fbw7 E3 ligase-mediating ubiquitination (35). Upon Notch receptor-ligand binding,
Jagged1 ligand undergoes a proteolytic cleavage, resulting in the
production of a free intracellular domain (JICD), which is reported
to reduce the stability of ICN1 through Fbw7-mediating
ubiquitination and degradation of the protein (36). In fact, Jagged1 was found to be
upregulated by HBx in our previous experiment (20). Thus, the undetectability of ICN1 in
the present study might be due to the increased JICD, which still
needs further investigation.
The relationship of Notch1, Notch4 with HBx
indicates a possible physical interaction between them. Actually,
we find that Notch1, but not Notch4, morphologically co-localizes
and physically interacts with HBx. Our result demonstrates that,
besides regulation of expression, the effects of HBx on Notch1
might also result from their interaction. HBx has been reported to
interact with various proteins to take part in the carcinogenesis
of HCC (37,38). The interaction of HBx with Notch1
in our case might lead to activation of Notch signaling or other
crosstalk signaling pathways, which would probably contribute to
the development of HBV-associated HCC.
Furthermore, we investigated the role of the
activated Notch signaling in HepG2X cells. After suppression of
Notch signaling, cell growth and proliferation of HepG2X is
significantly attenuated, while no apparent growth inhibition
occurs in the control HepG2-pc cells with a lower activity of Notch
signaling. Moreover, we found that suppression of Notch signaling
induces cell cycle arrest and cell apoptosis in HepG2X cells. Thus,
it can be concluded that HBx promotes cell growth and inhibits cell
apoptosis in HCC through its activation of Notch signaling.
Previous reports showed that Notch signaling can be activated by
several viral oncogenes, such as EBNA2 and human papillomavirus E6
and E7 proteins, and take part in the carcinogenesis of these
virus-associated cancers (39,40).
Thus, our finding indicates Notch signaling as a downstream pathway
activated by HBx in HBV-associated HCC.
In conclusion, the present study indicates that HBx
activates Notch signaling by its effects on Notch1 and Notch4 and,
therefore, recruits Notch signaling as a downstream pathway
contributing to its carcinogenic role in HBV-associated HCC.
Acknowledgements
The authors gratefully acknowledge the Department of
Pathology (Xijing Hospital, the Fourth Military Medical University)
for providing HCC tissue samples. The present study is supported by
the Military Medical Scientific Youth Cultivation Project (no.
13QNP053) and the Wuhan Young and Middle-aged Medical Backbone
Personnel Training Project.
References
|
1
|
Artavanis-Tsakonas S, Rand MD and Lake RJ:
Notch signaling: Cell fate control and signal integration in
development. Science. 284:770–776. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Davis RL and Turner DL: Vertebrate hairy
and Enhancer of split related proteins: Transcriptional repressors
regulating cellular differentiation and embryonic patterning.
Oncogene. 20:8342–8357. 2001. View Article : Google Scholar
|
|
3
|
Gao J, Liu J, Fan D, Xu H, Xiong Y, Wang
Y, Xu W, Wang Y, Cheng Y and Zheng G: Up-regulated expression of
Notch1 and Jagged1 in human colon adenocarcinoma. Pathol Biol
(Paris). 59:298–302. 2011. View Article : Google Scholar
|
|
4
|
Mazur PK, Riener MO, Jochum W, Kristiansen
G, Weber A, Schmid RM and Siveke JT: Expression and
clinicopathological significance of notch signaling and cell-fate
genes in biliary tract cancer. Am J Gastroenterol. 107:126–135.
2012. View Article : Google Scholar
|
|
5
|
Zhou M, Jin WY, Fan ZW and Han RC:
Analysis of the expression of the Notch3 receptor protein in adult
lung cancer. Oncol Lett. 5:499–504. 2013.PubMed/NCBI
|
|
6
|
Zhu H, Zhou X, Redfield S, Lewin J and
Miele L: Elevated Jagged-1 and Notch-1 expression in high grade and
metastatic prostate cancers. Am J Transl Res. 5:368–378.
2013.PubMed/NCBI
|
|
7
|
Jin HY, Zhang HY, Wang X, Xu J and Ding Y:
Expression and clinical significance of Notch signaling genes in
colorectal cancer. Tumour Biol. 33:817–824. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ozawa T, Kazama S, Akiyoshi T, Murono K,
Yoneyama S, Tanaka T, Tanaka J, Kiyomatsu T, Kawai K, Nozawa H, et
al: Nuclear Notch3 expression is associated with tumor recurrence
in patients with stage II and III colorectal cancer. Ann Surg
Oncol. 21:2650–2658. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ogawa R, Ishiguro H, Kimura M, Funahashi
H, Wakasugi T, Ando T, Shiozaki M and Takeyama H: NOTCH1 expression
predicts patient prognosis in esophageal squamous cell cancer. Eur
Surg Res. 51:101–107. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tiollais P, Pourcel C and Dejean A: The
hepatitis B virus. Nature. 317:489–495. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maguire HF, Hoeffler JP and Siddiqui A:
HBV X protein alters the DNA binding specificity of CREB and ATF-2
by protein-protein interactions. Science. 252:842–844. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kekulé AS, Lauer U, Weiss L, Luber B and
Hofschneider PH: Hepatitis B virus transactivator HBx uses a tumour
promoter signalling pathway. Nature. 361:742–745. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim CM, Koike K, Saito I, Miyamura T and
Jay G: HBx gene of hepatitis B virus induces liver cancer in
transgenic mice. Nature. 351:317–320. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shirakata Y, Kawada M, Fujiki Y, Sano H,
Oda M, Yaginuma K, Kobayashi M and Koike K: The X gene of hepatitis
B virus induced growth stimulation and tumorigenic transformation
of mouse NIH3T3 cells. Jpn J Cancer Res. 80:617–621. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang F, Zhou H, Yang Y, Xia X, Sun Q, Luo
J and Cheng B: Hepatitis B virus X protein promotes the growth of
hepatocellular carcinoma by modulation of the Notch signaling
pathway. Oncol Rep. 27:1170–1176. 2012.PubMed/NCBI
|
|
16
|
Wang F, Xia X, Wang J, Sun Q, Luo J and
Cheng B: Notch1 signaling contributes to the oncogenic effect of
HBx on human hepatic cells. Biotechnol Lett. 35:29–37. 2013.
View Article : Google Scholar
|
|
17
|
Sun Q, Wang R, Wang Y, Luo J, Wang P and
Cheng B: Notch1 is a potential therapeutic target for the treatment
of human hepatitis B virus X protein-associated HCC. Oncol Rep.
31:933–939. 2014.
|
|
18
|
Luo J, Zhou H, Wang F, Xia X, Sun Q, Wang
R and Cheng B: The hepatitis B virus X protein downregulates NF-κB
signaling pathways through decreasing the Notch signaling pathway
in HBx-transformed L02 cells. Int J Oncol. 42:1636–1643.
2013.PubMed/NCBI
|
|
19
|
Xu J, Yun X, Jiang J, Wei Y, Wu Y, Zhang
W, Liu Y, Wang W, Wen Y and Gu J: Hepatitis B virus X protein
blunts senescence-like growth arrest of human hepatocellular
carcinoma by reducing Notch1 cleavage. Hepatology. 52:142–154.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gao J, Chen C, Hong L, Wang J, Du Y, Song
J, Shao X, Zhang J, Han H, Liu J, et al: Expression of Jagged1 and
its association with hepatitis B virus X protein in hepatocellular
carcinoma. Biochem Biophys Res Commun. 356:341–347. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kato H, Taniguchi Y, Kurooka H, Minoguchi
S, Sakai T, Nomura-Okazaki S, Tamura K and Honjo T: Involvement of
RBP-J in biological functions of mouse Notch1 and its derivatives.
Development. 124:4133–4141. 1997.PubMed/NCBI
|
|
22
|
Gao J, Song Z, Chen Y, Xia L, Wang J, Fan
R, Du R, Zhang F, Hong L, Song J, et al: Deregulated expression of
Notch receptors in human hepatocellular carcinoma. Dig Liver Dis.
40:114–121. 2008. View Article : Google Scholar
|
|
23
|
Struhl G and Adachi A: Nuclear access and
action of notch in vivo. Cell. 93:649–660. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Das I, Craig C, Funahashi Y, Jung KM, Kim
TW, Byers R, Weng AP, Kutok JL, Aster JC and Kitajewski J: Notch
oncoproteins depend on gamma-secretase/presenilin activity for
processing and function. J Biol Chem. 279:30771–30780. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Benn J and Schneider RJ: Hepatitis B virus
HBx protein activates Ras-GTP complex formation and establishes a
Ras, Raf, MAP kinase signaling cascade. Proc Natl Acad Sci USA.
91:10350–10354. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shih WL, Kuo ML, Chuang SE, Cheng AL and
Doong SL: Hepatitis B virus X protein activates a survival
signaling by linking SRC to phosphatidylinositol 3-kinase. J Biol
Chem. 278:31807–31813. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tarn C, Zou L, Hullinger RL and Andrisani
OM: Hepatitis B virus X protein activates the p38 mitogen-activated
protein kinase pathway in dedifferentiated hepatocytes. J Virol.
76:9763–9772. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zeng Q, Li S, Chepeha DB, Giordano TJ, Li
J, Zhang H, Polverini PJ, Nor J, Kitajewski J and Wang CY:
Crosstalk between tumor and endothelial cells promotes tumor
angiogenesis by MAPK activation of Notch signaling. Cancer Cell.
8:13–23. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Weijzen S, Rizzo P, Braid M, Vaishnav R,
Jonkheer SM, Zlobin A, Osborne BA, Gottipati S, Aster JC, Hahn WC,
et al: Activation of Notch-1 signaling maintains the neoplastic
phenotype in human Ras-transformed cells. Nat Med. 8:979–986. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gao J, Dong Y, Zhang B, Xiong Y, Xu W,
Cheng Y, Dai M, Yu Z, Xu H and Zheng G: Notch1 activation
contributes to tumor cell growth and proliferation in human
hepatocellular carcinoma HepG2 and SMMC7721 cells. Int J Oncol.
41:1773–1781. 2012.PubMed/NCBI
|
|
31
|
Zhou L, Wang D, Li Q, Sun W, Zhang Y and
Dou K: The downregulation of Notch1 inhibits the invasion and
migration of HCC cells by inactivating the
cyclooxygenase-2/Snail/E-cadherin pathway in vitro. Dig Dis Sci.
58:1016–1025. 2013. View Article : Google Scholar
|
|
32
|
Hu Y, Li H, Qiu K, Li D, Zhou J, Hu Y and
Zhang F: Downregulation of Notch1 inhibits the invasion of human
HCC HepG2 and MHCC97H cells through the regulation of PTEN and FAK.
Int J Mol Med. 34:1081–1086. 2014.PubMed/NCBI
|
|
33
|
Zhou L, Zhang N, Song W, You N, Li Q, Sun
W, Zhang Y, Wang D and Dou K: The significance of Notch1 compared
with Notch3 in high metastasis and poor overall survival in HCC.
PLoS One. 8:e573822013. View Article : Google Scholar
|
|
34
|
Ahn S, Hyeon J and Park CK: Notch1 and
Notch4 are markers for poor prognosis of HCC. Hepatobiliary
Pancreat Dis Int. 12:286–294. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oberg C, Li J, Pauley A, Wolf E, Gurney M
and Lendahl U: The Notch intracellular domain is ubiquitinated and
negatively regulated by the mammalian Sel-10 homolog. J Biol Chem.
276:35847–35853. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim MY, Jung J, Mo JS, Ann EJ, Ahn JS,
Yoon JH and Park HS: The intracellular domain of Jagged-1 interacts
with Notch1 intracellular domain and promotes its degradation
through Fbw7 E3 ligase. Exp Cell Res. 317:2438–2446. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Choi YH, Kim HI, Seong JK, Yu DY, Cho H,
Lee MO, Lee JM, Ahn YH, Kim SJ and Park JH: Hepatitis B virus X
protein modulates peroxisome proliferator-activated receptor gamma
through protein-protein interaction. FEBS Lett. 557:73–80. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kalra N and Kumar V: The X protein of
hepatitis B virus binds to the F box protein Skp2 and inhibits the
ubiquitination and proteasomal degradation of c-Myc. FEBS Lett.
580:431–436. 2006. View Article : Google Scholar
|
|
39
|
Zimber-Strobl U and Strobl LJ: EBNA2 and
Notch signalling in Epstein-Barr virus mediated immortalization of
B lymphocytes. Semin Cancer Biol. 11:423–434. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Weijzen S, Zlobin A, Braid M, Miele L and
Kast WM: HPV16 E6 and E7 oncoproteins regulate Notch-1 expression
and cooperate to induce transformation. J Cell Physiol.
194:356–362. 2003. View Article : Google Scholar : PubMed/NCBI
|