Introduction
Melanoma is a highly aggressive neoplasm,
characterized by early metastatic spreading and poor response to
chemotherapy and radiotherapy. Despite the recent advances in the
treatment of the metastatic disease with the approval of monoclonal
antibodies that block immune checkpoints (ipilimumab, nivolumab and
pembrolizumab) and inhibitors of BRAF (vemurafenib and dabrafenib)
or MEK (trametinib), the prognosis of the advanced disease remains
poor. Melanoma progression is favoured by the formation of new
vessels from the pre-existing ones (angiogenesis), and by the
ability of tumour cells to invade the extracellular matrix (ECM)
and to form structures similar to blood vessels (vasculogenic
mimicry).
A key role in the angiogenic process is played by
members of the vascular endothelial growth factor (VEGF) family,
and in particular by the VEGF-A and the placenta growth factor
(PlGF) (1,2), which can be released by the tumour
cell itself. VEGF-A interacts with two membrane tyrosine kinase
receptors (3): VEGFR-1 (involved
in the activation of cell migration and in the modulation of the
signalling transduced by the VEGFR-2) and VEGFR-2 (mainly
responsible for the signal transduction that mediates the effects
of VEGF-A). Conversely, PlGF binds only to VEGFR-1, promoting
migration, proliferation and survival of endothelial cells.
The expression of PlGF is upregulated in several
types of human cancers and is associated with a poor prognosis
(4,5). PlGF is capable of transducing its own
signals through the phosphorylation of tyrosine residues within
VEGFR-1, which are distinct from those phosphorylated upon
stimulation of the receptor by VEGF-A (6). PlGF expression in the skin of
transgenic mice inoculated with syngeneic melanoma cells, favours
tumour growth and mobilization of endothelial and hematopoietic
stem cells. In the same murine model PlGF also increases the number
and size of melanoma-associated vessels and the formation of
pulmonary metastases (7).
Moreover, PlGF promotes tumour cell invasion of the ECM and
enhances the activity of selected matrix metalloproteinases
(7). Interestingly, PlGF plays
also a role in melanoma cell resistance to chemotherapy through a
pathway that involves NF-κB activation (8).
The VEGFR-1 is expressed in endothelial cells during
vessel formation and remodelling, in macrophages and in
myoepithelial cells, favouring cell migration and survival
(9–11). Moreover, it is frequently expressed
in a variety of human tumours where it predicts poor prognosis and
recurrence (1,9). In cancer cells, VEGFR-1 signalling
has been shown to inhibit apoptosis and to induce chemoresistance
(1,12,13).
Indeed, VEGFR-1 neutralization has been reported to prolong
survival of tumour-bearing mice (14). We previously demonstrated that PlGF
and VEGFR-1 are co-expressed in a large number of human melanoma
cell lines (15) and, together
with other groups, suggested that the interaction of PlGF with
VEGFR-1 might modulate cellular pathways important for melanoma
cell proliferation, apoptosis and invasiveness (7,15–18).
Another receptor, capable of binding VEGF-A and PlGF
is neuropilin-1 (NRP-1), a membrane protein devoid of tyrosine
kinase activity. Through the interaction with VEGF-A, NRP-1 acts as
a co-receptor, enhancing the signal transmitted by VEGFR-1 and
VEGFR-2 (2,4,19–21).
Moreover, although the intracellular domain of NRP-1 is not able to
directly transmit a signal, its interaction with intracellular
regulators allows NRP-1 to activate signal transduction pathways
independently of tyrosine kinase VEGF-A receptors (22). In this regard, PlGF has been
recently shown to activate MAPK and AKT signalling and promote
tumour cell survival by interacting with NRP-1 in the absence of
VEGFR-1 (23). Actually, a wide
variety of human malignancies (cell lines and tissue samples)
express NRP-1 and experimental evidence indicates an association
between the expression of NRP-1 and tumour growth, invasiveness,
angiogenesis and poor prognosis (24).
Given the key role played by VEGFs and VEGFRs in the
process of angiogenesis, intensive preclinical and clinical
investigation has been focused on the development of targeted drugs
that interfere with their activation or signal transduction
(25). To date, two angiogenic
monoclonal antibodies have been approved for cancer treatment:
bevacizumab, a humanized antibody that targets VEGF-A, preventing
its interaction with both VEGFR-2 and VEGFR-1, and ramucirumab, a
fully human antibody against VEGFR-2. Several small molecules that
interact with the catalytic domain of VEGFRs inhibiting their
tyrosine kinase activity, are also available. With few exceptions,
these agents are mostly used in combination with chemotherapy for
the treatment of a variety of solid tumours (26).
Unfortunately, the benefits obtained from
anti-angiogenic therapies have short duration, due to acquired
resistance. The mechanisms responsible for resistance to
anti-VEGF-A therapies (26,27)
include, among others, the formation of blood vessels by mechanisms
alternative to angiogenesis (intussusception, co-option,
vasculogenic mimicry), the production of angiogenic factors
different from VEGF-A (e.g., PlGF) and the high expression of
VEGFR-1 (in tumour or endothelial cells and macrophages) or NRP-1
(in tumour cells). Interestingly, although melanoma is a highly
vascularised tumour and potentially amenable to treatment with
anti-angiogenic agents, none of the anti-VEGF-A or VEGFR inhibitors
have been approved so far for the therapy of this malignancy.
In this study we investigated whether PlGF and NRP-1
cooperate in promoting melanoma aggressiveness and in the
acquisition of resistance to anti-VEGF-A therapies. Our results
indicate that in melanoma cells lacking VEGFR-1, PlGF is able to
stimulate ECM invasion and vasculogenic mimicry through the sole
activation of NRP-1. We also found that plasma levels of PlGF are
higher in patients with metastatic melanoma than in healthy
subjects and further increase during treatment with a
bevacizumab-containing chemotherapy regimen.
Materials and methods
Melanoma cell lines and culture
conditions
The origin and culture conditions of the human
umbilical vein endothelial cells (HUVECs), used as control, and of
the human melanoma cell lines utilised in the study (GR-Mel,
derived from a primary tumour, M14 and WM266-4, both derived from
meta-static lesions), as well as their characterization regarding
the expression of VEGFR-1 and NRP-1, have been described elsewhere
(15). The cell clones M14-N and
M14-C were derived from the M14 cell line, as previously described
(28,29). The M14-N clone expresses NRP-1 and
secretes high levels of VEGF-A and MMP-2, but lacks VEGFR-2
expression (28). The control
M14-C clone is devoid of the expression of VEGFR-2 and NRP-1 and
secretes lower amounts of VEGF-A and MMP-2 (28,29).
Culture media and reagents were purchased from Sigma-Aldrich (St.
Louis, MO, USA) and fatty acid-free bovine serum albumin (BSA) was
from Roche Diagnostics GmbH (Mannheim, Germany). The recombinant
human PlGF used in the functional assays was from R&D Systems,
Inc. (Minneapolis, MN, USA).
Western blot analysis
VEGFR-1 and NRP-1 expression in melanoma cells was
evaluated in cell extracts by western blot analysis as described
(28). The anti-human NRP-1 mouse
monoclonal antibody (A-12), the rabbit polyclonal antibodies
anti-human VEGFR-1 (C17) and anti-human β-tubulin (H-235, used as a
loading control) were purchased from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA) and utilized at the final concentration of
0.2 μg/ml.
Migration and invasion assays
In vitro migration assays were performed
using Boyden chambers equipped with 8-μm pore diameter
polycarbonate filters (Nuclepore; Whatman Inc., Clifton, NJ, USA),
coated with 5 μg/ml gelatin (30).
Briefly, melanoma cells were collected from continuous cultures,
washed, suspended in migration medium (1 μg/ml heparin/0.1% BSA in
RPMI-1640) and loaded (1.5×105 cells) into the upper
compartment of the Boyden chambers. Migration medium, with or
without 50 ng/ml PlGF, was added to the lower compartment and the
chambers were incubated at 37°C in a CO2 atmosphere for
18 h. The filters were then removed from the chambers and cells
were fixed in ethanol and stained in crystal violet. The migrated
cells, attached to the lower surface of the filters, were counted
under the microscope. Twelve high-magnification microscopic fields
(magnification, ×100), randomly selected on triplicate filters,
were scored for each experimental condition.
In vitro ECM invasion by melanoma cells was
analysed in Boyden chambers as described for the migration assay,
except that polycarbonate filters were coated with 20 μg of the
commercial basement membrane matrix Matrigel (Trevigen,
Gaithersburg, MD, USA) (18) and
1×105 cells/chamber were allowed to invade for 2 h.
In selected experiments, migration or invasion
assays were performed in the presence of antibodies against PlGF
(mouse monoclonal anti-human antibody, MAB264 from R&D Systems,
Inc.), NRP-1 (rabbit polyclonal anti-human antibody, H286 from
Santa Cruz Biotechnology, Inc.), and VEGFR-1 (goat polyclonal
anti-human antibody; AF321) and VEGFR-2 (goat polyclonal anti-human
antibody; AF357) (both from R&D Systems, Inc.) or the
corresponding control immunoglobulins (mouse IgG1 isotype control
from R&D Systems, Inc.). Melanoma cells were pre-incubated with
the antibody under investigation for 30 min at room temperature in
a rotating wheel and, then, loaded in the Boyden chambers without
removing the antibody.
ELISA quantification of VEGF and PlGF
levels
For the evaluation of PlGF secretion by melanoma
cell lines, semi-confluent cell cultures were incubated for 24 h in
0.1% BSA/RPMI-1640 medium without serum. Thereafter, culture
supernatants were collected and concentrated at least 10-fold in
Centriplus concentrators (Amicon, Beverly, MA, USA). Cells were
detached from the flasks with a PBS/EDTA solution and the total
cell number/culture was recorded to normalise cytokine
secretion.
For the evaluation of PlGF and VEGF-A levels in the
plasma of melanoma patients and healthy donors (see below), plasma
was separated from peripheral blood and cryopreserved at −80°C.
The amounts of VEGF or PlGF present in the culture
medium or in plasma samples were determined using commercial ELISA
kits (R&D Systems, Inc.), following the instructions from the
manufacturer.
Differentiation of melanoma cells in
tubule-like structures
The analysis of tubule-like structures on Matrigel
was performed as previously described (29). Melanoma cells were suspended in
complete culture medium (1×105 cells in 0.5 ml),
dispensed onto 100 μl of solidified Matrigel (diluted 1:3 in
serum-free RPMI-1640 medium) and incubated for 6 h at 37°C in a 5%
CO2 environment. Afterwards, the plates were
photographed using a Leica inverted microscope equipped with a
Canon digital camera (PowerShot G5). The formation of tubule-like
structures was quantified by counting the number of cell
intersections in ten different microscopic fields for each
experimental group (×50 magnification).
Studies in patients with metastatic
melanoma
Plasma levels of VEGF-A and PlGF were measured in
eight healthy donors and eight melanoma patients enrolled in a
phase II study evaluating bevacizumab (10 mg/kg days 1 and 15, q28)
in combination with paclitaxel (60 mg/m2, days 1, 8 and
15, q28) and carboplatin (AUC 2, days 1, 8 and 15, q28), as a
second-line treatment for metastatic melanoma. The study protocol
was approved by the Ethics Committee of the ‘Istituto Dermopatico
dell'Immacolata’-IRCCS, at the 02/12/08 meeting (Protocol number
#133/EC/2008, Reference number: Register of the Ethics Committee
247/1; Promoter: IDI-IRCCS, Oncology Division, Professor Marchetti;
EudraCT code: 2008-06191-30; Protocol: IDI-ONC-3-20080901). Written
informed consent was obtained from all the subjects (patients and
controls).
The inclusion criteria were as follows: age between
18 and 75 years; histologically confirmed metastatic melanoma
progressing after one line of chemotherapy not containing platinum
analogues; measurable metastatic disease according to the Response
Evaluation Criteria in Solid Tumours (RECIST); ECOG PS ≤2; life
expectancy of more than 3 months; adequate blood parameters
(leukocytes >4,000/mm3, neutrophils
>2,000/mm3, platelets >100,000/mm3,
haemoglobin >10 g/dl, serum creatinine <2.5 mg/dl, total
bilirubin/GOT/GPT <1.5x the upper limit).
The exclusion criteria were as follows: presence of
brain metastases, a history of other cancers diagnosed within 5
years (except basal cell skin cancer or carcinoma in situ of
the cervix); previous treatment with platinum analogues and/or
bevacizumab; concomitant immunotherapy; radiation therapy performed
within 28 days before treatment; a history of myocardial
infarction, congestive heart failure or heart disease and/or
uncontrolled, active infections; acute and/or chronic HIV,
hepatitis and/or tuberculosis infections; peripheral neuropathy
(higher than grade 2, according to the NCIC-CTC criteria);
contraindication to the use of corticosteroids (unstable diabetes
mellitus, active stomach ulcer); baseline LDH level >1.5x the
upper limit of the normal range; pregnancy or breast feeding.
Blood samples were collected during the first cycle
of therapy on day 1 before bevacizumab and chemotherapy
administration, on day 8 before chemotherapy administration and on
day 15 before bevacizumab and chemotherapy administration.
Results
NRP-1 enhances PlGF-mediated ECM invasion
in melanoma cells expressing VEGFR-1
We have previously demonstrated in a murine model
that PlGF promotes the invasiveness of melanoma cells through the
activation of its tyrosine kinase receptor VEGFR-1 (7). In order to investigate the
requirement of NRP-1 for this PlGF function, we initially evaluated
the ability of PlGF to stimulate ECM invasion using melanoma cell
lines that we had previously characterized for VEGFR-1 and NRP-1
expression by RT-PCR analysis (15) (Fig.
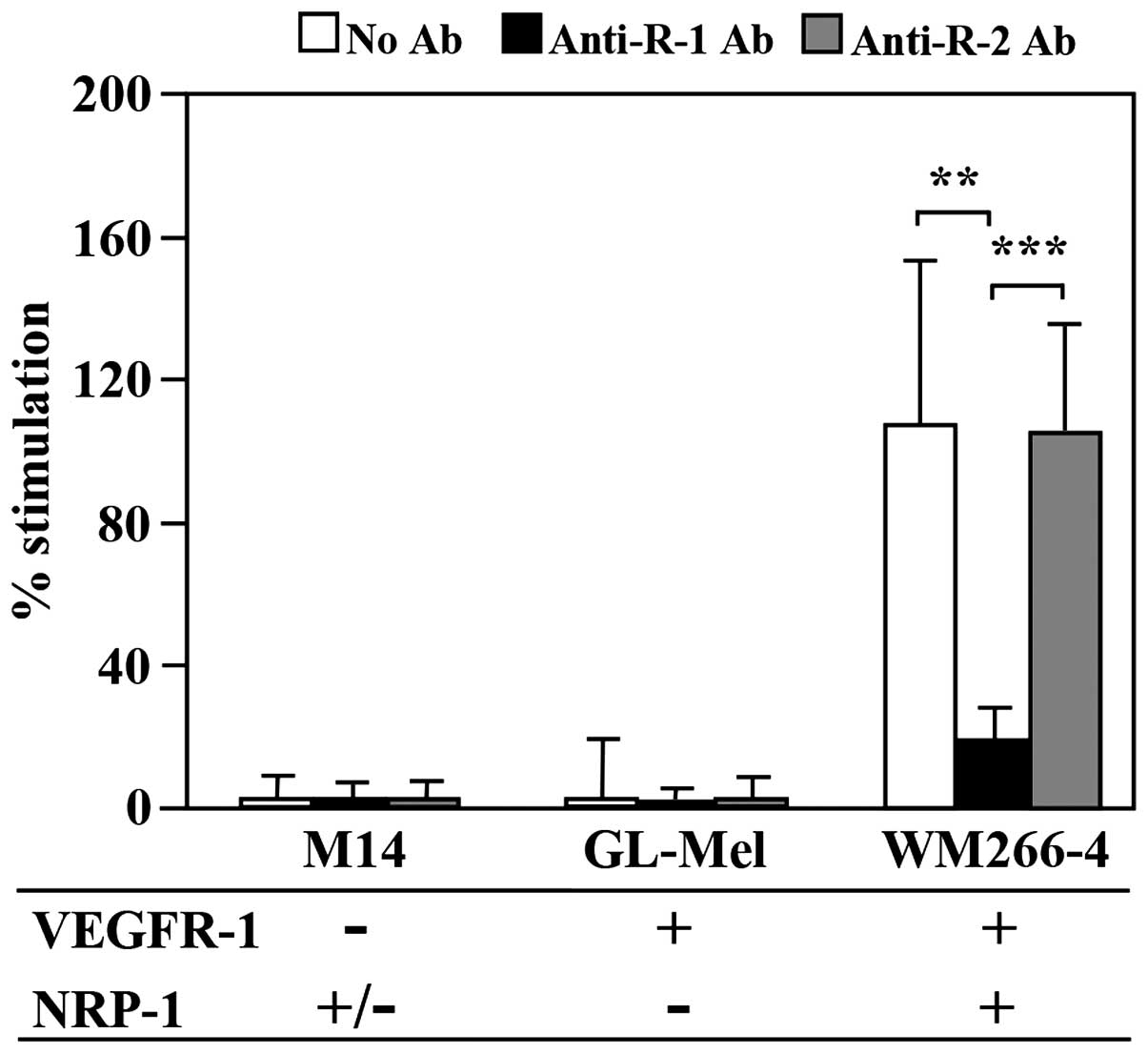
1). M14 cells, lacking VEGFR-1 and expressing very low levels
of NRP-1, and GL-Mel cells, expressing VEGFR-1 but lacking NRP-1,
did not respond to PlGF. Conversely, WM266-4 cells, expressing both
VEGFR-1 and NRP-1, efficiently migrated through the ECM when
exposed to the growth factor. In these cells a complete abrogation
of melanoma cell invasiveness was observed when the assay was
carried out in the presence of a blocking antibody against VEGFR-1,
whereas a blocking antibody against VEGFR-2, used as a control, did
not have any effect on PlGF-induced invasion (Fig. 1). These results suggested that PlGF
strongly induced ECM invasion only when melanoma cells expressed
high levels of NRP-1, besides VEGFR-1.
In VEGFR-1 negative melanoma cells PlGF
promotes ECM invasion and vasculogenic mimicry through the
interaction with NRP-1
We then investigated whether NRP-1 acts only as
co-receptor for VEGFR-1 or is also able to initiate by itself a
signal transduction after PlGF binding in the absence of VEGFR-1.
To this end, we used two previously described cell clones derived
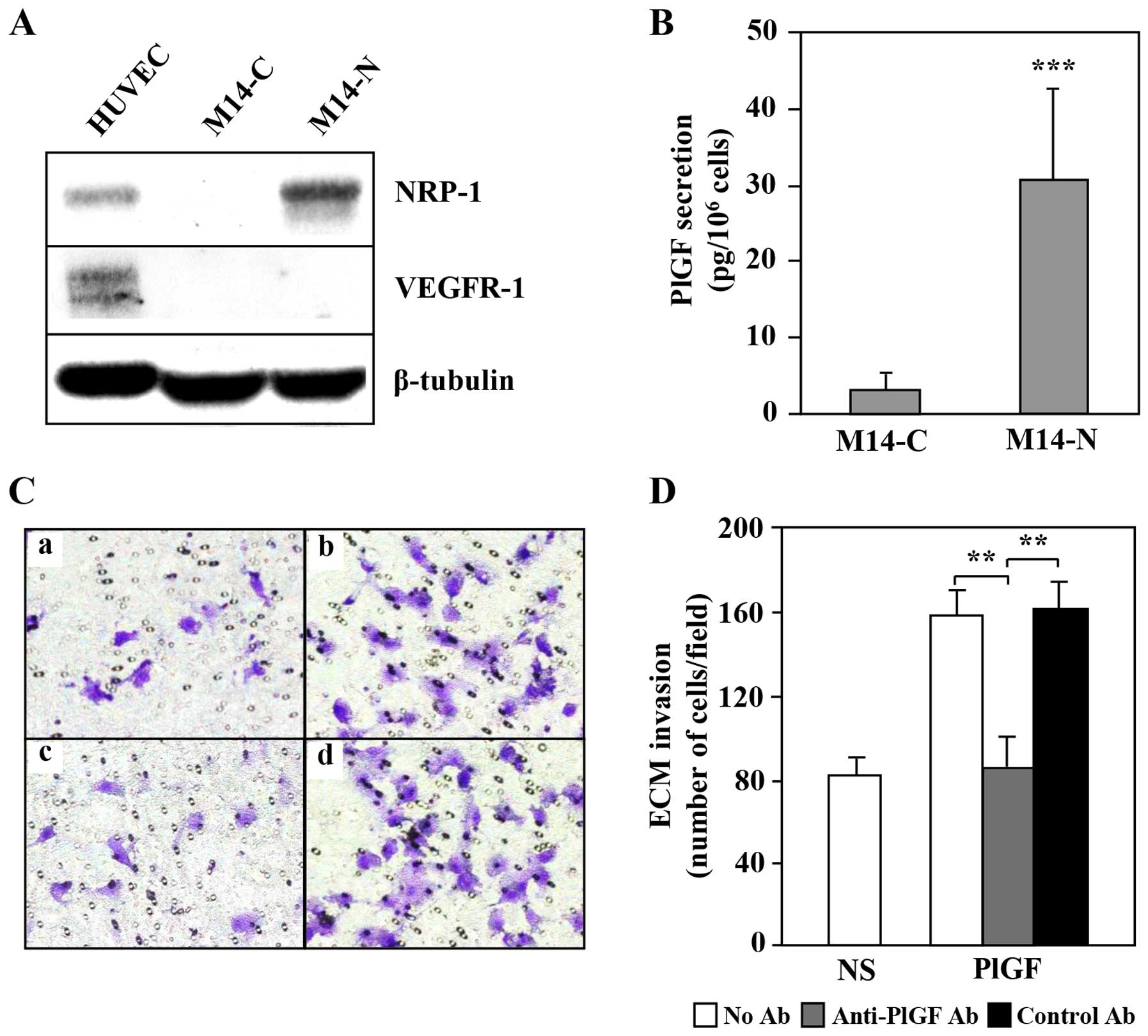
from the M14 melanoma cell line: the M14-N cells, characterized by
an aggressive phenotype, and the M14-C cells with a negligible
invasive capability (28,31). The M14-N cells lack VEGFR-1 and
express high levels of NRP-1 protein, whereas the M14-C cells do
not express either VEGFR-1 or NRP-1 (Fig. 2A). Evaluation of PlGF secretion in
the culture supernatants indicated an increased secretion of this
cytokine by M14-N cells as compared to M14-C cells (Fig. 2B). Moreover, PlGF specifically
induced ECM invasion by M14-N cells, since an anti-PlGF antibody
completely reversed this effect, whereas an unrelated control
antibody did not affect the ECM invasion triggered by this cytokine
(Fig. 2C and D).
The invasiveness of M14-N cells is strongly hampered
by NRP-1 neutralization (28),
making it difficult to demonstrate a direct activation of NRP-1 by
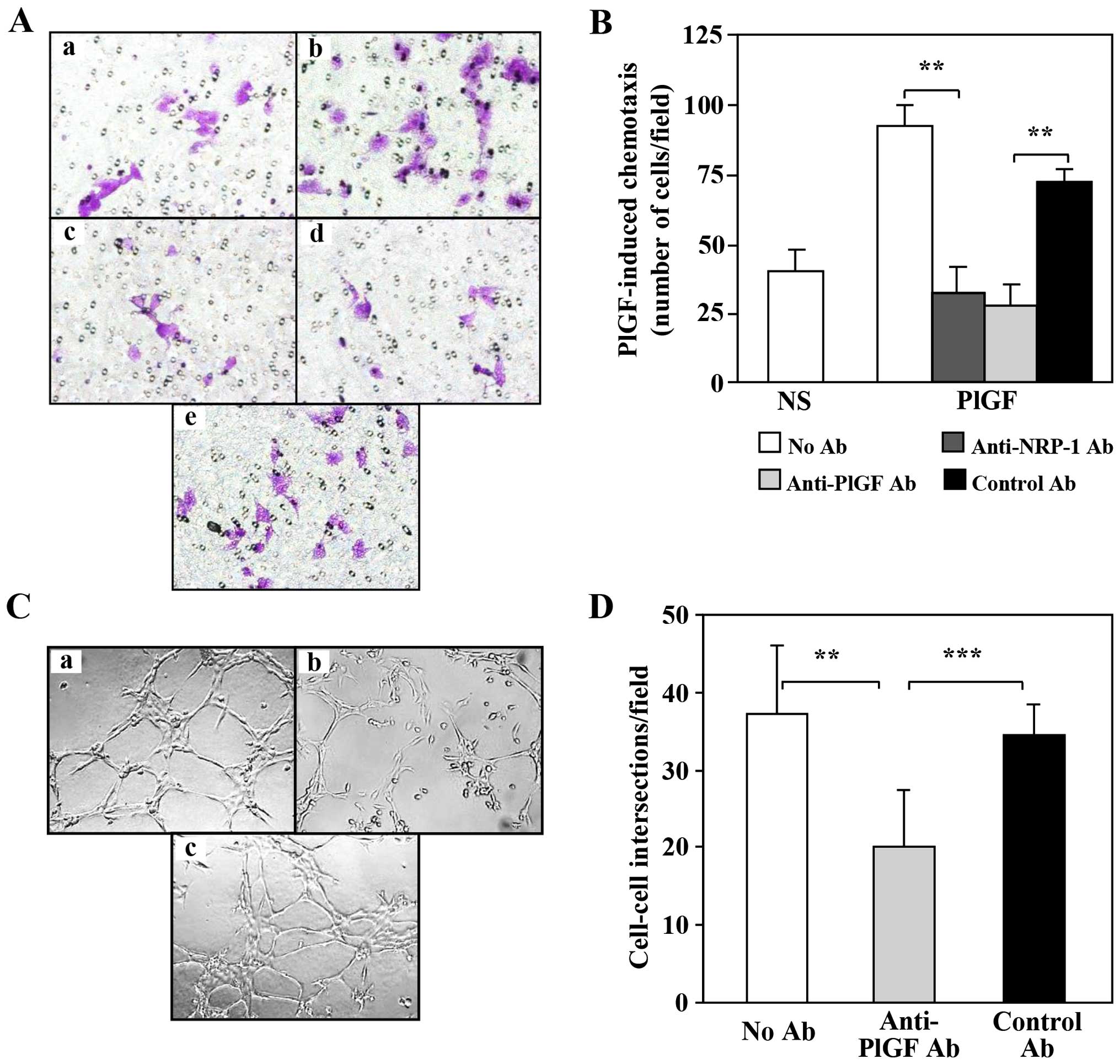
PlGF. Therefore, the ability of PlGF to activate NRP-1 in M14-N
cells was tested using a chemotaxis assay, in which the background
migration was very low and the stimulation by this cytokine
strongly induced migration (Fig. 3A
and B). It should be noted that due to the highly invasive
phenotype of M14-N cells, for the invasion assay a short incubation
time was sufficient (2 h), whereas the migration assay required a
longer incubation (18 h). Blocking antibodies to PlGF or to the
NRP-1 were equally able to completely abrogate PlGF-induced
chemotaxis.
A characteristic of highly aggressive cancers, and
in particular of melanoma (32),
is the ability to form tubule-like structures (vasculogenic
mimicry), which provide neoplastic cells with nutrients and
facilitate their haematogenous spreading. M14-N cells are able to
form tubular structures and this property is strongly dependent on
NRP-1 activity (29). Herein we
demonstrate that PlGF is directly involved in the formation of
tubule-like structures (Fig. 3C and
D). In fact, an anti-PlGF blocking antibody markedly reduced
cell-to-cell intersections, which were instead unaffected by the
treatment with a control antibody (Fig. 3C and D). Thus, the effects of PlGF
on vasculogenic mimicry of M14-N cells likely require the
activation of NRP-1.
All these data strongly suggested the presence of an
autocrine PlGF/NRP-1 loop in highly aggressive melanoma cells.
Analysis of plasma levels of PlGF and
VEGF-A in patients with metastatic melanoma
We next investigated whether high PlGF levels could
be present in the plasma of patients with metastatic melanoma, and
whether the level of this cytokine could be affected by a
chemotherapy regimen including bevacizumab to target angiogenesis.
To this end, we took advantage from a phase II study carried out to
evaluate bevacizumab in combination with carboplatin and paclitaxel
as a second-line therapy in patients with metastatic melanoma.
From March 2009 to May 2011, 51 patients with
metastatic melanoma progressing after a first line of chemotherapy
were examined for eligibility. On the basis of the exclusion
criteria, 42 patients (21 with secondary brain injury; 10 with ECOG
PS ≥3; 8 with no measurable target lesions; 2 with previous heart
attack and 1 with previous ictus cerebri) were not enrolled
in the study. In addition, one patient withdrew informed consent at
the time of the first drug administration. Therefore, only eight
patients, five males and three females with a median age of 56
years (38–66), were available for the analysis of VEGF-A and PlGF
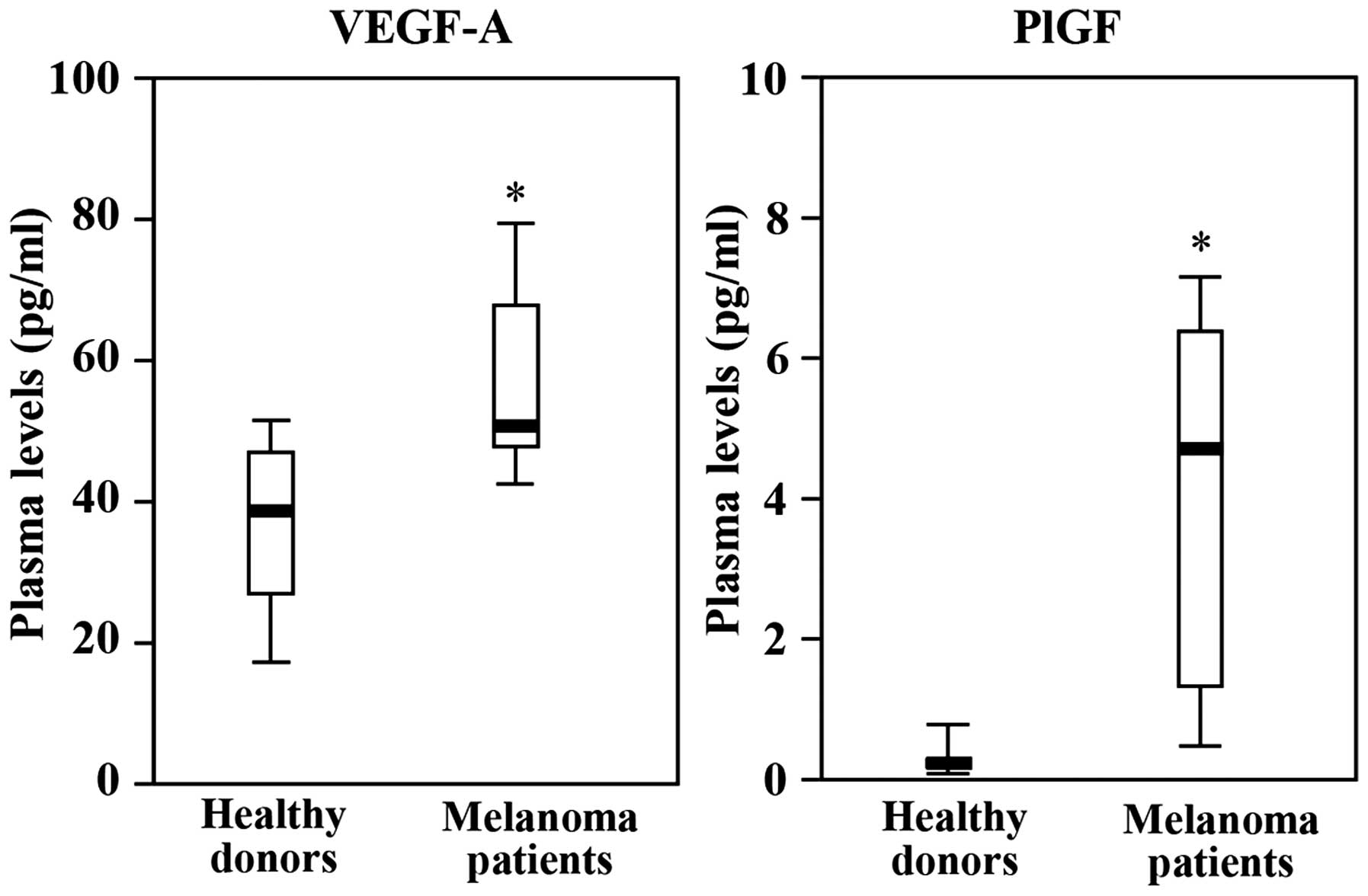
levels in the blood.
Plasma levels of VEGF-A and PlGF in the eight
melanoma patients were determined on day 1, 8 and 15 of the first
cycle of therapy. Blood samples were collected just before the
administration of the drugs. VEGF-A and PlGF levels detected in the
plasma collected from patients on day 1 (i.e., baseline values)
were compared with those of age- and gender-matched healthy donors.
The results showed significant higher amounts of VEGF-A and PlGF in
the patients as compared to the healthy controls. Remarkably, we
observed a 20-fold difference in the levels of PlGF between
melanoma patients and healthy volunteers (Fig. 4).
For each of the eight patients, VEGF-A and PlGF
plasma levels detected on day 8 and 15 were then compared with
baseline values (day 1). The results indicated that after a single
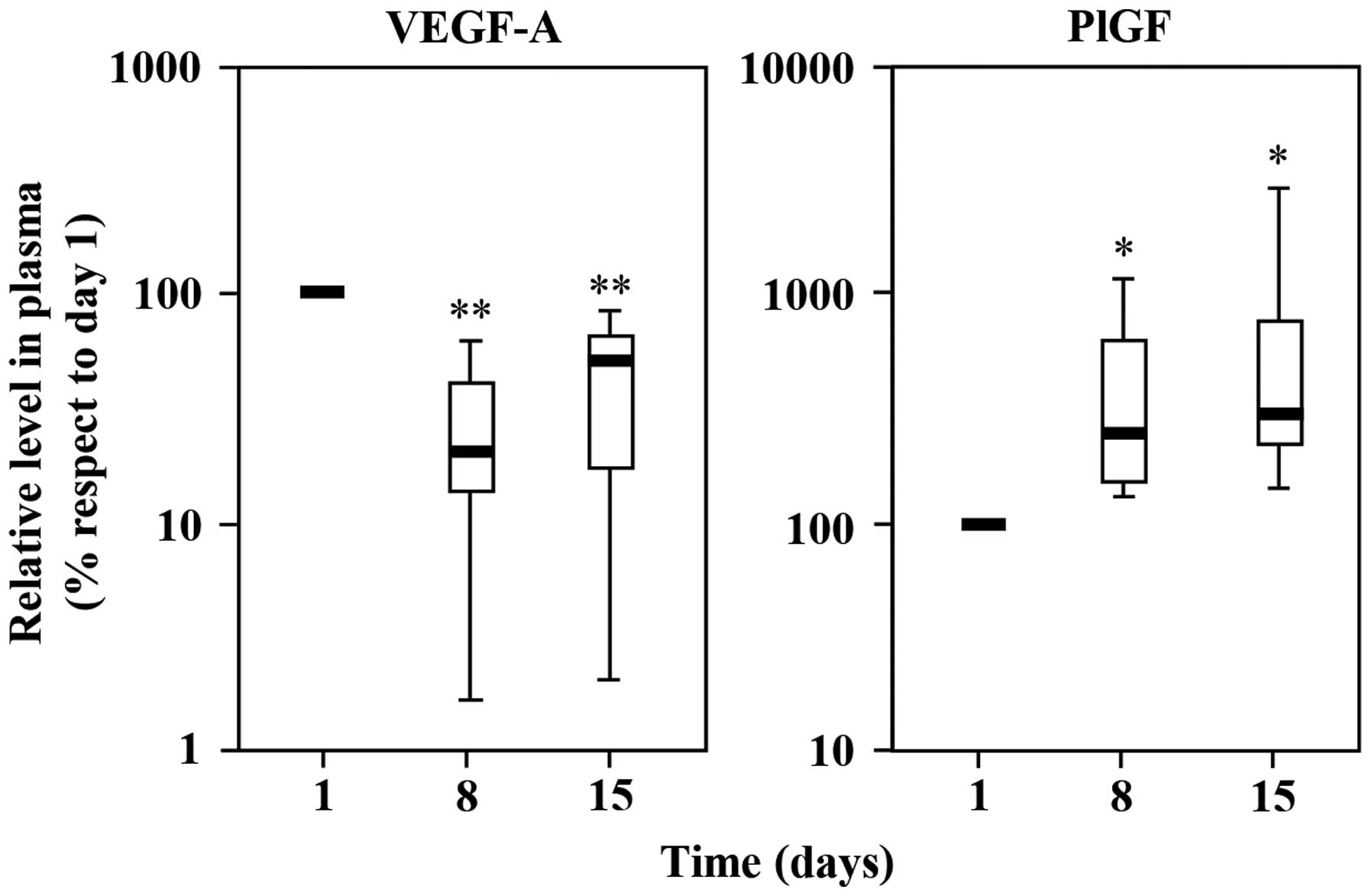
administration of bevacizumab plus chemotherapy, VEGF-A markedly
decreased (day 8) and started to increase again on day 15, even
though its levels remained significantly below the values detected
before starting the treatment cycle (Fig. 5). On the other hand, plasma PlGF
significantly increased on day 8 (2.3-fold increase) and remained
high until the end of the cycle (3-fold increase) (day 15; Fig. 5).
Overall, the results indicated that patients with
metastatic melanoma show measurable levels of PlGF in the plasma
and that treatment with bevacizumab induces a further increase of
this cytokine.
Discussion
In the present study we demonstrate for the first
time that PlGF promotes ECM invasion and vasculogenic mimicry of
human melanoma cells through the activation of NRP-1 and
independently of VEGFR-1. These findings strongly suggest a role
for PlGF and NRP-1 in melanoma aggressiveness and in the activation
of compensatory pro-angiogenic mechanisms that may contribute to
innate or acquired resistance to anti-angiogenic therapies
targeting VEGF-A.
The involvement of PlGF in melanoma progression has
been previously suggested (7,15–18,33)
and confirmed by studies performed in a murine transgenic model
where high PlGF expression levels were selectively induced in the
skin (7). On the other hand,
NRP-1, initially considered only a co-receptor of tyrosine kinase
receptors for different ligands, has been more recently shown to
play an important role in the activation of specific signal
transduction pathways independently of other membrane receptors
(23,34–36).
Our previous data indicate that NRP-1, upon interaction with
VEGF-A, may contribute to the melanoma metastatic potential even in
the absence of VEGFR-2, stimulating tumour invasiveness and
vasculogenic mimicry, which facilitates the haematogenous spreading
of cancer cells (28,29). These effects require the triggering
of signal transduction pathways involving AKT (28), specific gene expression (31), integrin activation (29) and the secretion of
metalloproteinases that degrade the ECM, such as metalloproteinases
2 and 9 (28,29,37).
Even though disease progression is highly dependent
on angiogenesis, melanoma may undergo phenotypic changes which
limit the efficacy of anti-VEGF-A agents. Here we demonstrate that
in melanoma cells NRP-1 may be activated by PlGF also in the
absence of VEGFR-1. Both PlGF and NRP-1 are, therefore, involved in
the induction of mechanisms that may result in tumour progression
and resistance to anti-angiogenic therapies.
In our melanoma model, we found that cells
expressing NRP-1 produce a higher amount of PlGF as compared to the
NRP-1 negative cells. PlGF is directly involved in the invasive
phenotype of M14-N cells, since neutralizing antibodies reverted
ECM invasion in response to the growth factor. Moreover, the
chemotactic response of NRP-1 positive cells to exogenous PlGF was
abrogated by anti-NRP-1 antibodies, suggesting that a direct
interaction of PlGF with NRP-1 is required for PlGF-mediated
melanoma invasiveness. Thus, in VEGFR-1 negative cells NRP-1
behaves as a primary receptor capable of transmitting the signal
deriving from its binding to PlGF.
Noteworthy, we also found increased levels of PlGF
in the plasma of patients with metastatic melanoma as compared to
healthy donors. These patients were enrolled in a phase II clinical
trial in which bevacizumab was administered in combination with the
chemotherapeutic agents paclitaxel and carboplatin. Drug treatment
was followed by a transient decrease of VEGF-A and a parallel
increase of PlGF plasma levels. In patients with non-small cell
lung cancer, treated with a bevacizumab-carboplatin-paclitaxel
regimen, a decrease in VEGF-A was also reported and associated with
clinical benefit (38). Due to the
small number of melanoma patients enrolled in the clinical study,
we cannot draw any definitive conclusion about the predictive value
of VEGF-A decline and PlGF increase in the plasma of patients.
Nevertheless, our results encourage further studies in a larger
number of patients to evaluate the role of plasma PlGF as a
potential marker of melanoma resistance to anti-VEGF-A
treatment.
High levels of PlGF can result in tumour progression
through autocrine or paracrine mechanisms. PlGF can be produced not
only by tumour cells but also by cells of the tumour stroma
(39). Indeed, in a murine in
vivo model, the increased plasma levels of PlGF in response to
an anti-angiogenic therapy were shown to derive from the host
tissues and not from the tumour itself (40). Moreover, using human melanoma cell
lines secreting both VEGF and PlGF, we found that treatment with
bevacizumab did not cause any modulation of PlGF release in the
culture supernatants (data not shown). Therefore, in patients
treated with an anti-VEGF-A therapy high plasma levels of PlGF,
produced by the tumour or by the stroma, can favour melanoma
spreading as long as cancer cells express a significant amount of
NRP-1, even in the absence of VEGFR-1.
Considerable experimental evidence allows
hypothesizing a potential role for PlGF or NRP-1 as therapeutic
targets. Differently from VEGF-A, PlGF plays a marginal role in
physiological conditions in adults, but is involved in the
induction of the angiogenic switch in pathological conditions, in
the mobilization of hematopoietic precursor stem cells from the
bone marrow and in the growth and migration of cancer cells
(41). Moreover, in preclinical
animal models monoclonal antibodies specific for PlGF inhibit the
growth and metastasis in different types of tumours and increase
the efficacy of chemotherapy without causing significant side
effects (4,6,41).
Concerning NRP-1 as therapeutic target, this
receptor is expressed in endothelial, highly aggressive melanoma
and immune cells and, therefore, is involved in all the biological
processes crucial for melanoma progression: angiogenesis,
vasculogenic mimicry and tumour evasion from the control of the
immune system. Moreover, a high-affinity monoclonal antibody
targeting NRP-1 has been shown to inhibit migration of human
endothelial cells and tumor formation in animal models (42,43).
It also inhibited breast cancer cell proliferation and enhanced
chemosensitivity of human non-small cell lung, kidney, prostate
cancer and other carcinoma cells, by interfering with
integrin-dependent survival pathways (44–46).
Thus, the targeting of NRP-1 seems to be a valuable strategy for
combination therapies, including anti-angiogenic agents, as
reviewed (47).
In conclusion, in this study we have shown that PlGF
promotes ECM invasiveness and vasculogenic mimicry of melanoma
cells, and that these functions are principally mediated by PlGF
activation of NRP-1. Moreover, we found that the plasma levels of
PlGF in melanoma patients are higher than in healthy donors, and
that these levels tend to further increase in the course of a
bevacizumab-containing chemotherapy regimen. PlGF production and
NRP-1 expression may, therefore, increase tumour aggressiveness and
contribute to melanoma escape from anti-VEGF-A therapies. Thus, the
blockade of PlGF interaction with its receptors, VEGFR-1 and, more
importantly, NRP-1, represents a therapeutic strategy that may
enhance the efficacy or counteract resistance to inhibitors of
VEGF-A mediated pathways.
Acknowledgements
This study was supported by the Italian Ministry of
Health and in part by the ‘Associazione Italiana per la Ricerca sul
Cancro’ (AIRC) to GG (AIRC 2013 IG 14042).
References
|
1
|
Fischer C, Mazzone M, Jonckx B and
Carmeliet P: FLT1 and its ligands VEGFB and PlGF: drug targets for
anti-angiogenic therapy? Nat Rev Cancer. 8:942–956. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roskoski R Jr: VEGF receptor
protein-tyrosine kinases: structure and regulation. Biochem Biophys
Res Commun. 375:287–291. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lohela M, Bry M, Tammela T and Alitalo K:
VEGFs and receptors involved in angiogenesis versus
lymphangiogenesis. Curr Opin Cell Biol. 21:154–165. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dewerchin M and Carmeliet P: Placental
growth factor in cancer. Expert Opin Ther Targets. 18:1339–1354.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim KJ, Cho CS and Kim WU: Role of
placenta growth factor in cancer and inflammation. Exp Mol Med.
44:10–19. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Autiero M, Waltenberger J, Communi D,
Kranz A, Moons L, Lambrechts D, Kroll J, Plaisance S, De Mol M,
Bono F, et al: Role of PlGF in the intra- and intermolecular cross
talk between the VEGF receptors Flt1 and Flk1. Nat Med. 9:936–943.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marcellini M, De Luca N, Riccioni T,
Ciucci A, Orecchia A, Lacal PM, Ruffini F, Pesce M, Cianfarani F,
Zambruno G, et al: Increased melanoma growth and metastasis
spreading in mice overexpressing placenta growth factor. Am J
Pathol. 169:643–654. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Levati L, Ruffini F, Muzi A, Umezawa K,
Graziani G, D'Atri S and Lacal PM: Placenta growth factor induces
melanoma resistance to temozolomide through a mechanism that
involves the activation of the transcription factor NF-κB. Int J
Oncol. 38:241–247. 2011.
|
|
9
|
Schwartz JD, Rowinsky EK, Youssoufian H,
Pytowski B and Wu Y: Vascular endothelial growth factor receptor-1
in human cancer: concise review and rationale for development of
IMC-18F1 (Human antibody targeting vascular endothelial growth
factor receptor-1). Cancer. 116(Suppl): 1027–1032. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Adini A, Kornaga T, Firoozbakht F and
Benjamin LE: Placental growth factor is a survival factor for tumor
endothelial cells and macrophages. Cancer Res. 62:2749–2752.
2002.PubMed/NCBI
|
|
11
|
Zhou Y, Bellingard V, Feng K-T, McMaster M
and Fisher SJ: Human cytotrophoblasts promote endothelial survival
and vascular remodeling through secretion of Ang2, PlGF, and
VEGF-C. Dev Biol. 263:114–125. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu Y, Hooper AT, Zhong Z, Witte L, Bohlen
P, Rafii S and Hicklin DJ: The vascular endothelial growth factor
receptor (VEGFR-1) supports growth and survival of human breast
carcinoma. Int J Cancer. 119:1519–1529. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu Y, Zhong Z, Huber J, Bassi R, Finnerty
B, Corcoran E, Li H, Navarro E, Balderes P, Jimenez X, et al:
Anti-vascular endothelial growth factor receptor-1 antagonist
antibody as a therapeutic agent for cancer. Clin Cancer Res.
12:6573–6584. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fragoso R, Pereira T, Wu Y, Zhu Z,
Cabeçadas J and Dias S: VEGFR-1 (FLT-1) activation modulates acute
lymphoblastic leukemia localization and survival within the bone
marrow, determining the onset of extramedullary disease. Blood.
107:1608–1616. 2006. View Article : Google Scholar
|
|
15
|
Lacal PM, Failla CM, Pagani E, Odorisio T,
Schietroma C, Falcinelli S, Zambruno G and D'Atri S: Human melanoma
cells secrete and respond to placenta growth factor and vascular
endothelial growth factor. J Invest Dermatol. 115:1000–1007. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Graells J, Vinyals A, Figueras A, Llorens
A, Moreno A, Marcoval J, Gonzalez FJ and Fabra A: Overproduction of
VEGF concomitantly expressed with its receptors promotes growth and
survival of melanoma cells through MAPK and PI3K signaling. J
Invest Dermatol. 123:1151–1161. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Graeven U, Fiedler W, Karpinski S, Ergün
S, Kilic N, Rodeck U, Schmiegel W and Hossfeld DK:
Melanoma-associated expression of vascular endothelial growth
factor and its receptors FLT-1 and KDR. J Cancer Res Clin Oncol.
125:621–629. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lacal PM, Ruffini F, Pagani E and D'Atri
S: An autocrine loop directed by the vascular endothelial growth
factor promotes invasiveness of human melanoma cells. Int J Oncol.
27:1625–1632. 2005.PubMed/NCBI
|
|
19
|
Grünewald FS, Prota AE, Giese A and
Ballmer-Hofer K: Structure-function analysis of VEGF receptor
activation and the role of coreceptors in angiogenic signaling.
Biochim Biophys Acta. 1804:567–580. 2010. View Article : Google Scholar
|
|
20
|
Neagoe PE, Lemieux C and Sirois MG:
Vascular endothelial growth factor (VEGF)-A165-induced prostacyclin
synthesis requires the activation of VEGF receptor-1 and -2
heterodimer. J Biol Chem. 280:9904–9912. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Allain B, Jarray R, Borriello L, Leforban
B, Dufour S, Liu WQ, Pamonsinlapatham P, Bianco S, Larghero J,
Hadj-Slimane R, et al: Neuropilin-1 regulates a new VEGF-induced
gene, Phactr-1, which controls tubulogenesis and modulates
lamel-lipodial dynamics in human endothelial cells. Cell Signal.
24:214–223. 2012. View Article : Google Scholar
|
|
22
|
Chittenden TW, Claes F, Lanahan AA,
Autiero M, Palac RT, Tkachenko EV, Elfenbein A, Ruiz de Almodovar
C, Dedkov E, Tomanek R, et al: Selective regulation of arterial
branching morphogenesis by synectin. Dev Cell. 10:783–795. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Snuderl M, Batista A, Kirkpatrick ND, Ruiz
de Almodovar C, Riedemann L, Walsh EC, Anolik R, Huang Y, Martin
JD, Kamoun W, et al: Targeting placental growth factor/neuropilin 1
pathway inhibits growth and spread of medulloblastoma. Cell.
152:1065–1076. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ellis LM: The role of neuropilins in
cancer. Mol Cancer Ther. 5:1099–1107. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Holmes K, Roberts OL, Thomas AM and Cross
MJ: Vascular endothelial growth factor receptor-2: structure,
function, intra-cellular signalling and therapeutic inhibition.
Cell Signal. 19:2003–2012. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
van Beijnum JR, Nowak-Sliwinska P,
Huijbers EJ, Thijssen VL and Griffioen AW: The great escape; the
hallmarks of resistance to antiangiogenic therapy. Pharmacol Rev.
67:441–461. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lambrechts D, Lenz HJ, de Haas S,
Carmeliet P and Scherer SJ: Markers of response for the
antiangiogenic agent bevacizumab. J Clin Oncol. 31:1219–1230. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ruffini F, D'Atri S and Lacal PM:
Neuropilin-1 expression promotes invasiveness of melanoma cells
through vascular endothelial growth factor receptor-2-dependent and
-independent mechanisms. Int J Oncol. 43:297–306. 2013.PubMed/NCBI
|
|
29
|
Ruffini F, Graziani G, Levati L, Tentori
L, D'Atri S and Lacal PM: Cilengitide downmodulates invasiveness
and vasculogenic mimicry of neuropilin 1 expressing melanoma cells
through the inhibition of αvβ5 integrin. Int J Cancer.
136:E545–E558. 2015. View Article : Google Scholar
|
|
30
|
Orecchia A, Lacal PM, Schietroma C, Morea
V, Zambruno G and Failla CM: Vascular endothelial growth factor
receptor-1 is deposited in the extracellular matrix by endothelial
cells and is a ligand for the alpha 5 beta 1 integrin. J Cell Sci.
116:3479–3489. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ruffini F, Tentori L, Dorio AS, Arcelli D,
D'Amati G, D'Atri S, Graziani G and Lacal PM: Platelet-derived
growth factor C and calpain-3 are modulators of human melanoma cell
invasiveness. Oncol Rep. 30:2887–2896. 2013.PubMed/NCBI
|
|
32
|
Seftor RE, Hess AR, Seftor EA, Kirschmann
DA, Hardy KM, Margaryan NV and Hendrix MJ: Tumor cell vasculogenic
mimicry: From controversy to therapeutic promise. Am J Pathol.
181:1115–1125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Escudero-Esparza A, Martin TA,
Douglas-Jones A, Mansel RE and Jiang WG: PGF isoforms, PLGF-1 and
PGF-2 and the PGF receptor, neuropilin, in human breast cancer:
prognostic significance. Oncol Rep. 23:537–544. 2010.PubMed/NCBI
|
|
34
|
Banerjee S, Sengupta K, Dhar K, Mehta S,
D'Amore PA, Dhar G and Banerjee SK: Breast cancer cells secreted
platelet-derived growth factor-induced motility of vascular smooth
muscle cells is mediated through neuropilin-1. Mol Carcinog.
45:871–880. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dhar K, Dhar G, Majumder M, Haque I, Mehta
S, Van Veldhuizen PJ, Banerjee SK and Banerjee S: Tumor
cell-derived PDGF-B potentiates mouse mesenchymal stem
cells-pericytes transition and recruitment through an interaction
with NRP-1. Mol Cancer. 9:2092010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pellet-Many C, Frankel P, Evans IM, Herzog
B, Jünemann-Ramírez M and Zachary IC: Neuropilin-1 mediates PDGF
stimulation of vascular smooth muscle cell migration and signalling
via p130Cas. Biochem J. 435:609–618. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Frank A, David V, Aurelie TR, Florent G,
William H and Philippe B: Regulation of MMPs during melanoma
progression: from genetic to epigenetic. Anticancer Agents Med
Chem. 12:773–782. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tamiya M, Tamiya A, Yamadori T, Nakao K,
Asami K, Yasue T, Otsuka T, Shiroyama T, Morishita N, Suzuki H, et
al: Phase2 study of bevacizumab with carboplatin-paclitaxel for
non-small cell lung cancer with malignant pleural effusion. Med
Oncol. 30:6762013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jayasinghe C, Simiantonaki N and
Kirkpatrick CJ: Cell type- and tumor zone-specific expression of
pVEGFR-1 and its ligands influence colon cancer metastasis. BMC
Cancer. 15:1042015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bagley RG, Ren Y, Weber W, Yao M,
Kurtzberg L, Pinckney J, Bangari D, Nguyen C, Brondyk W, Kaplan J,
et al: Placental growth factor upregulation is a host response to
antiangiogenic therapy. Clin Cancer Res. 17:976–988. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fischer C, Jonckx B, Mazzone M, Zacchigna
S, Loges S, Pattarini L, Chorianopoulos E, Liesenborghs L, Koch M,
De Mol M, et al: Anti-PlGF inhibits growth of
VEGF(R)-inhibitor-resistant tumors without affecting healthy
vessels. Cell. 131:463–475. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liang WC, Dennis MS, Stawicki S, Chanthery
Y, Pan Q, Chen Y, Eigenbrot C, Yin J, Koch AW, Wu X, et al:
Function blocking antibodies to neuropilin-1 generated from a
designed human synthetic antibody phage library. J Mol Biol.
366:815–829. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pan Q, Chanthery Y, Liang WC, Stawicki S,
Mak J, Rathore N, Tong RK, Kowalski J, Yee SF, Pacheco G, et al:
Blocking neuropilin-1 function has an additive effect with
anti-VEGF to inhibit tumor growth. Cancer Cell. 11:53–67. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xin Y, Li J, Wu J, Kinard R, Weekes CD,
Patnaik A, Lorusso P, Brachmann R, Tong RK, Yan Y, et al:
Pharmacokinetic and pharmacodynamic analysis of circulating
biomarkers of anti-NRP1, a novel antiangiogenesis agent, in two
phase I trials in patients with advanced solid tumors. Clin Cancer
Res. 18:6040–6048. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jia H, Cheng L, Tickner M, Bagherzadeh A,
Selwood D and Zachary I: Neuropilin-1 antagonism in human carcinoma
cells inhibits migration and enhances chemosensitivity. Br J
Cancer. 102:541–552. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zeng F, Luo F, Lv S, Zhang H, Cao C, Chen
X, Wang S, Li Z, Wang X, Dou X, et al: A monoclonal antibody
targeting neuropilin-1 inhibits adhesion of MCF7 breast cancer
cells to fibronectin by suppressing the FAK/p130cas signaling
pathway. Anticancer Drugs. 25:663–672. 2014.PubMed/NCBI
|
|
47
|
Graziani G and Lacal PM: Neuropilin-1 as
therapeutic target for malignant melanoma. Front Oncol. 5:1252015.
View Article : Google Scholar : PubMed/NCBI
|