Introduction
Malignant gliomas are the most common primary brain
tumor in the central nervous system. Gliomas are classified by the
2007 World Health Organization into four histopathologic grades
based on the degree of malignancy (1). Current therapeutic strategies for
gliomas consist of surgical resection, radiotherapy and
chemotherapy. However, the prognosis of patients suffering from
malignant glioma remains poor (2,3).
There is a pivotal need for novel molecular targets and approaches
to treat this fatal disease. Over the past several decades, genetic
studies have implicated number of protein coding genes in the
formation and progression of malignant gliomas; however, the
function of non-coding RNAs, especially long non-coding RNAs
(lncRNAs), in glioma pathogenesis remains largely unknown.
LncRNAs are a class of non-coding RNA transcripts
longer than 200 nucleotides with little or no protein-coding
capacity (4). Recently, increasing
evidence has revealed that more genomic sequences are transcribed
into lncRNAs than protein-coding RNAs (5). In addition, a number of studies have
shown that significant numbers of lncRNAs are involved in a variety
of biological processes through multiple regulation of mechanisms
(6–9). MEG3, identified as a member of
lncRNAs, is an imprinted gene with maternal expression which
encodes a non-coding RNA (10).
Dysregulation of MEG3 has been found in various human tumors
including hepatocellular carcinoma, renal cell carcinoma and
neuroblastoma (11,12). Moreover, MEG3 expression was lost
in the majority of clinically non-functioning human pituitary
tumors, and it suppressed cancer cell growth, stimulated
p53-mediated transcriptional activation, and selectively activated
p53 target genes (13,14).
MEG3 expression is under epigenetic control, and the
loss of MEG3 expression due to aberrant CpG methylation of MEG3 has
been observed in ovarian cancer (15). Previous studies suggest that DNA
methyltransferase 1 (DNMT1) overexpression may be responsible for
the aberrant DNA methylation of tumor-related genes in gliomas
(16). In addition,
5-aza-2-deoxycytidine (5-AzadC), a DNMT inhibitor, induced
re-expression of MEG3 due to demethylation of MEG3 in
hepatocellular cancer (17).
It was hypothesized that epigenetic control of MEG3
by DNA methylation would result in the discovery of novel and
specific epigenetic regulators of gliomas. The present study
determines that DNA methylation mediates the repression of MEG3
transcription in glioma. In addition, experimental evidence
suggests that hypermethylation of MEG3 mediated by DNMT1 is a
molecular cause for glioma.
Materials and methods
Materials
Dulbecco's modified Eagle's medium (DMEM) and fetal
bovine serum (FBS) were obtained from Hyclone (Logan, UT, USA).
Lipofectamine 2000, TRIzol® reagent was purchased from
Invitrogen (Carlsbad, CA, USA). M-MLV Reverse Transcriptase was
purchased from Promega (Madison, WI, USA; cat. M1705). All other
chemicals were obtained from Sigma (St. Louis, MO, USA). The
antibodies used were as follows: anti-DNMT1 (1:500 dilution; Santa
Cruz Biotechnology, Inc.); anti-MDM2 (1:500 dilution; Boster,
China); anti-p53 (1:500 dilution; Boster); anti-β-actin (1:2,000
dilution; Santa Cruz Biotechnology, Inc.). DNA extraction kit was
acquired from Axygen. Streptavidin peroxidase (SP)
immunohistochemical kit was acquired from Zhongshan Biotechnology
Corp. (Beijing, China).
Patients and tissue samples
Tissue samples from normal brain tissues and human
glioma tumors were collected from the Neurosurgery Department of
The Second Affiliated Hospital of Anhui Medical University (Hefei,
China). Samples were collected and snap-frozen in liquid nitrogen
immediately and preserved in −80°C until use, and their
histological type was further confirmed using the standard
hematoxylin and eosin staining according to the WHO criteria. A
total of 71 samples were used for this study including
primary-grade pilocytic astrocytomas or grade II astrocytomas (WHO
I/II, n=23), grade III anaplastic astrocytomas or grade IV
glioblastoma multiforme (WHO III/IV, n=36) and normal brain tissues
derived from the temporal lobes and saddle area of the patients
with arachnoid cyst after surgery (n=12), both glioma patients and
controls were similar with respect to sex and age. This study was
approved by the Research Ethics Committee of The Second Affiliated
Hospital of Anhui Medical University. Informed consent was obtained
from all the patients.
Immunohistochemistry
We studied 62 glioma patients who had been
surgically treated in Department of Neurosurgery, The Second
Affiliated Hospital of Anhui Medical University, Hefei, China. For
immunohistochemistry, resected glioma tissues were fixed in 10%
formalin solution and embedded in paraffin. Histological slices of
3 mm were prepared, then deparaffined in xylene, and dehydrated
with ethanol. Endogenous peroxidase was blocked with 0.3%
H2O2 in methanol for 20 min at room
temperature (RT). Following antigen retrieval, the sections were
blocked with 5% BSA for 20 min at RT and then probed with 1:500
rabbit anti-DNMT1 at 4°C overnight. After washing, the sections
were incubated with biotinylated goat anti-rabbit immunoglobulins
at RT for 1 h, and visualized using the peroxidase conjugated
streptavidin and diaminobenzidine, followed by counterstaining with
Mayer's hematoxylin. Results of the immunohistochemical (IHC)
staining were evaluated by a pathologist blinded to all clinical
data. The criterion for IHC scoring was that a specimen with
>10% positive staining tumor cells should be considered
positive, and our IHC data for pharyngeal cancer were classed as
positive or negative accordingly.
Cell culture
U251, U87 and A172 human glioma cell lines were
obtained from the Chinese Academy of Sciences Cell Bank. Cells were
maintained in DMEM supplemented with 10% heat-inactivated FBS and
100 U/ml penicillin/streptomycin at 37°C in humidified atmosphere
of 5% CO2.
5-aza-2′-deoxycytidine treatment
U251 and U87 glioma cells were seeded overnight in
culture dishes, 5-AzadC (Sigma-Aldrich) was added and was refreshed
every 24 until 72 h treatment finished. The medium containing PBS
only was regarded as a control.
Methylation-specific polymerase chain
reaction
The methylation status of the MEG3 promoter region
was determined by methylation-specific PCR(MSP) using
bisulfite-modified DNA. Genomic DNA was extracted using the QIAamp
DNA mini kit (Axygen). Two primer sets were used to amplify the
promoter region of the MEG3 gene that incorporated a number of CpG
sites, one specific for the methylated sequence (MEG3-M, forward,
5′-TATGAGTTGTAAGCGGTAGAGTTC-3′; reverse,
5′-TACGAACTTAACGAAAAAAAATCAT-3′) and the other for the unmethylated
sequence (MEG3-U, forward, 5′-GAATATGAGTTGTAAGTGGTAGAGTTT-3′;
reverse, 5′-TACAAACTTAACAAAAAAAAATCATACT-3′). The primers used in
the present study detect specifically the promoter sequence of the
MEG3 gene rather than that of the MEG3 pseudogene. For MSP reaction
we used the HotStarTaq Master Mix kit (Qiagen). PCR reactions were
carried out in a final 25 μl volume containing: HotStarTaq Master
mix (final concentration 1X PCR buffer, 2.5 U Hot Star Taq DNA
polymerase, 200 μm of each dNTP and 1.5 mM MgCl2), 0.4
μM primers and bisulphite-modified DNA (~30 ng). Amplification was
performed in a thermocycler with the following conditions: 94°C for
2 min, cycled at 94°C for 30 sec, 54°C or 50°C for 30 sec, and 72°C
for 45 sec (36 cycles) followed by extension at 72°C for 7 min. MSP
experiments were performed at least in duplicate.
RNA interference (RNAi) analysis
RNAi experiments in glioma cells were performed by
forward transfection in cultured glioma cells (2×105
cells per 200 mm2 dish) using Lipofectamine RNAiMax
(Invitrogen) according to the manufacturer's protocol. For DNMT1,
MDM2 and p53 immunoblotting, glioma cells were cultured in
serum-free DMEM for 12 h and then subjected to reverse transfection
with RNAiMax in Opti-MEM. Small interfering RNA (siRNA)
oligonucleotides against DNMT1 genes or scrambled sequences were
synthesized by the Shanghai GenaPharma Corp. The following siRNA
sequences were used: si-DNMT1 (human), 5′-GGGACUGUGUCUCUGUUAUTT-3′
(sense) and 5′-AUAACAGAGACACAGUCCCTT-3′ (antisense); si-control
with scrambled sequence (negative control siRNA having no perfect
matches to known human genes), 5′-UUCUCCGAACGUGUCACGUTT-3′ (sense)
and 5′-ACGUGACACGUUCGGAGAATT-3′ (antisense). Transfection was
allowed to proceed for various times and cells were processed for
different assays. The siRNA transfection efficiency of
Lipofectamine RNAiMax in cells was determined by the BLOCK-iT Alexa
FluorR Red Fluorescent Oligo protocol (Invitrogen). Significance of
differences was assessed using paired one tailed t-test.
Real-time PCR analysis
Total RNA was extracted from glioma specimen or
glioma cells using TRIzol reagent (Invitrogen). Real-time
quantitative PCR analysis was performed using SYBR Green Master Mix
kit on Thermo Fisher connect Real-Time PCR platform. In brief, each
PCR reaction mixture containing 10 μl of 2X SYBR GreenMaster Mix, 1
μl of sense and antisense primers (5 μmol/μl) and 1 μl of cDNA (10
ng), was run for 40 cycles with denaturation at 95°C for 15 sec,
annealing at 60°C for 30 sec, and extension at 72°C for 30 sec, in
a total volume of 20 μl. For relative quantification,
2−ΔΔCT was calculated and used as an indication of the
relative expression levels, which was calculated by subtracting CT
values of the control gene from the CT values of MEG3 and DNMT1.
Real-time PCR was carried out under a standard protocol using the
following primers: DNMT1 (forward, 5′-CGGCTTCAGCACCTCATTTG-3′;
reverse, 5′-AGGTCGAGTCGGAATTGCTC-3′), MEG3 (forward,
5′-ATCATCCGTCCACCTCCTTGTCTTC-3′; reverse,
5′-GTATGAGCATAGCAAAGGTCAGGGC-3′). GAPDH was applied as an internal
control. The primer sequences of GAPDH were
5′-AGCAAGAGCACAAGAGGAAG-3′ and 5′-GGTTGAGCACAGGGTACTTT-3′.
MTT assay
U251 and U87 glioma cells were trypsinized,
resuspended, seeded into a 96-well plate with a concentration of
2,000 cells per well, and incubated at 37°C 3 days post-siRNA
transfection. The number of viable cells was measured at daily
intervals (at 12, 24, 48 and 72 h). At each time-point, 10 μl of 5
mg/ml MTT (Dingguo Biotechnology) was added, and incubation was
continued for 4 h. Then the medium was removed carefully and 150 μl
of DMSO was added at the end of incubation. The absorbance was
measured at 592 nm on the spectrophotometer.
Colony formation assay
A total of 200 U251 and U87 glioma cells were seeded
in 6-well plates after 3 days of siRNA transfection. The medium was
changed at regular time intervals. After 11 days of culture at
37°C, the natural colonies were washed with PBS and fixed with 4%
paraformaldehyde for 30 min at room temperature. The colonies were
then stained with Giemsa for 10 min, washed with water and
air-dried. The total number of colonies with >50 cells was
counted under fluorescence microscopy.
Cell apoptosis assay
For cell apoptosis assay, cells were plated into
6-well plates at 1×105 cells per well. Several days
after siRNA transfection or 5-AzadC treatment, the cells were
harvested by trypsinization and washed with PBS. Annexin
V-fluorescein isothiocyanate and PI double-staining (BD
Biosciences) was used to detect and quantify cellular apoptosis by
FCM. All the tests were performed in triplicate.
Western blotting
U251 and U87 glioma cells were lysed with RIPA lysis
buffer (Beyotime, China). Whole extracts were prepared, and protein
concentrations were determined using the BCA protein assay kit
(Boster). Whole-cell extracts (20 or 40 μg) were then fractionated
by electrophoresis through an 8 or 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Gels were
run at a 120 V for 2 h before transfer onto a PVDF membrane
(Millipore Corp., Billerica, MA, USA). After blocking against
non-specific protein binding, nitrocellulose blots were incubated
for 1 h with primary antibodies diluted in TBS/Tween-20 (0.075%
Tween-20) containing 3% Marvel. Anti-DNMT1, anti-MDM2 and anti-p53
were diluted 1:400. Following incubation with the primary antibody,
blots were washed three times in TBS/Tween-20 before incubation for
1 h with goat anti-mouse or mouse anti-rabbit horseradish
peroxidase conjugated antibody at a 1:10,000 dilution in
TBS/Tween-20 containing 5% milk. After extensive washing in
TBS/Tween-20, the blots were rinsed with distilled water and
proteins were detected using the enhanced chemiluminescence system.
Proteins were visualized with ECL-chemiluminescent kit (ECL-plus,
Thermo Scientific).
Statistical analysis
The data are expressed as mean ± SD of three
independent experiments. Statistical analysis was performed using
the Student's t-test and the one-way analysis of variance (ANOVA).
Pearson's test was performed to calculate the association between
DNMT1 and MEG3 expression. Significance was defined as
P<0.05.
Results
MEG3 methylation status and loss of MEG3
expression in glioma
To determine the expression levels of MEG3 in glioma
samples and cell lines, total RNAs were extracted from glioma
tissues at low-grade (grade I and II), high-grade (grade III and
IV), and normal brain tissue samples, and the expression level of
MEG3 was analyzed using qRT-PCR. As shown in Fig. 1A, the levels of MEG3 expression
were significantly decreased in glioma tissues compared with normal
brain tissues. To explore whether there is any association between
the downregulation of MEG3 and the pathogenesis of glioma, we
detected the expression of MEG3 in glioma samples with different
grades and found marked downregulation in high-grade glioma samples
and to a lesser degree, decreased in low-grade glioma samples, as
compared to normal brain tissues, which gave clues that MEG3 may be
involved in the disease pathogenesis in glioma (Fig. 1B). Of note, 3 glioma cell lines
displayed significantly downregulated expression of MEG3 relative
to that in normal brain tissues (Fig.
1C).
To investigate the mechanism underlying the
decreased MEG3 expression, we analyzed whether MEG3 promoter region
hypermethylation is responsible for the downregulation of MEG3
expression. Methylation-specific PCR analysis indicated that the
promoter region of MEG3 from glioma tissues in which MEG3
expression was downregulated was strongly methylated, whereas
normal brain tissues and low-grade glioma tissues in which MEG3
expression was present had unmethylated MEG3 promoter region
(Fig. 1D). These results suggest
that methylation of MEG3 promoter likely contributes to the loss of
MEG3 expression in high-grade glioma.
MEG3 downregulation due to the
hypermethylation of its promoter region is restored after treatment
of glioma cells with the DNMT inhibitor 5-AzadC
To further confirm that the loss of MEG3 expression
was caused by promoter methylation in U87 and U251 glioma cells
in vitro, the DNMT inhibitor 5-AzadC was used to inhibit
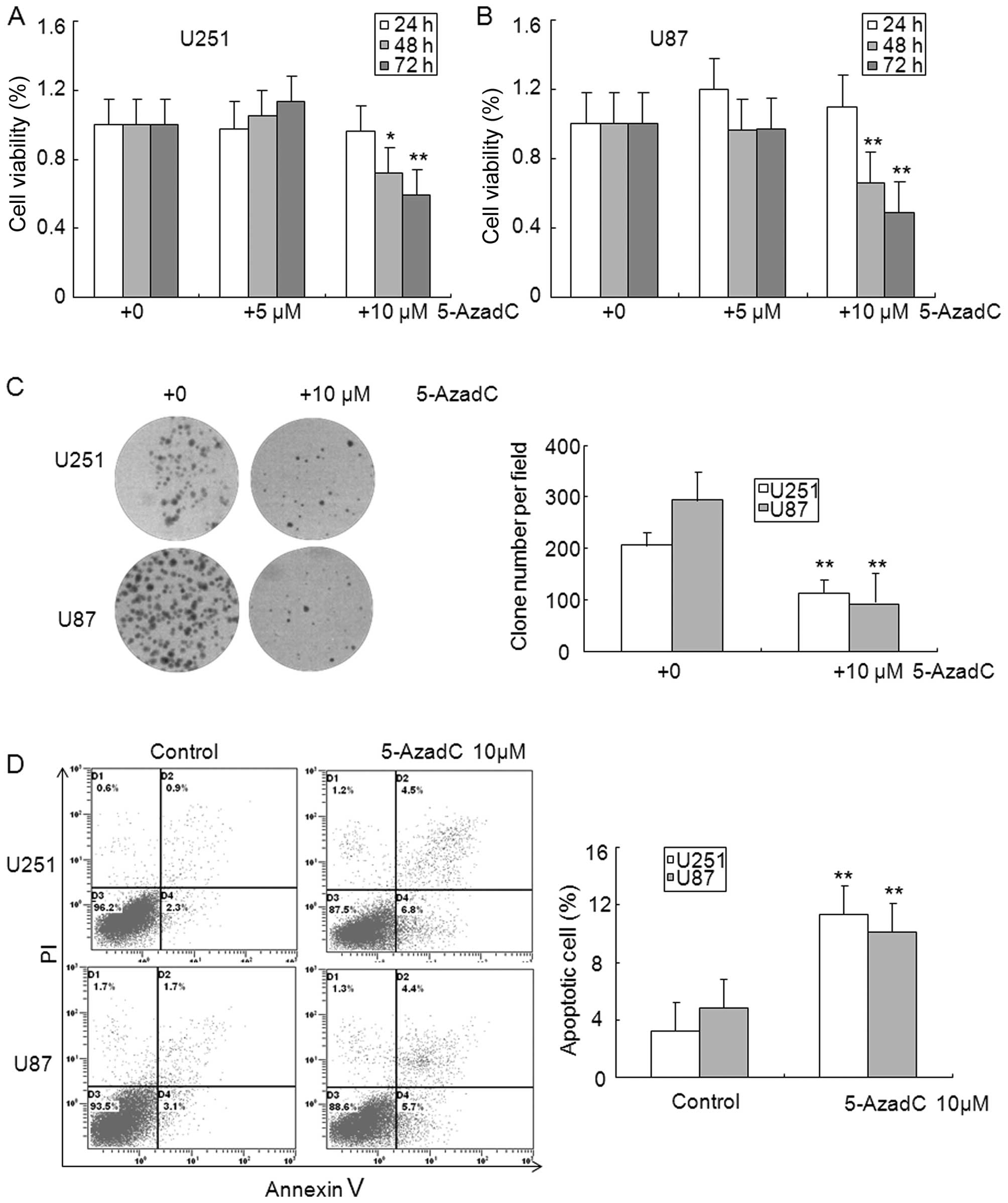
methylation. As illustrated in Fig. 2A
and B, treatment with 10 μM 5-AzadC markedly inhibited the
expression of DNMT1, and upregulated the expression of MEG3,
however, expression of DNMT1 and MEG3 did not show significant
changes in 5 μM 5-AzadC treated glioma cells. In addition, aberrant
hypermethylation of MEG3 was diminished in 5-AzadC treated glioma
cells compared with control cells (Fig. 2C). The MTT assay showed that
exposure with 5-AzadC for 48 and 72 h significantly inhibited the
cell viability of U251 and U87 glioma cells (Fig. 3A and B). The clone formation assay
was performed to further confirm the effect of 5-AzadC on glioma
cell proliferation, and data indicated that 5-AzadC treatment
significantly inhibited the number of clones in U251 and U87 glioma
cells (Fig. 3C). Reduction of
glioma cell growth by 5-AzadC treatment suggests that these cells
may undergo programmed cell death. To test this hypothesis, we
analyzed cell apoptosis induced by 5-AzadC using Annexin V and PI
double staining. As shown in Fig.
3D, 5-AzadC treatment significantly induced apoptosis of U251
and U87 glioma cells compare to control cells. These results
suggest that the decreased expression of MEG3 is related to
reversible epigenetic mechanisms such as DNA promoter region
methylation in glioma.
DNMT1 is involved in hypermethylation of
MEG3 in glioma cells
To gain insights into the possible involvement of
DNMT1 in glioma, we examined the expression of DNMT1 in glioma
tissues. As shown in Fig. 4A, the
levels of DNMT1 mRNA expression were significantly upregulated in
gliomas tissues compared with normal brain tissues. The levels of
DNMT1 mRNA expression were positively correlated to glioma grades,
with lower grades exhibiting upregulation >3-fold and higher
grades demonstrating a >5-fold increase in DNMT1 expression
(Fig. 4B). These data provided us
with experimental evidence that DNMT1 expression was upregulated in
a graded manner in gliomas, with higher grades showing highest
expression. In addition, immunohistochemical analyses showed that
DNMT1 expression was significantly increased in high-grade glioma,
and low-grade glioma compared with normal brain tissues, suggesting
this gene is associated with glioma malignancy (Fig. 4C). Next, we investigated whether
levels of DNMT1 mRNA expression was inversely correlated with MEG3
expression in glioma tissues. A statistically significant inverse
correlation was observed between DNMT1 mRNA and MEG3 (Fig. 4D), supporting the role of DNMT1 in
expression of MEG3.
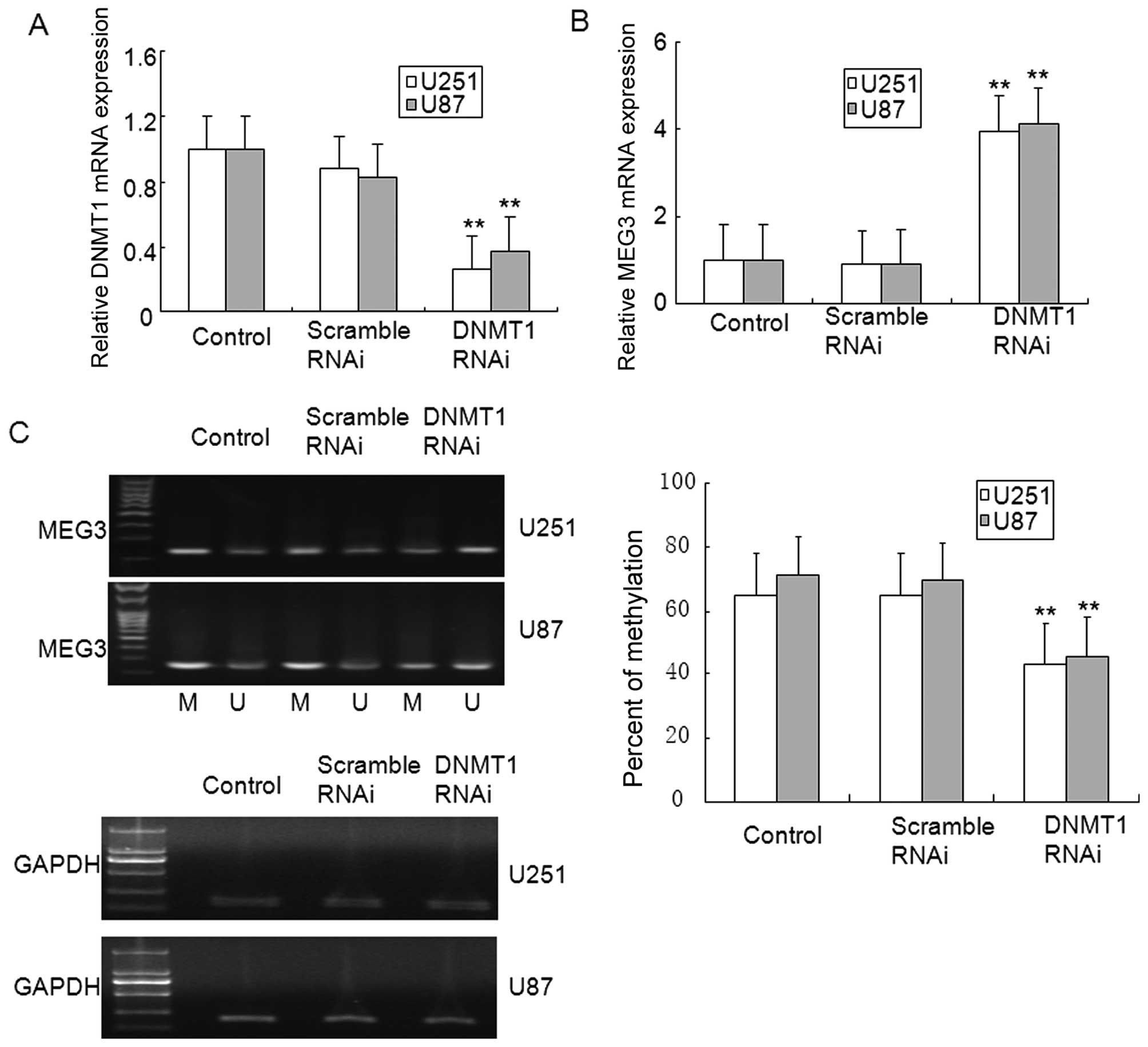
To determine the role of DNMT1 in glioma, the
expression of DNMT1 was inhibited by DNMT1 RNAi. Data from the MTT
assay showed that transfection with DNMT1 RNAi, but not scrambled
RNAi for 48 and 72 h reduced the cell viability of U251 and U87
glioma cells (Fig. 5A and B). The
results from the clone formation assay verified the MTT results,
and the finding that DNMT1 RNAi inhibited clone formation in U251
and U87 glioma cells (Fig. 5C).
The apoptosis of glioma cells was also observed in DNMT1 knockdown
U251 and U87 glioma cells (Fig.
5D). In addition, DNMT1 knockdown in U251 and U87 glioma cells
ameliorated MEG3 methylation, and restored MEG3 mRNA expression
(Fig. 6A–C). These results suggest
that MEG3 hypermethylation was dependent on DNMT1 in glioma.
The downregulation of MEG3 due to
hypermethylation of MEG3 contributes to the inhibition of p53
pathway in glioma cells
To study the potential signaling pathways affected
by MEG3 methylation, the protein levels of both MDM2 and p53 were
examined in glioma cells. As illustrated in Fig. 7A, treatment with 5-AzadC reduced
the expression levels of DNMT1 and MDM2 proteins in U251 and U87
glioma cells, whereas upregulated the levels of p53 protein
expression. Of note, the repression of MDM2 expression mediated by
MEG3 may be correlated with enhanced expression of p53 protein.
Finally, the effect of DNMT1 silencing on the
activation of p53 pathway was examined in glioma cells. As
illustrated in Fig. 7B, knockdown
of DNMT1 significantly repressed the expression of DNMT1 and MDM2
protein compared to control or scrambled U251 and U87 glioma cells,
however, the expression levels of p53 protein were induced. These
results suggest that an aberrant methylation occurred in MEG3
promoter contributes to the decrease in p53 expression by the
activation of MDM2 in gliomas.
Discussion
Malignant glioma is the most common and fatal brain
tumor world-wide (18). In
addition to conventional therapeutic strategies, targeted therapies
are currently identified to interfere with the transduction of
pivotal signaling pathways or to suppress the function of tumor
specific molecules in malignant glioma. It is widely accepted that
the future treatment options for glioma will greatly benefit from
our improved understanding of the complex molecular mechanism in
gliomas.
Maternally expressed gene 3 (MEG3), an imprinted
gene, is a tumor suppressor gene located in chromosome 14q32
(19). MEG3 is downregulated in a
number of cancer types, suggesting that MEG3 is a potential tumor
suppressor gene. In the present study, our findings revealed that
MEG3 expression was markedly decreased in glioma tissues, and
several glioma cell types compared with normal brain tissues. Loss
of MEG3 expression can result from epigenetic modification, such as
microRNA or promoter hypermethylation (20,21).
Hypermethylation mostly occurs at the promoter of genes that are
involved in processes resulting in tumor formation and progression
and has been shown for diverse types of genes related to tumor
suppression, DNA repair, cell cycle regulation, apoptosis,
invasion, and migration. Recently, several reports have shown that
loss of MEG3 expression and promoter hypermethylation of MEG3 were
observed in several types of human tumors, including pituitary
adenomas, neuroblastomas, pheochromocytomas, Wilms tumors, and
other carcinomas (12,22). In the present study, marked
hypermethylation of MEG3 and the loss of MEG3 expression was found
in high grade gliomas. These results suggest that the
downregulation of MEG3 expression probably attributed, at least in
part, to epigenetic modification by the hypermethylation of MEG3
promoter in gliomas in vivo.
5-AzadC is a nucleoside analog of cytidine in which
the 5-carbon of the pyrimidine ring is replaced by a nitrogen.
Following incorporation into DNA, 5-AzadC covalently binds to DNMTs
through the nitrogen in the 5-position of the modified pyrimidine
and traps it for proteosomal degradation, leading to inhibition of
the activity of the maintenance DNA methyltransferase DNMT1
(23,24). A recent study by Tsai et al
showed that treatment of cancer cells with DNMT inhibitors resulted
in sustained changes in gene expression and critical signaling
pathways involved in tumorigenesis (25). Our present results showed that
treatment of glioma cells with 5-AzadC reversed the methylation of
the MEG3, and restored MEG3 expression. Zhang et al reported
that overexpression of MEG3 suppresses DNA synthesis, colony
formation in meningioma cells (21). In addition, forced expression of
MEG3 inhibited the proliferation and induced apoptosis in glioma
cells (26). In the present study,
we demonstrated that 5-AzadC treatment inhibited the growth of
glioma cells, and resulted in apoptosis of glioma cells. These
findings suggest that 5-AzadC inhibits hypermethylation of MEG3 and
subsequently induces MEG3 expression, followed by blocking the
growth and inducing the apoptosis of glioma cells.
DNA methylation involves transfer of a methyl group
to the 5-carbon in cytosine of a CpG dinucleotide by DNA
methyl-transferases (DNMTs) - DNMT1, DNMT3A, and DNMT3B. DNMT1 is
the main enzyme responsible for maintaining the methylation pattern
onto daughter strands after DNA replication (27). DNMT1 is frequently upregulated in
various human cancers such as hepatic, prostate, and gliomas
(28–30). In this study, we found that the
upregulation of DNMT1 expression associated with gliomas malignancy
is observed in gliomas specimens. Selective depletion of DNMT1 has
been shown to induce demethylation of promoters and re-expression
of the silenced genes, including tumor suppressor genes or tumor
suppressive miRNAs, due to hypermethylation (31,32).
Here, we demonstrated that knockdown of DNMT1 inhibited the growth
of glioma cells and induced apoptosis of glioma cells, and reversed
the methylation of MEG promoter and subsequently restored MEG3
expression. These findings suggest that the upregulation of DNMT1
promotes MEG3 hypermethylation with the downregulation of MEG3
expression, followed by promotion of the growth of glioma
cells.
Tumor suppression is a cellular defense mechanism
preventing the neoplastic transformation of normal cells, and the
tumor suppressor p53 has a central role in the initiation and
progression of tumors (33). P53
levels are drastically lowered due to rapid degradation via the
ubiquitin-proteasome pathway. The ubiquitination of p53 is mainly
regulated by MDM2, referred to as an E3 ubiquitin ligase. A
decrease in MDM2 protein level was observed in non-small cell lung
cancer cells transfected with MEG3 (34). Overexpression of MEG3 resulted in a
significant increase in p53 protein levels and dstrongly stimulated
p53-dependent transcription. Moreover, MDM2 levels were
downregulated in cells transfected with MEG3, suggesting that MDM2
suppression contributes, at least in part, to p53 accumulation
induced by MEG3 (14). In the
present report, we provide two lines of evidence suggesting that
epigenetic silencing of MEG3 results in the repression of p53
signal pathways and subsequently regulates the growth of glioma
cells. First, the inhibition of DNMT1 by 5-AzadC, or its specific
downregulation by siRNA reversed the MEG3 promoter methylation and
restored MEG3 expression. Second, treatment with 5-AzadC or
knockdown of DNMT1 repressed the expression of MDM2 and induced the
activation of p53 pathways. MEG3 is known to suppress the
activation of MDM2 pathways, and following the activation of p53
pathway. Therefore, the downregulation of MEG3 expression in glioma
cells results from the occurrence of aberrant hypermethylation in
the MEG3 promoter. Such downregulation of MEG3 contributes to MDM2
pathways, and subsequently represses the p53 pathway.
In conclusion, our findings from the present study
strongly suggest MEG3 as a candidate tumor suppressor gene
associated with the pathogenesis and progression of human gliomas.
The expression of MEG3 could be restored via epigenetic
modification, which may suggest therapeutic potential for the
treatment of gliomas. To our knowledge, this is the first report on
hypermethylation of MEG3 as a mechanism of the development of
gliomas. MEG3/Gtl2 is a single-copy gene, these isoform expression
patterns are tissue and cell type specific. Twelve different MEG3
gene transcripts were generated by alternative splicing, using
different exons in the middle of the RNA (35). Further investigation of the
structure-function relationship of MEG3 is necessary to provide
more information associated with the pathogenesis of human gliomas
to reveal novel mechanisms to broaden our knowledge of the
involvement of non-coding RNAs in human gliomas biology, and
eventually to develop new therapeutic strategies for gliomas.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 81402078), Natural Science
Foundation of Anhui Province (no. 1508085MH194).
Abbreviations:
|
lncRNAs
|
long non-coding RNAs
|
|
MEG3
|
maternally expressed gene 3
|
|
DNMT1
|
DNA methyltransferase 1
|
|
5-AzadC
|
5-aza-2-deoxycytidine
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
FBS
|
fetal bovine serum
|
|
SP
|
streptavidin peroxidase
|
|
IHC
|
immunohistochemical
|
|
MSP
|
methylation-specific PCR
|
|
RNAi
|
RNA interference
|
|
SiRNA
|
small interfering RNA
|
|
DNMTs
|
DNA methyltransferases
|
References
|
1
|
Wang Y and Jiang T: Understanding high
grade glioma: Molecular mechanism, therapy and comprehensive
management. Cancer Lett. 331:139–146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, et al; European Organisation for Research and Treatment
of Cancer Brain Tumour and Radiation Oncology Groups; National
Cancer Institute of Canada Clinical Trials Group. Effects of
radiotherapy with concomitant and adjuvant temozolomide versus
radiotherapy alone on survival in glioblastoma in a randomised
phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet
Oncol. 10:459–466. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wilusz JE, Sunwoo H and Spector DL: Long
noncoding RNAs: Functional surprises from the RNA world. Genes Dev.
23:1494–1504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nagano T and Fraser P: No-nonsense
functions for long noncoding RNAs. Cell. 145:178–181. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu Y, Zhang L, Zhang L, Wang Y, Li H, Ren
X, Wei F, Yu W, Liu T, Wang X, et al: Long non-coding RNA HOTAIR
promotes tumor cell invasion and metastasis by recruiting EZH2 and
repressing E-cadherin in oral squamous cell carcinoma. Int J Oncol.
46:2586–2594. 2015.PubMed/NCBI
|
|
7
|
Wang Y, Chen W, Yang C, Wu W, Wu S, Qin X
and Li X: Long non-coding RNA UCA1a (CUDR) promotes proliferation
and tumorigenesis of bladder cancer. Int J Oncol. 41:276–284.
2012.PubMed/NCBI
|
|
8
|
Ørom UA, Derrien T, Beringer M, Gumireddy
K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q,
et al: Long noncoding RNAs with enhancer-like function in human
cells. Cell. 143:46–58. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tian D, Sun S and Lee JT: The long
noncoding RNA, Jpx, is a molecular switch for X chromosome
inactivation. Cell. 143:390–403. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kobayashi S, Wagatsuma H, Ono R, Ichikawa
H, Yamazaki M, Tashiro H, Aisaka K, Miyoshi N, Kohda T, Ogura A, et
al: Mouse Peg9/Dlk1 and human PEG9/DLK1 are paternally expressed
imprinted genes closely located to the maternally expressed
imprinted genes: Mouse Meg3/Gtl2 and human MEG3. Genes Cells.
5:1029–1037. 2000. View Article : Google Scholar
|
|
11
|
Anwar SL, Krech T, Hasemeier B, Schipper
E, Schweitzer N, Vogel A, Kreipe H and Lehmann U: Loss of
imprinting and allelic switching at the DLK1-MEG3 locus in human
hepatocellular carcinoma. PLoS One. 7:e494622012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Astuti D, Latif F, Wagner K, Gentle D,
Cooper WN, Catchpoole D, Grundy R, Ferguson-Smith AC and Maher ER:
Epigenetic alteration at the DLK1-GTL2 imprinted domain in human
neoplasia: Analysis of neuroblastoma, phaeochromocytoma and Wilms'
tumour. Br J Cancer. 92:1574–1580. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gejman R, Batista DL, Zhong Y, Zhou Y,
Zhang X, Swearingen B, Stratakis CA, Hedley-Whyte ET and Klibanski
A: Selective loss of MEG3 expression and intergenic differentially
methylated region hypermethylation in the MEG3/DLK1 locus in human
clinically nonfunctioning pituitary adenomas. J Clin Endocrinol
Metab. 93:4119–4125. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou Y, Zhong Y, Wang Y, Zhang X, Batista
DL, Gejman R, Ansell PJ, Zhao J, Weng C and Klibanski A: Activation
of p53 by MEG3 non-coding RNA. J Biol Chem. 282:24731–24742. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sheng X and Li J, Yang L, Chen Z, Zhao Q,
Tan L, Zhou Y and Li J: Promoter hypermethylation influences the
suppressive role of maternally expressed 3, a long non-coding RNA,
in the development of epithelial ovarian cancer. Oncol Rep.
32:277–285. 2014.PubMed/NCBI
|
|
16
|
Hervouet E, Vallette FM and Cartron PF:
Impact of the DNA methyltransferases expression on the methylation
status of apoptosis-associated genes in glioblastoma multiforme.
Cell Death Dis. 1:e82010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Braconi C, Kogure T, Valeri N, Huang N,
Nuovo G, Costinean S, Negrini M, Miotto E, Croce CM and Patel T:
microRNA-29 can regulate expression of the long non-coding RNA gene
MEG3 in hepatocellular cancer. Oncogene. 30:4750–4756. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Furnari FB, Fenton T, Bachoo RM, Mukasa A,
Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al:
Malignant astrocytic glioma: Genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou Y, Zhang X and Klibanski A: MEG3
noncoding RNA: A tumor suppressor. J Mol Endocrinol. 48:R45–R53.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yan J, Guo X, Xia J, Shan T, Gu C, Liang
Z, Zhao W and Jin S: MiR-148a regulates MEG3 in gastric cancer by
targeting DNA methyltransferase 1. Med Oncol. 31:8792014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang X, Gejman R, Mahta A, Zhong Y, Rice
KA, Zhou Y, Cheunsuchon P, Louis DN and Klibanski A: Maternally
expressed gene 3, an imprinted noncoding RNA gene, is associated
with meningioma pathogenesis and progression. Cancer Res.
70:2350–2358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao J, Dahle D, Zhou Y, Zhang X and
Klibanski A: Hyper-methylation of the promoter region is associated
with the loss of MEG3 gene expression in human pituitary tumors. J
Clin Endocrinol Metab. 90:2179–2186. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kuo HK, Griffith JD and Kreuzer KN:
5-Azacytidine induced methyltransferase-DNA adducts block DNA
replication in vivo. Cancer Res. 67:8248–8254. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lengauer C, Kinzler KW and Vogelstein B:
DNA methylation and genetic instability in colorectal cancer cells.
Proc Natl Acad Sci USA. 94:2545–2550. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tsai HC, Li H, Van Neste L, Cai Y, Robert
C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, et al:
Transient low doses of DNA-demethylating agents exert durable
antitumor effects on hematological and epithelial tumor cells.
Cancer Cell. 21:430–446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang P, Ren Z and Sun P: Overexpression of
the long non-coding RNA MEG3 impairs in vitro glioma cell
proliferation. J Cell Biochem. 113:1868–1874. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jurkowska RZ, Jurkowski TP and Jeltsch A:
Structure and function of mammalian DNA methyltransferases.
ChemBioChem. 12:206–222. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kreth S, Thon N, Eigenbrod S, Lutz J,
Ledderose C, Egensperger R, Tonn JC, Kretzschmar HA, Hinske LC and
Kreth FW: O-methylguanine-DNA methyltransferase (MGMT) mRNA
expression predicts outcome in malignant glioma independent of MGMT
promoter methylation. PLoS One. 6:e171562011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakagawa T, Kanai Y, Ushijima S, Kitamura
T, Kakizoe T and Hirohashi S: DNA hypermethylation on multiple CpG
islands associated with increased DNA methyltransferase DNMT1
protein expression during multistage urothelial carcinogenesis. J
Urol. 173:1767–1771. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saito Y, Kanai Y, Nakagawa T, Sakamoto M,
Saito H, Ishii H and Hirohashi S: Increased protein expression of
DNA methyltransferase (DNMT) 1 is significantly correlated with the
malignant potential and poor prognosis of human hepatocellular
carcinomas. Int J Cancer. 105:527–532. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bian EB, Zhao B, Huang C, Wang H, Meng XM,
Wu BM, Ma TT, Zhang L, Lv XW and Li J: New advances of DNA
methylation in liver fibrosis, with special emphasis on the
crosstalk between microRNAs and DNA methylation machinery. Cell
Signal. 25:1837–1844. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Foltz G, Yoon JG, Lee H, Ryken TC,
Sibenaller Z, Ehrich M, Hood L and Madan A: DNA
methyltransferase-mediated transcriptional silencing in malignant
glioma: A combined whole-genome microarray and promoter array
analysis. Oncogene. 28:2667–2677. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lu KH, Li W, Liu XH, Sun M, Zhang ML, Wu
WQ, Xie WP and Hou YY: Long non-coding RNA MEG3 inhibits NSCLC
cells proliferation and induces apoptosis by affecting p53
expression. BMC Cancer. 13:4612013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang X, Rice K, Wang Y, Chen W, Zhong Y,
Nakayama Y, Zhou Y and Klibanski A: Maternally expressed gene 3
(MEG3) noncoding ribonucleic acid: Isoform structure, expression,
and functions. Endocrinology. 151:939–947. 2010. View Article : Google Scholar :
|