Introduction
Prostate cancer is the most frequently diagnosed
male malignancy and one of the leading causes of death among men in
western countries, and its incidence has been rising in China
during the past few years. However, the precise molecular
mechanisms underlying the progression of prostate cancer are still
underexplored. It is generally considered that the carcinogenesis
of prostate is a consequence of genetic and epigenetic
modifications, converting normal prostate glandular epithelium to
pre-neoplastic lesions and invasive carcinoma at the final stage.
It is essential to develop more specific diagnostic and prognostic
biomarkers for predicting the severity of prostate cancer, which
demands a better understanding of the molecular pathogenesis of
prostate cancer (1).
Recent studies suggest that microRNAs (miRNAs) are
significantly altered in prostate cancer, suggesting that miRNAs
are involved in the progression of prostate cancer. As naturally
occurring small non-coding RNAs (≈22 nucleotides), miRNAs bind to
the complementary sites in the 3′ untranslated regions (3′ UTRs) of
targeting mRNAs and further regulate either translational
inhibition or mRNA degradation. miRNAs have been implicated in
controlling a wide range of cellular processes such as cell cycle,
proliferation and apoptosis, differentiation and development and
metabolism. Importantly, aberrant expression of miRNAs can cause
diverse human disorders, especially malignancies. In fact, miRNAs
act as either tumor suppressors or oncogenes, and participate in
the development of a variety of malignancies in a tissue-specific
manner. Distinct miRNAs that are either upregulated or
downregulated in prostate cancer have been identified in a few
microarray assays by comparing with normal prostate tissue.
Considerable miRNA profiling data indicate a relationship of miRNA
expression patterns with carcinogenesis or degree of cancer
differentiation in prostate cancer. The specific miRNA expression
patterns might be useful biomarkers for early detection and
prognostic assessment of prostate cancer (2–4).
However, the precise role of specific miRNAs in regulating the
carcinogenesis of prostate cancer is still largely unclear.
The mir-17-92 cluster, which is also
designated as oncomir-1, is one of the best-characterized
oncogenic miRNAs (5). Human
miR-17-92 is located within intron 3 of the C13orf25
gene at 13q31.3, a region frequently amplified in many
hematopoietic and solid malignancies (6). The distinct six mature miRNAs encoded
by mir-17-92 are classified into three separate miRNAs
families, the miR-17 family (including miR-17,
miR-18 and miR-20), the miR-19 family
(including miR-19a and miR-19b) and the miR-92
family.
The mir-17-92 cluster is expressed in
embryonic cells and essential in normal development. Dysregulation
of the mir-17-92 cluster has been found in multiple
hematopoietic and solid malignancies such as those derived from
breast, lung, pancreas, kidney, colon, liver, stomach and prostate
(7–13). In general, the mir-17-92
cluster functions as oncogenes and promotes carcinogenesis by
negatively regulating tumor suppressors and/or genes that control a
variety of biological behaviors such as cell cycle, cell
proliferation and survival (8,14).
The mir-17-92 cluster has been ascribed an
oncogenic role in prostate cancer. In primary prostate cancer
tissues, miR-17 and miR-20 are overexpressed compared
to that in benign prostate tissues (7). Moreover, miR-20 expression is
significantly higher in patients with a high Gleason score than
patients with a low score (15).
The individual member of the mir-17-92 cluster has a
characteristic expression profile in different prostate cancer cell
lines (16). Overexpression of
miR-20 decreases apoptosis in a prostate cancer cell line,
while inhibition of miR-20 by an antisense oligonucleotide
results in increased cell death after doxorubicin treatment
(17). Recent studies have
demonstrated that the aberrant expression of miRNAs, including
miR-17, miR-20, miR-92, miR-106a/b,
miR-125 and miR-145 is involved in prostate cancer
onset, progression, and metastasis (18). Nevertheless, the precise role of
the miR-17-92 cluster in modulating malignant progression of
prostate cancer is poorly understood.
In the present study, we investigated the functional
roles of the mir-17-92 cluster in prostate cancer cells and
examine its significance as a therapeutic target. The expression
pattern of the androgen-independent prostate cancer cells was quite
different from that of the androgen-dependent cells. Forced
introduction of the miR-17-92 cluster into DU145
androgen-independent prostate cancer cells affected a variety of
biological behaviors of cells. The stimulatory function of this
miRNA cluster in prostate cancer cell growth, migration and
invasion and chemo-resistance were observed. Activated protein
kinase B (PKB, also known as AKT) and
extracellular-signal-regulated kinase (ERK) signaling pathways
played critical roles in these cellular processes. Collectively, we
provided evidence that the mir-17-92 cluster functioned as
an oncogene in prostate cancer and could possess potential for
targeted therapy in human prostate cancer.
Materials and methods
Tissue culture
Cells were cultured in RPMI-1640 media supplemented
with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml
streptomycin and 2 mM glutamine. All cell lines were maintained in
a humidified atmosphere containing 5% CO2 at 37ºC.
MG-132 (cat. no. 474790) was purchased from Millipore. Cisplatin
(cat. no. 029K1426) was purchased from Sigma-Aldrich. Puromycin
(cat. no. J593) was purchased from Amresco.
Transfection
The miR-17-92 cluster overexpression vector
was constructed on a backbone of MSCV vector containing GFP (gift
from Professor Yong Zhao). Phoenix A packaging cells were
transfected with MSCV-GFP-miR-17-92 or control vector
(MSCV-GFP) by FuGene HD (Roche, Shanghai, China). Virus
supernatants were collected and target cells were infected with the
virus supernatants. For obtaining stable expressing cells, the
cells were selected for two weeks in the presence of puromycin (5
μg/ml).
RNA extraction
Total RNA was isolated using the TRIZol reagent
(Tiangen Biotech, Co., Ltd., Beijing, China) according to the
manufacturer's instructions. RNA yield and purity were determined
spectrophotometrically at 260–280 nm and the integrity of RNA was
verified by NanoDrop 1000 (Thermo Fisher Scientific, Co., Ltd.,
Shanghai, China).
Quantitative reverse-transcriptase
polymerase chain reaction (qRT-PCR)
Total RNA (2 μg) was reverse-transcribed with
SuperScript M-MLV (Promega, Shanghai, China) according to the
manufacturer's instructions. Triplicates were performed for all
qRT-PCR reactions with a LightCycler 480 System (Roche). Primers
were designed using Primer-BLAST (PubMed) and synthesized from
Invitrogen (Beijing, China). The cDNAs were amplified using 2X
LC480 SYBR-Green I Master Mix (Roche). Data were analyzed with the
Pfafflmethod that provides a means for quantification of a target
gene transcript in comparison to a reference gene (19). Primer sequences are as follows:
BIM: 5′-ACAGAGCCACAAGAC AGGAGCCC-3′ and
5′-CGCCGCAACTCTTGGGCGAT-3′; CCND1:
5′-CACTTTCAGTCCAATAGGTGTAG-3′ and 5′-TTCTTCTTGACTGGCACG-3′;
PTEN: 5′-TGGGGAAGT AAGGACCAGAGA-3′ and
5′-TGAGGATTGCAAGTTCCGCC-3′; PHLPP2:
5′-GTACGCAAGGGAAAGACCCA-3′ and 5′-AGCAAGGGAGTATTGCCGTC-3′.
qRT-PCR analysis of mature miRNA
expression
Total RNA (2 μg) was polyadenylated with ATP by
E. coli poly(A) polymerase (PAP; New England Biolabs, Ltd.,
Beijing, China). After phenol-chloroform extraction and ethanol
precipitation, the polyadenylated RNAs were dissolved in
diethyl-pyrocarbonate (DEPC)-treated water and reverse-transcribed
with M-MLV and universal RT primer sequence. After a dilution of
1:20, the cDNAs were amplified using 2X LC480 SYBR-Green I Master
Mix. The mean threshold cycle (Ct) was determined from triplicates.
U6 expression was used as an internal control. Data were
analyzed with the Pfafflmethod (19).
Western blot analysis
Whole-cell extracts and nuclear/cytoplasm fractions
were prepared according to the standard procedures. Proteins were
fractionated on an SDS-PAGE gel and transferred to nitrocellulose
membranes. After blocking, membranes were probed with different
antibodies (Abs). Membranes were then washed and incubated with an
appropriate secondary Abs. Proteins were detected and scanned with
an Odyssey system (LI-COR Biosciences, Lincoln, NE, USA). Abs
against RelA (sc-372X), p105/p50 (sc-7178X), p100/p52 (sc-298),
RelB (sc-226X), c-Rel (sc-70), cyclin D1 (sc-20044), and Lamin A/C
(sc-20681) were purchased from Santa Cruz Biotechnology. Abs
against ERK1/2 (#4695), p-ERK1/2 (#4370S), ERCC1 (#5437S), AKT
(#4691), p-AKT (Ser473, #4060), p-AKT (Thr308, #2965), BAD (#9239),
BIK (#4592), BIM (#2933), BID (#2002), Bcl-xl (#2764S), Mcl-1
(#4572), PTEN (#9559) and integrin β-1 (#9699S) were obtained from
Cell Signaling Technology. α-tubulin (AJ1034a) and β-actin Abs
(AO1215a) were purchased from Abgent. IRDye 680CW (926-32222) and
IRDye 800CW secondary Abs (926-32210) were obtained from LI-COR
Biosciences.
In-cell western assay
Cells were seeded in a 96-well flat-bottom
polystyrene plate. After culturing for 24 h, medium was removed and
the cells were fixed in phosphate-buffered saline (PBS) containing
3.7% formaldehyde for 20 min at room temperature. The cells were
washed three times with PBS containing 0.1% Triton X-100, and
blocked in PBS containing 5% milk for 1.5 h at room temperature.
The cells were then incubated with rabbit ERK1/2 or p-ERK1/2 Abs in
the blocking buffer for 2 h at room temperature and subsequently
washed with PBS containing 0.1% Tween-20. Infrared anti-rabbit
IRDye 800CW secondary Abs in the blocking buffer was then added and
incubated for 1 h at room temperature in the dark. The plates were
then imaged on an Odyssey infrared scanner using microplate2
settings with sensitivity of 5.0 in the 800-nm wavelength channels.
The fluorescence intensity was measured by Odyssey software and
α-tubulin expression was used as an internal control.
Real-time cell growth assay
Cell growth was performed with the xCELLigence RTCA
instrument (Roche) using E-plate. In this assay, impedance for
indicated times was continuously monitored by the system, and the
value was indicated as ‘Cell Index’ which was determined by the
number of cells seeded, the overall size and morphology of the
cells, and the degree to which the cells interact with the sensor
surface. The electrical impedance of each well was measured at
regular intervals by the system. Data were collected and analyzed
by RTCA software 1.2 (20).
Cell cycle analysis
After washed in ice-cold PBS twice, cells were fixed
with ice-cold 70% ethanol overnight at 4ºC. Fixed cells were
centrifuged and counted. Cells (1×106) were then
re-suspended in 2 ml solution containing 40 μg/ml propidium iodide
(PI; Sigma-Aldrich) and 100 μg/ml RNaseA (Invitrogen) for 30 min at
37ºC. Samples were analyzed by FACSCalibur™ (BD Biosciences,
Shanghai, China) within 1 h after preparation. After gating out
cell debris and fixation of artifacts, flow cytometric analysis
allowed for the discrimination of DNA contents.
Ki-67 assay
Cells were cultured in RPMI-1640 media supplemented
with 10% FBS on coverslides. After continuous culture for 24, 48
and 72 h, the slides were fixed in cold 4% paraformaldehyde for 30
min, and treated with 1% Triton X-100 PBS solution for 10 min.
Then, 1X PBS with 10% FBS was used to block the cells for 45 min at
room temperature. Cells were incubated with a Ki-67 antibody (1:100
diluted; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Following
1X PBS washing, sections were incubated for 30 min using the
secondary Abs (rabbit anti-mouse IgA-B, GK500705; GTVision™ III).
Then, the 3,3-diaminobenzine (DAB) was used to visualize the
immune-reactive products. Results were carried out by the system
microscope IX71 (Olympus, Tokyo, Japan).
Terminal nucleotidyl transferase-mediated
nick end labeling (TUNEL) assay
Cells were cultured on coverslides for 24, 48 and 72
h in a humidified incubator at 37ºC and 5% CO2.
According to the manufacturer's instructions of TUNEL system kit
(Roche), the slides were fixed by cold 4% paraformaldehyde for 30
min. Following 1X PBS washing, 3% H2O2
methanol solution was used to block the slides for 10 min at 20ºC.
Then the slides were treated using the 1% Triton PBS solution for 2
min on ice after PBS washing. Avoiding light, 50 μl of TUNEL
reaction solution were applied to incubate the cells on slides for
60 min at 37ºC. Following PBS washing, the signals of TUNEL were
converted using peroxidase (POD) for 30 min at 37ºC, and the
sections were treated with DAB for 3 min at room temperature.
Results were examined by the light system microscope IX71.
Cell migration assay
Cell migration was also performed with the
xCELLigence RTCA instrument according to a previous report
(20). In this assay, CIM-plate
assembled with upper chamber and lower chamber was used. RPMI-1640
media (180 μl) supplemented with 10% FBS was added in each well on
the lower chamber. Cells were suspended in RPMI-1640 media free of
FBS, 10,000 cells/well were added on the upper chamber. After
attachment, cell migration towards lower chamber containing 10% FBS
media was continuously monitored by the system, and data were
collected and analyzed by RTCA software 1.2.
Scratch healing assay
The confluent cell layers were scratched with a line
using a sterile pipette tip of 200 μl and washed three times with
PBS. The scratched area was then imaged continuously at
magnifications of ×10 with the light system microscope IX71. The
migratory distance is used to measure the migratory ability of
cells.
Cell invasion assay
Cell invasion was also performed with the
xCELLigence RTCA instrument. In this assay, CIM-plate with an upper
chamber and a lower chamber was used. RPMI-1640 (180 μl)
supplemented with 10% FBS was added in each well in the lower
chamber. Cells were suspended in RPMI-1640 free of FBS, 10,000
cells/well were added on the upper chamber. For cell invasion
assay, wells of the upper chamber were pre-coated with Matrigel
(cat. no. 356234; BD Bioscience) for at least 4 h. After
attachment, cells invaded through Matrigel towards the lower
chamber containing 10% FBS media were continuously monitored by the
system, and data were collected and analyzed by RTCA software
1.2.
Gelatinase zymography
Gelatinase zymography was performed in an 8%
SDS-PAGE gel in the presence of 0.1% gelatin under non-reducing
conditions. Culture media were mixed with sample buffer and loaded
for SDS-PAGE with Tris-glycine SDS buffer. Samples were not boiled
before electrophoresis. Following electrophoresis, the gels were
washed twice in 2.5% Triton X-100 for 30 min at room temperature to
remove SDS. The gels were then incubated at 37ºC overnight in
substrate buffer containing 50 mM Tris-HCl and 10 mM
CaCl2 at pH 8.0 and stained with 0.5% Coomassie blue
R250 in 50% methanol and 10% glacial acetic acid for 30 min and
destained. Upon renaturation of the enzyme, the gelatinases
digested the gelatin in the gel to produce clear bands against an
intensely stained background.
Statistical analysis
Data were expressed as mean ± SD. Differences were
analyzed by the Student's t-test. Sample sizes were chosen to
produce statistically unambiguous results. A P-value of ≤0.05 was
considered significant, a P-value of ≤0.01 and ≤0.001,
respectively, was considered as highly significant.
Results
Distinct miR-17-92 expression pattern in
prostate cancer cell lines
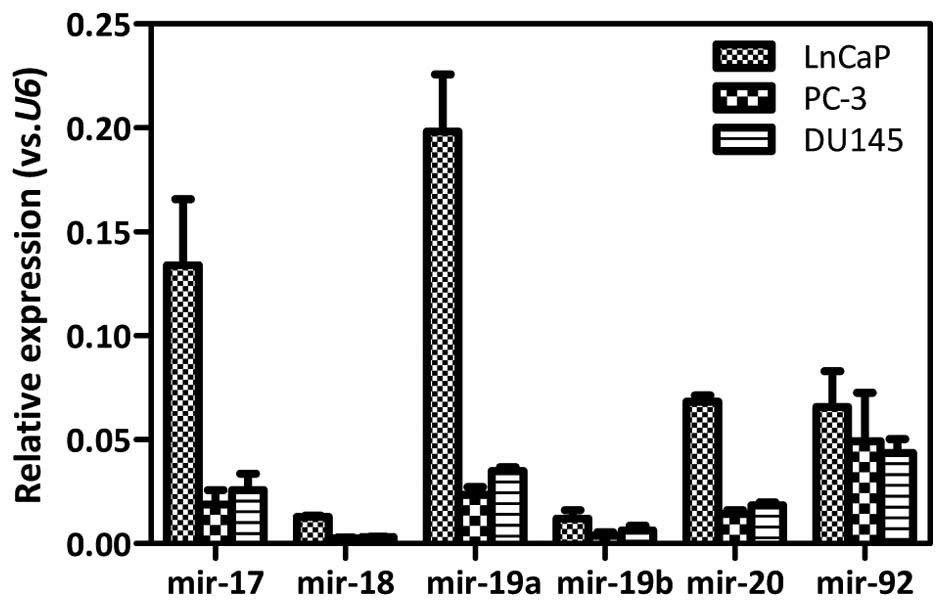
The expression of the miR-17-92 cluster was
determined in androgen-dependent LnCaP and androgen-independent
DU145 as well as PC-3 prostate cancer cell lines by qRT-PCR
analysis. The expression levels of individual miR-17-92
members in DU145 cells were comparable to that in PC-3 cells. The
expression of miR-17 (P=0.0006), miR-18
(P<0.0001), miR-19a (P=0.0005), miR-19b
(P=0.0575), and miR-20 (P<0.0001) was increased ~3- to
6-fold in LnCaP cells, as compared with that of DU145 cells.
Similarly, the expression of miR-17 (P=0.004), miR-18
(P<0.0001), miR-19a (P=0.0004), miR-19b
(P=0.0121), and miR-20 (P<0.0001) was increased ~4- to
8-fold in LnCaP cells, as compared with that of PC-3 cells.
However, no significant difference in the miR-92 expression
was detected among these three cell lines (Fig. 1). Therefore, the expression of the
miR-17-92 cluster was much more abundant in
androgen-dependent LnCaP cells than that in androgen-independent
cells. The expression levels of the miR-17-92 cluster were
near equivalent between the androgen-independent DU145 and PC-3
prostate cancer cells.
Introduction of the miR-17-92 cluster
into prostate cancer cells
To understand the functions of miR-17-92 on
the biological behavior of DU145 prostate cancer cells, plasmid
overexpressing the miR-17-92 cluster was constructed and
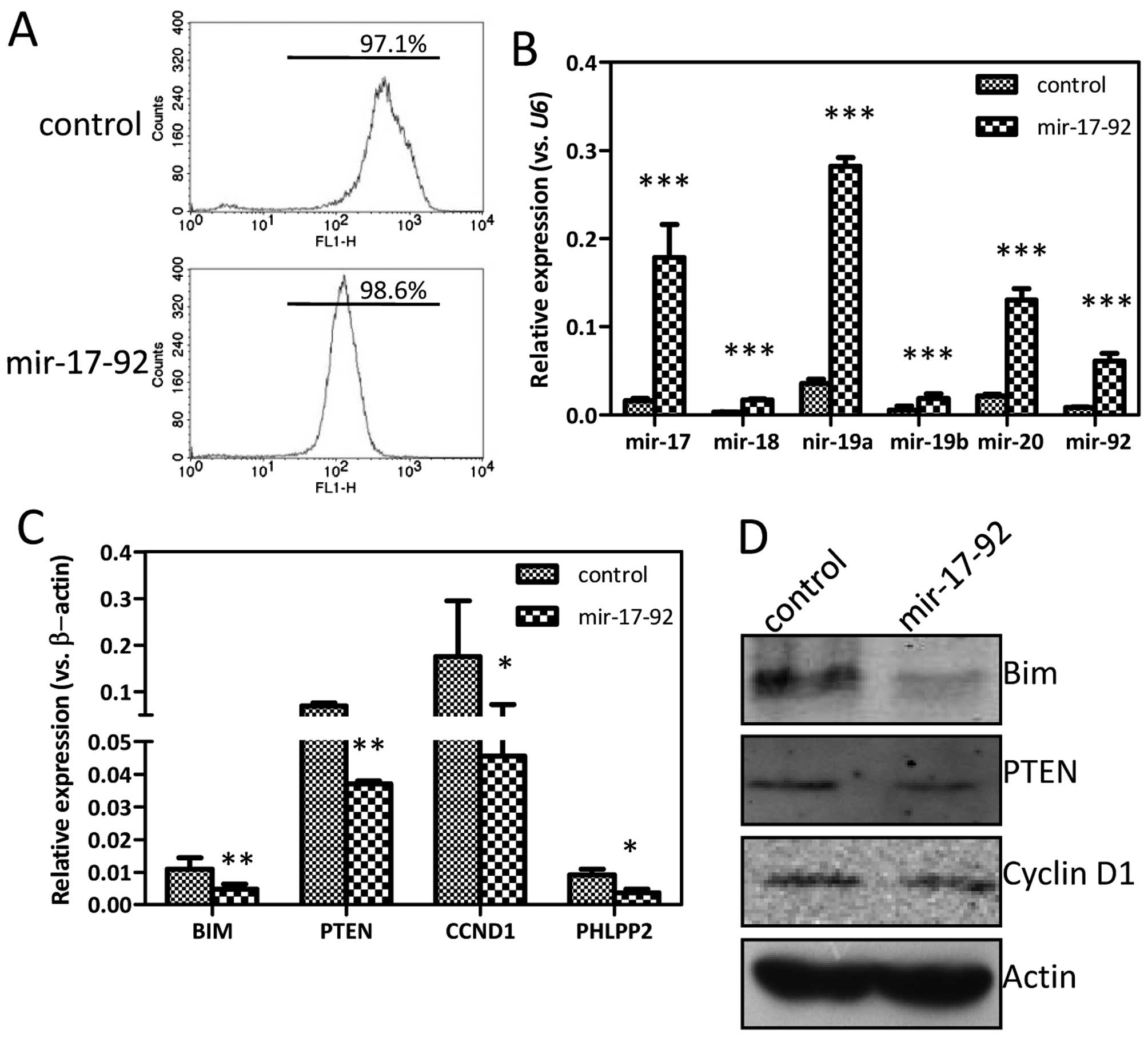
transfected into DU145 cells. The individual monoclones were
subsequently selected by puromycin for two weeks. The percentages
of GFP-positive cells in the established DU145-control and
DU145-miR-17-92 cell lines were 97.1 and 98.6%,
respectively, detected by flow cytometry (Fig. 2A). The selected monoclone was
further expanded and examined for the miR-17-92 expression
by qRT-PCR analysis. The expression of every miR-17-92
family member was markedly increased in the DU145-miR-17-92 cells
compared to that in DU145-control cells, 11-fold for miR-17
(P<0.0001), 5-fold for miR-18 (P<0.0001), 8-fold for
miR-19a (P<0.0001), 3-fold for miR-19b (P=0.0096),
6-fold for miR-20 (P=0.0001), and 7-fold for miR-92
(P<0.0001), indicating a successful introduction of the
miR-17-92 cluster (Fig.
2B). The expression of some well-known target genes of the
miR-17-92 cluster, such as BIM, PTEN,
CCND1 and PHLPP2 was evidently reduced in the
DU145-miR-17-92 cells compared to that of DU145-control cells,
albeit with different levels (Fig.
2C). In line with the qRT-PCR results, the expression of BIM,
PTEN and cyclin D1 was reduced at the protein level detected by the
western blotting (Fig. 2D).
Therefore, it is shown that the DU145 prostate cancer cell line
overexpressing the miR-17-92 cluster was established
successfully.
miR-17-92 overexpression promotes DU145
cell growth
The cell growth affected by the miR-17-92
overexpression was monitored dynamically using a real-time
xCELLigence system. The cell growth of DU145 cells overexpressing
miR-17-92 was much faster than that of the DU145-control
cells and statistically significant difference were found between
the two established cell lines during the 72-h continuous
monitoring (Fig. 3A). The cell
cycle analysis and cellular DNA content measurement were detected
by flow cytometry, and no obvious differences were found between
the DU145-control and DU145-miR-17-92 cells in any of the three
phases (G0-G1, S and G2-M)
(Fig. 3B). To investigate whether
the miR-17-92 overexpression affects the proliferation
capability of the DU145 cells, a Ki-67 cell proliferation assay was
carried out. The frequencies of Ki-67-positive cells in the
DU145-control group were 56.57±1.68, 85.48±0.26 and 90.85±2.08% at
24, 48 and 72 h, respectively; while those in the DU145-miR-17-92
group were 73.64±0.68, 93.43±1.23 and 97.36±0.86% (Fig. 3C). The cellular proliferation of
the DU145-miR-17-92 cells was higher at all time-points although, a
statistically significant difference in the frequency of
Ki-67-positive cells between the two cell lines was only observed
at 24 h. A TUNEL assay was performed to quantitatively examine
apoptotic cells. As showed in Fig.
3D, both the DU145-control and DU145-miR-17-92 cells underwent
apoptosis in a time-dependent manner. The percentages of apoptotic
cells in the DU145-control group were 6.76±0.09, 14.51±0.86 and
20.73±1.64% at 24, 48 and 72 h, respectively; while those in the
DU145-miR-17-92 group were 1.86±0.15, 7.90±0.40 and 4.92±0.48%.
Apoptosis was markedly reduced in the DU145-miR-17-92 cells and
there was a statistically significant difference in the frequencies
of apoptotic cells at all time-points between the two cell lines.
Thus, it is shown that miR-17-92 overexpression promoted
cell growth in DU145 prostate cancer cells. The diminished cellular
apoptosis together with the increased cellular proliferation
contributed together to the enhanced growth upon the introduction
of the miR-17-92 cluster into DU145 cells.
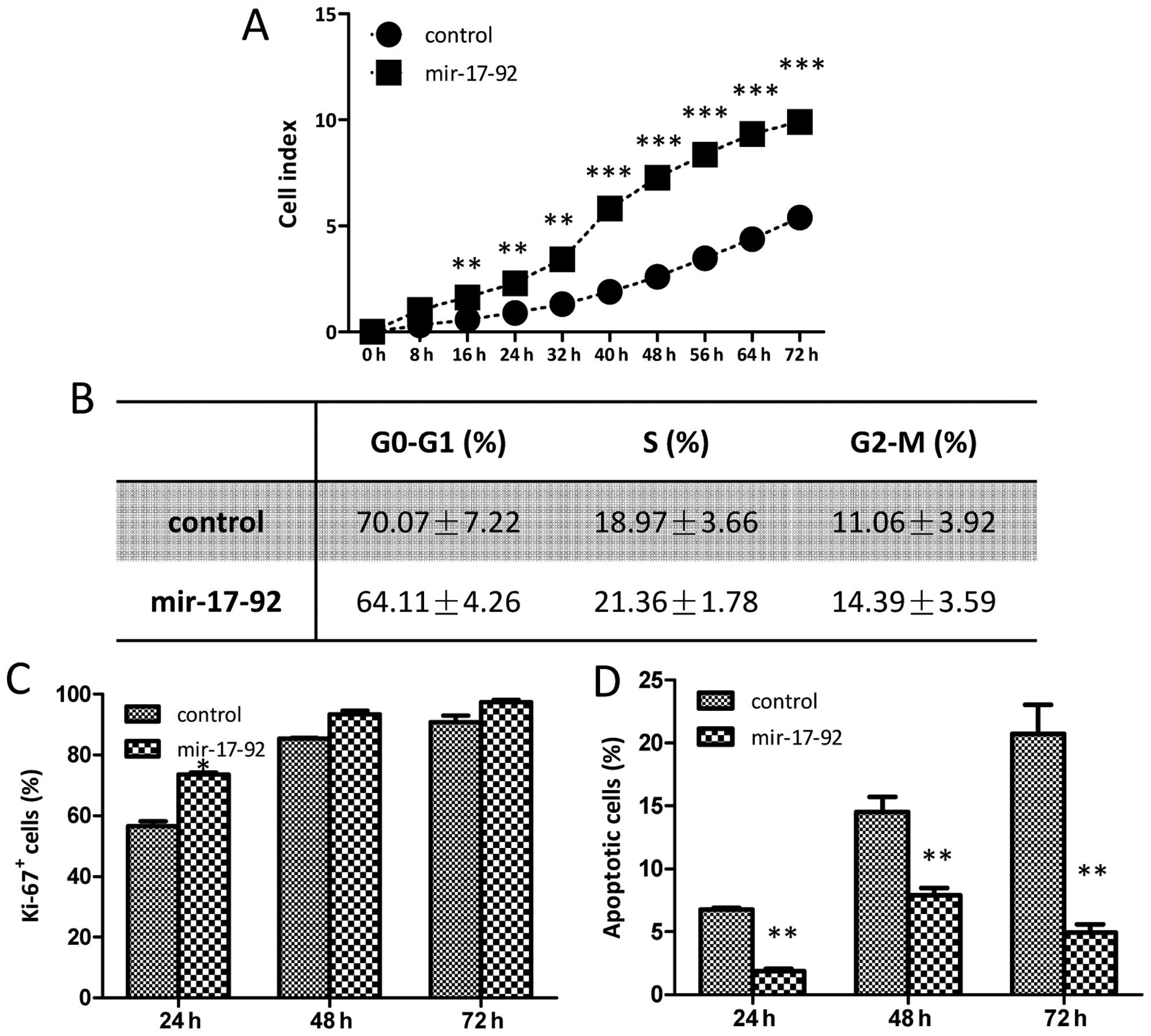 | Figure 3Overexpression of the
miR-17-92 cluster promotes DU145 cell growth. (A) The cell
growth curves between the DU145-control and DU145-miR-17-92 cells
were detected by xCELLigence system using E-plate. Each plate was
inoculated with 8,000 cells, and the cell growth was detected
during a 72-h continuous monitoring. (Student's t-test,
*P<0.05, **P<0.01,
***P<0.001). (B) The cell cycle analysis between the
two established cell lines was examined by flow cytometry. The
table presents the data of three phases
(G0-G1, S and G2-M). (C) The Ki-67
assay was carried out to quantitatively evaluate the proliferation
capabilities. The bar chart represents the frequencies of Ki-67
positive cells of the two established cell lines. Significant
differences are indicated (Student's t-test, *P<0.05,
**P<0.01). (D) The TUNEL assay was carried out to
quantitatively evaluate the apoptotic cells. The bar chart
represents the percentages of apoptotic cells of the two
established cell lines. Significant differences are indicated
(Student's t-test, *P<0.05,
**P<0.01). |
miR-17-92 regulates apoptosis-related and
proliferation-related protein
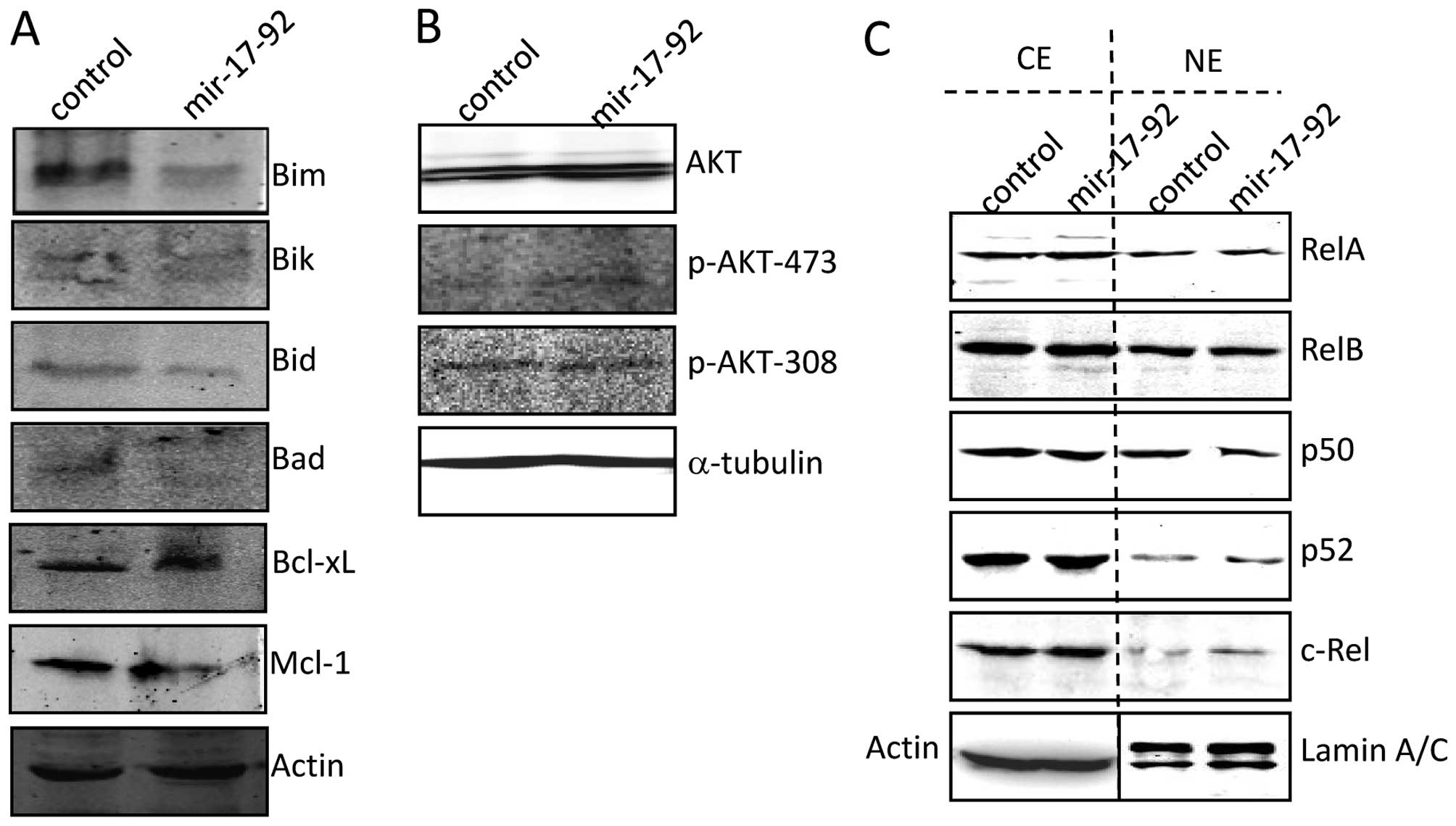
As shown in Fig.
4A, the expression of pro-apoptotic proteins such as BIM, BIK,
BID and BAD was distinctly reduced in the DU145-miR-17-92 cells
compared to that of the DU145-control cells. However, no
significant difference in anti-apoptotic proteins such as Bcl-xL
and Mcl-1 were found between the two cell lines. The AKT signaling
molecules were examined. The expression level of total AKT was
comparable in the whole-cell extracts of the DU145-control and
DU145-miR-17-92 cells. However, miR-17-92 overexpression
induced clear phosphorylation of AKT at Ser 473, but not at Thr 308
(Fig. 4B), which was correlated
with the inhibited PTEN expression (Fig. 2D). The expression of RelA and p50,
contributing to the canonical NF-κB activity, was comparable
between the DU145-miR-17-92 and DU145-control cells at both
cytoplasmic and nuclear fractions. The expression of RelB and p52,
contributing to the non-canonical NF-κB activity, also showed no
difference between the two cell lines (Fig. 4C). Therefore, the NF-κB signaling
was not affected by miR-17-92 overexpression in DU145 cells.
Collectively, forced introduction of the miR-17-92 cluster
into DU145 cells suppressed the expression of pro-apoptotic related
protein, particularly BIM, which functioned critically in apoptosis
resistance. Moreover, forced introduction of the miR-17-92
cluster into DU145 cells activated the AKT signaling due to PTEN
inhibition, which contributed at least partially to the increased
cellular proliferation.
miR-17-92 overexpression induces ERK1/2
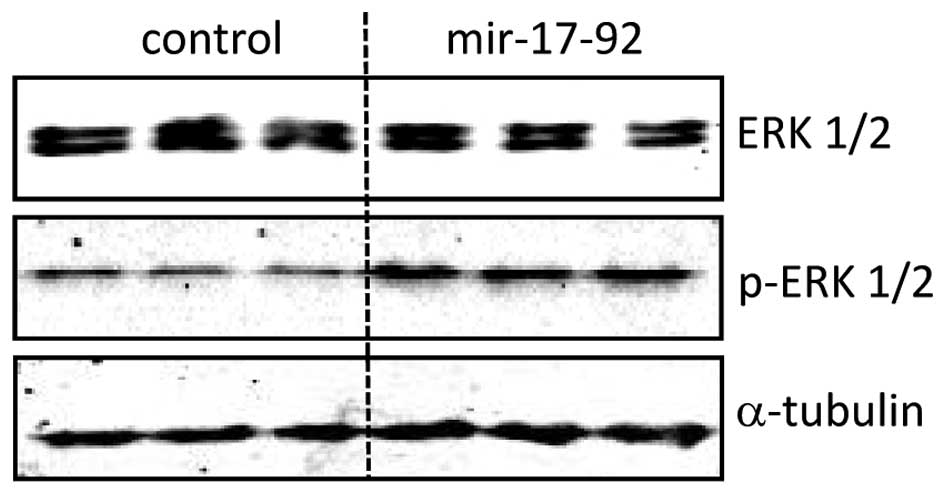
phosphoration in DU145 cells
The protein expression level of total ERK1/2 was
comparable between the DU145-miR-17-92 and DU145-control cells.
However, miR-17-92 overexpression in DU145 cells induced
continuous phosphorylation of ERK1/2 (p-ERK1/2) detected by western
blot analysis (Fig. 5). In
addition, the expression of total ERK1/2 and p-ERK1/2 was analyzed
quantitatively in situ by an In-Cell western assay (data not
shown). The ERK1/2 expression in the DU145-miR-17-92 cells was
similar to that of the DU145-control cells. In line with the
results from western blot analysis, the expression of p-ERK1/2 was
increased ~3-fold (P<0.0001) in the DU145-miR-17-92 cells as
compared to that in the DU145-control cells (data not shown).
Therefore, overexpression of the miR-17-92 cluster in DU145
prostate cancer cells induces the phosphorylation of ERK1/2 without
affecting basal ERK1/2.
miR-17-92 overexpression enhances
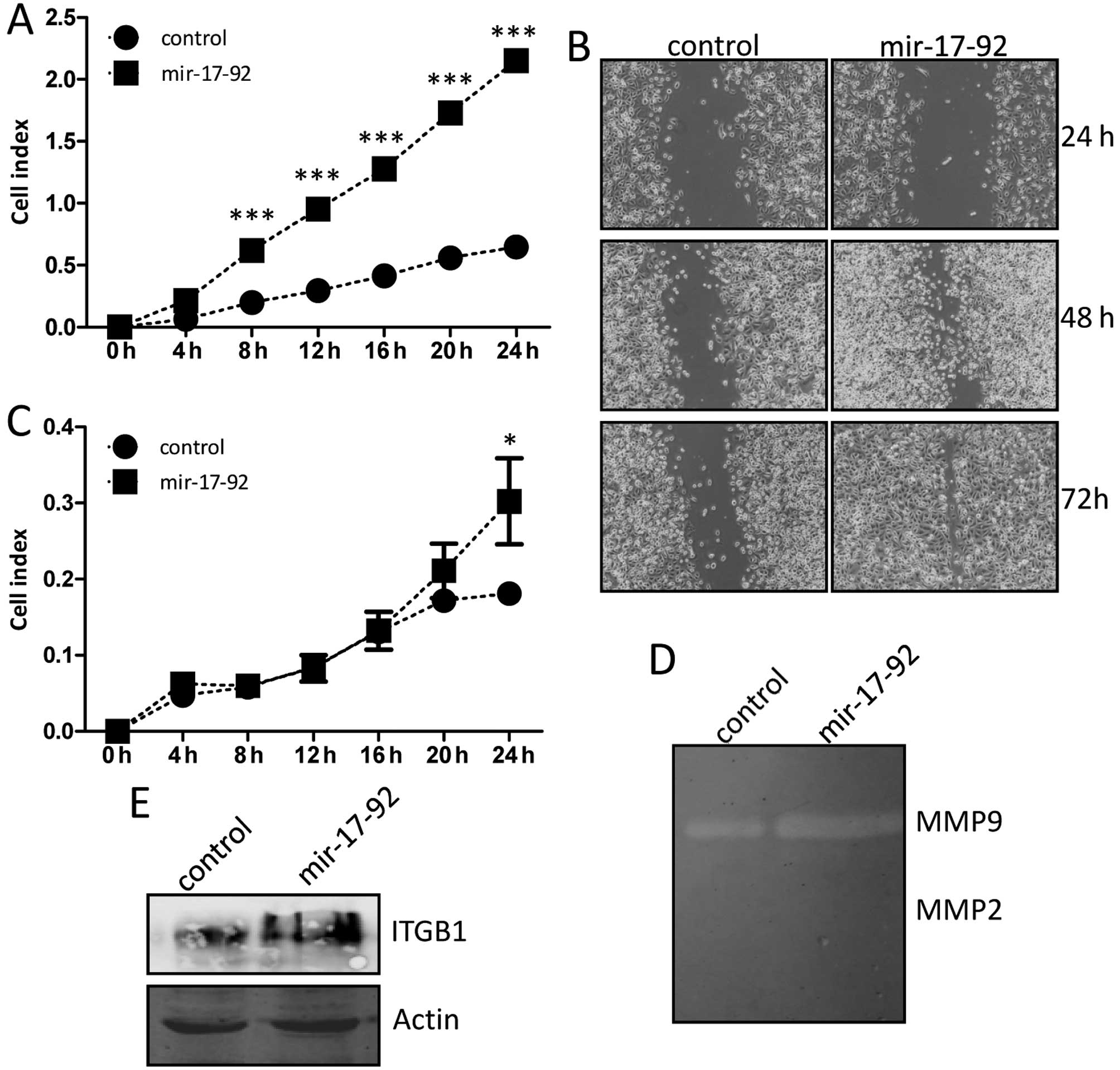
migration and invasion abilities of DU145 cells
The migration ability was examined dynamically by a
real-time xCELLigence system using CIM-plates. The DU145 cells
overexpressing miR-17-92 migrated markedly faster than that
of the DU145-control cells during the 24-h continuous monitoring,
and there were significantly statistical difference in the
migration assay between the two established cell lines (Fig. 6A). The in vitro scratch
assay was also carried out to evaluate quantitatively the migration
ability of cells. A scratched cell monolayer was generated in both
cell lines and images were captured after culturing for 24, 48 and
72 h. At the 48 and 72-h time-point of the assay, it was shown that
the DU145-miR-17-92 cells migrated from the edge towards the center
of the scratch more rapidly than that of the DU145-control cells
(Fig. 6B), indicating better
migratory ability. The invasion ability was also examined by the
real-time xCELLigence system using matrigel (dilution at
1:40)-coated CIM-plates. The DU145 cells overexpressing
miR-17-92 invaded through matrigel faster than that of the
DU145-control cells at the 24 h time-point, and there was
statistically significant difference between the two established
cell lines (Fig. 6C). The gelatin
zymography experiment was further performed to explore the relative
amounts of active and inactive gelatinase (MMP-2 or MMP-9). As
showed in Fig. 6D, the MMP-2
activity was not detected in either of the cell lines, and the
MMP-9 activity was slightly increased in the DU145-miR-17-92 cells
compared to that of the DU145-control cells, indicating the
enhanced invasion ability of cells. Notably, integrin β-1
expression at the protein level was clearly induced in the
DU145-miR-17-92 cells compared to that of the DU145-control cells
(Fig. 6E). Thus, these results
indicated that miR-17-92 overexpression promoted the
migration and invasion of DU145 prostate cancer cells, and the
upregulated integrin β-1 was implicated in these processes.
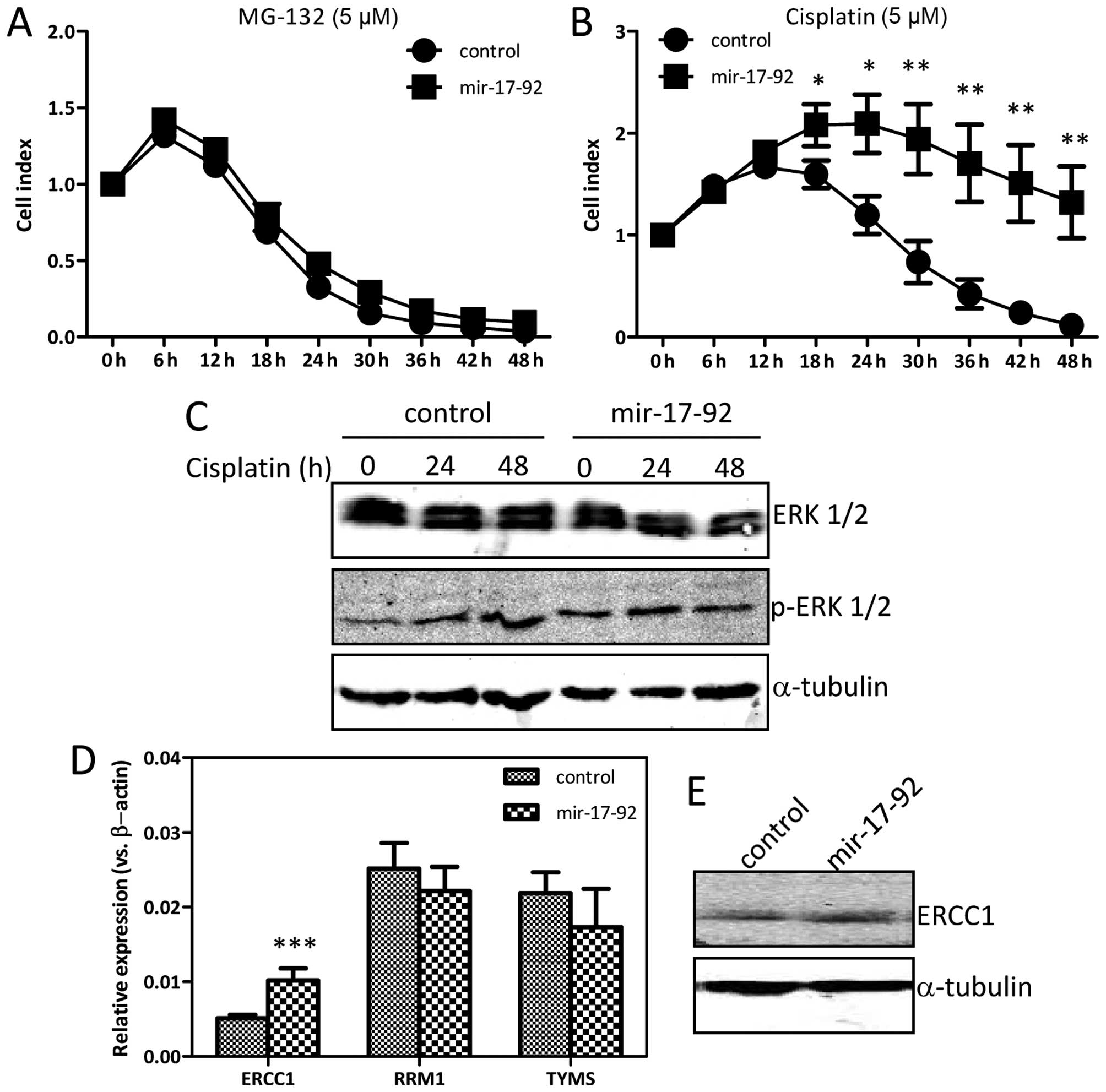
miR-17-92 overexpression affects
cisplatin-sensitivity
The effects of MG-132 and cisplatin on the
DU145-miR-17-92 and the DU145-control cells were monitored
dynamically by the xCELLigence system. MG-132, a proteasome
inhibitor, caused considerable cell death in both cell lines in a
time-dependent manner (Fig. 7A).
The cisplatin-treatment led to significant cell death in
DU145-control cells in a time-dependent manner (Fig. 7B). Interestingly, the
DU145-miR-17-92 cells were relatively resistant to the
cisplatin-treatment as compared with the DU145-control cells at the
time-point of 18 h (P=0.0272), 24 h (P=0.0105), 30 h (P=0.0064), 36
h (P=0.0054), 42 h (P=0.0047) and 48 h (P=0.0044). Overexpressing
the miR-17-92 cluster in DU145 cells increased
IC50 of cisplatin from 3.45 to 5.88 μM. During
cisplatin-treatment, the protein level of basal ERK1/2 remained
high and unchanged in both cell lines (Fig. 7C). The expression level of p-ERK1/2
was increased gradually in a time-dependent manner in the
DU145-control cells upon cisplatin-treatment. However, the
expression of p-ERK1/2 in the DU145-miR-17-92 cells, which was
already at a high level in the untreated condition, was not
influenced and remained high upon cisplatin-treatment. The mRNA
level of ERCC1, RRM1 and TYMS was analyzed by
qRT-PCR. As shown in Fig. 7D, the
ERCC1 mRNA level was increased ~2-fold (P=0.0001) in
DU145-miR-17-92 cells as compared to that in DU145-control cells,
while the RRM1 and TYMS mRNA levels were similar in
the two cell lines. The expression of ERCC1 was also increased at
the protein level in the DU145-miR-17-92 cells as compared to that
in the DU145-control cells (Fig.
7E). Collectively, overexpression of the miR-17-92
cluster in DU145 prostate cancer cells caused chemo-resistance to
cisplatin, at least partially due to the sustained and high level
of ERK1/2 phosphoration. In addition, the continuously activated
AKT signaling together with upregulated ERCC1 also contributed to
development of cisplatin-resistance.
Discussion
In the present study, the miR-17-92 cluster
regulation of diverse cellular behavior of prostate cancer cells
was systematically examined. The expression levels of the
miR-17-92 cluster in the androgen-independent cells were
much lower than that in the androgen-dependent prostate cancer
cells. Forced expression of the miR-17-92 cluster into the
DU145 androgen-independent cells interfered with multiple
biological events. The stimulatory function of the miR-17-92
cluster in prostate cancer cell growth, migration, invasion and
acquired cisplatin-resistance were clearly evident in
vitro.
The miR-17-92 cluster is widely expressed in
different cell types and essential for many developmental and
pathogenic processes. The expression of individual members of the
miR-17-92 cluster in the three prostate cancer cell lines
was analyzed by qRT-PCR. In line with a previous report (16), we found that the expression pattern
of the miR-17-92 cluster was clearly different between the
androgen-dependent and androgen-independent prostate cancer cell
lines. The expression levels of miR-17, miR-18,
miR-20 and miR-19 in the androgen-dependent LnCaP
cells were much higher than that in the androgen-independent DU145
and PC-3 cells. However, the expression pattern of the two
androgen-independent prostate cancer cell lines, DU145 and PC-3,
was quite similar according to the present study, albeit with
varied levels. Therefore, the expression of the miR-17-92
cluster was negatively correlated with the AR expression status in
prostate cancer cell lines, suggesting that the miR-17-92
cluster is possibly involved in the progression from
androgen-dependent into androgen-independent stage due to certain,
yet unknown, reasons.
To identify the contribution of the miR-17-92
cluster to tumorigenesis in prostate cancer, the miR-17-92
cluster was successfully transfected into the androgen-independent
DU145 cells, in which the miR-17-92 cluster was minimally
expressed. The mRNA expression of a number of well-known targets
including PTEN, BIM, CCND1 and PHLPP2
was inhibited in the miR-17-92-overexpressing DU145 cells.
In the study, we observed significant enhancement of prostate
cancer cell growth on the introduction of the miR-17-92
cluster and found that the improved cellular proliferation together
with the diminished apoptosis both contributed to the rapid cell
growth.
Located in the third intron of the C13orf25
gene, the miR-17-92 cluster is regulated by transcription
factors such as c-Myc, E2F family members, and STAT3, each of which
are frequently activated in cancer. Previous evidence suggests that
the growth stimulatory effect of the miR-17-92 cluster is
partially attributable to the unbalance between cellular
proliferation and apoptosis. Our findings here are similar with
observations in a variety of hematopoietic and solid malignancies
(21). Conditionally active the
miR-17-92 cluster in lymphocytes increases proliferation and
reduces activation-induced cell death (22). Leukemia expressing increased levels
of the miR-17-19 construct enhances cellular proliferation
associated with reduced expression of the cyclin-dependent kinase
inhibitor, CDKN1/p21, which is a direct target of
miR-17 and a potent negative regulator of the G1-S
checkpoint of cell cycle progression (23,24).
The miR-17-92 cluster also conveys significant stimulatory
activity in cell growth of lung and hepatocellular cancer cells
in vitro (25,26). Overexpression of the
miR-17-92 cluster enhances cholangiocarcinoma growth in
hairless outbred mice (27). Gain-
and loss-of-function assays demonstrate that miR-17 affects the
proliferation of gastric cancer cells both in vitro and
in vivo (28).
Overexpression of miR-17 in gastric cancer cells is
correlated with the amplification of several
proliferation-associated oncogene such as MYC, CCNE1,
ERBB2 and FGFR2. Knockdown of miR-17
suppresses the proliferative potential of KATO-III gastric cancer
cells, which is coupled with markedly decreased phosphorylated
ERK1/2 levels (29). In fact, a
variety of cell proliferation-associated and apoptosis-associated
genes such as MAPK, E2F and JAK, AKT,
STAT, Bcl-2, XIAP, PIK3R3 are
potentially targeted by the miR-17-92 cluster in a
tissue-specific manner (30). In
the present study, we showed that the total AKT was not changed
upon introduction of the miR-17-92 cluster into DU145 cells;
however, the phosphorylated AKT (p-AKT) at Ser 473 was induced.
Similarly, the induced phosphorylation of ERK1/2 (p-ERK1/2) was
also observed, though the total ERK levels remained unchanged.
Taken together, these data suggested that overexpession of the
miR-17-92 cluster in DU145 prostate cancer cells increased
cellular proliferation, the activated AKT and ERK1/2 signaling
pathways were implicated in the process.
Many studies have also reported oncogenic activity
of the miR-17-92 cluster through suppression of apoptotic
genes. In non-small cell lung cancer cells, prostaglandin E2
upregulates the MYC gene followed by elevation of the
miR-17-92 expression, which reduces PTEN expression and
enhances apoptosis resistance (31). Antisense-mediated inhibition of
miR-17 and miR-20 induces clear apoptosis in the
miR-17-92-overexpressing lung cancer cells, which is
partially related to the induction of E2F1 (32). Suppression of miR-17 in
thyroid cancer cells leads to complete cell growth arrest,
presumably due to caspase activation resulting in apoptosis
(33). The high-level
amplification of the miR-17-92 in glioblastoma inhibits
apoptosis by targeting CDKN1A, E2F1, PTEN and
CTGF (34). The survival
effect of miR-19 is mostly mediated by the increased
PI3K/AKT signaling, largely due to the posttranscriptional
repression of PTEN (35,36). miR-92 levels in colon cancer
cells have been shown to correlate negatively with reduced
apoptosis and with BIM expression (12). A potential anti-apoptotic role for
miR-20 has been found in PC-3 prostate cancer cell line
(17). In the present study, we
observed that overexpressing of the miR-17-92 cluster in
DU145 cells led to clear reduction of apoptosis, which was coupled
with the suppressed expression of certain pro-apoptotic proteins
such as BIM, whose dysregulation affects apoptosis in diverse
cancer cells. BIM is a direct target of multiple members of the
miR-17-92 cluster and other related miRNAs. As a
pro-apoptotic protein, BIM regulates cell death in various settings
through its ability to antagonize anti-apoptotic protein such as
Bcl-2 (37). Inhibited PTEN
expression also contributed to the apoptotic resistance. PTEN is a
crucial negative regulator of the highly oncogenic pro-survival
PI3K/AKT signaling pathway, and is predominantly targeted by
miR-19. In response to a variety of extracellular signals,
the PI3K/AKT pathway elicits diverse cellular responses to promote
cell survival, rapid proliferation and cell growth. Other molecular
events regulating the balance of the cellular proliferation and
apoptosis such as the NF-κB signaling, which is regulated by the
miR-17-92 cluster (38,39),
were not affected in the prostate cancer cells according to the
present study. Thus, we presented evidence that overexpession of
the miR-17-92 cluster in DU145 prostate cancer cells
promoted cell growth, the activated AKT and ERK1/2 signaling, and
dysregulated expression of apoptosis-related protein played
important roles. It is also conceivable that additional pathways
regulated by miR-17-92 cluster could synergize together to
promote cell growth. The miR-17-92 cluster is indeed a key
component of the complex regulatory signaling network for cellular
proliferation and apoptosis in prostate cancer cells.
Furthermore, we showed that overexpression of the
miR-17-92 cluster substantially enhanced the migration and
invasion of DU145 prostate cancer cells. Our findings are in line
with previous reports that the miR-17-92 cluster plays a
role in cancer invasion and metastasis (26,40).
Overexpression of miR-17 in MCF-7 breast cancer cells
renders the ability of invasiveness and migration by targeting the
HBP1/β-catenin pathway. Downregulation of endogenous miR-17
in MDA-MB-231 cells suppresses the migration and invasion (41). In addition, miR-17 can
enhance the migration ability of melanoma cells by inhibiting the
translation of ETV1 (42).
miR-19 facilitates gastric cancer cell migration and
invasion through targeting the antagonist of c-myc-MXD1 (43). In contrast, forced expression of
miR-92 suppresses peritoneal dissemination of ovarian cancer
in vivo by inhibiting integrin α-5 expression (44). For the first time, we provided
evidence that the miR-17-92 cluster played a positive role
in regulating the migration and invasion of prostate cancer cells,
and therefore functioned as an oncogene in prostate cancer
cells.
Diverse molecular regulators, including adhesion
receptor families, receptor tyrosine kinase, cytoskeleton protein,
adapters and signaling molecules, are involved in the regulation of
migration and invasion of cancer cells. Interestingly, we found
that the expression of integrin β-1 was upregulated in the
DU145-miR-17-92 cells. Integrin β-1, encoded by the ITGB1
gene, belongs to the family of heterodimeric transmembrane cell
surface receptors that contain 18 α and 8 β subunits and bridge
crosstalk between cell-cell and cell-extracellular matrix (ECM).
Integrin β-1 activation is a key regulator in the switch from
cellular dormancy to metastatic growth in vitro and in
vivo. Overexpression of integrin β-1 has been found in various
epithelial malignancies during invasion, angiogenesis and
metastasis. In some human solid tumors, increased expression of
integrin β-1 correlates with enhanced metastatic potential and
shortened patient survival (45,46).
Integrin β-1 and integrin-induced autophosphorylation of focal
adhesion kinase (FAK) are increased in prostate cancer cells in
primary prostate cancer and lymph node metastases. Increased
integrin β-1 activation in prostate cancer cells correlates with
metastatic potential in vivo. Using the integrin β-1
neutralizing mAbs 33B6 inhibits the phosphorylation of integrin β-1
downstream effectors, FAK and AKT, leading to increased apoptosis
(47). The activation of integrin
β-1 is likely through an inside-out mechanism, which induces
conformational changes of integrin β-1 in the absence of ECM
ligands. Diverse mechanisms contribute to the inside-out
activation, including interaction with its regulatory molecules,
biochemical modification of the integrins, and regulation by
extracellular growth factors. Activation of integrin β-1 could also
be regulated by the PI3K/AKT signaling pathway (48). Further studies on the mechanisms by
which integrin β-1 become constitutively activated upon
introduction of the miR-17-92 cluster into the prostate cancer
cells are warranted.
Besides its role in tumor onset and progression,
the miR-17-92 cluster can enhance chemo-resistance. In the
present study, we have also illustrated that overexpression of the
miR-17-92 cluster caused severe chemo-resistance to
cisplatin. Treated with cisplatin, DU145-miR-17-92 cells had less
cell death and a much higher IC50 value than that of
DU145-control cells. Cisplatin exerts its anticancer effects by
disrupting DNA structure in cell nuclei through the formation of
intra-strand and inter-strand cross-links. Cisplatin-based
chemotherapy has been widely used to treat a variety of cancers,
but dose limiting toxicities or intrinsic and acquired resistance
often reduced its clinical benefit in several types of cancer
including prostate cancer (49,50).
Many mechanisms are involved in developing cisplatin resistance
such as change in the expression levels of certain miRNAs,
cytoplasmic translocation of p21 by phosphorylation, and activation
of the PI3K/AKT signaling pathway. Among those, the PI3K/AKT
signaling pathway plays a specific role (51). Nevertheless, the involvement of the
miR-17-92 cluster in developing cisplatin-resistance is
still contradictory. For example, miR-17 and miR-20
are downregulated in cisplatin-resistant A549/DDP (cisplatin) cells
compared with A549 non-small lung cancer cells by miRNA microarray
profiling analysis. Inhibition of miR-17 and miR-20
increases cisplatin-resistance while over-expression of
miR-17 and miR-20 decreases cisplatin-resistance in
A549 cells (52). In the present
study, overexpression of the miR-17-92 cluster activated the
AKT signaling pathway due to inhibition of PTEN, which could in
part explain the development of cisplatin-resistance in DU145
cells.
ERK1/2 signaling pathway is shown to be associated
with various cellular processes such as differentiation,
proliferation, transformation and apoptosis. Anti-proliferative and
apoptotic effects of cisplatin have been attributed to activation
of ERK1/2 signaling in various cell lines. Cisplatin treatment
results in dose- and time-dependent activation of ERK1/2, which is
important for the induction of cisplatin-induced apoptosis in HeLa
and A549 cells (53).
Cisplatin-induced ERK1/2 activation is an upstream regulator of
p53, which leads to DNA damage caused by cisplatin (54). In the present study,
cisplatin-treatment induced the phosphorylaion of ERK1/2 in a
time-depend manner while the total ERK levels remained high and
unchanged in DU145 control cells, correlated with the
time-dependent apoptosis. Interestingly, the phosphorylaion of
ERK1/2 was constantly high during the cisplatin-treatment in
DU145-miR-17-92 cells, suggesting that the continuously activated
ERK signaling also contributed to cisplatin resistance.
High ERCC1 expression in cancer cells is associated
with lower cisplatin sensitivity and is a potential indicator of
response to cisplatin and prognosis (55,56).
Accumulation of ERK1/2 phosphorylation can increase ERCC1
expression to protect hepatoma and melanoma cells from DNA damage
(57,58). siRNA-mediated silencing of ERCC1
increases the cisplatin sensitivity in PC-3 and DU145 prostate
cancer cell lines (59). In DU145
prostate cancer cells, overexpressing the miR-17-92 cluster
caused upregulated ERCC1 expression, which was also responsible for
the acquired cisplatin-resistance. The expression level of the
miR-17-92 cluster in prostate cancer cells could be
considered as a potential biomarker in platinum-based
therapies.
These findings here establish a key tumor-promoting
role of the miR-17-92 cluster in the carcinogenesis of
prostate cancer. The miR-17-92 cluster plays a crucial role
in cell growth of the DU145 prostate cancer cells due to regulation
of cellular proliferation and apoptosis. Given the potent effects
on cell growth, migration, invasion, and chemo-sensitivity exerted
by the miR-17-92 cluster, miRNAs belonging to the cluster
represent attractive targets for cancer therapy.
Acknowledgements
The present was supported by the National Natural
Science Foundation of China (grant no. 81172433 to F.G.) and the
Natural Science Foundation of Jiangsu Provincial (grant no.
BK20151211 to F.G.).
References
|
1
|
Saini S, Majid S and Dahiya R: Diet,
microRNAs and prostate cancer. Pharm Res. 27:1014–1026. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Porkka KP, Pfeiffer MJ, Waltering KK,
Vessella RL, Tammela TL and Visakorpi T: MicroRNA expression
profiling in prostate cancer. Cancer Res. 67:6130–6135. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu J, Getz G, Miska EA, Alvarez-Saavedra
E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA,
et al: MicroRNA expression profiles classify human cancers. Nature.
435:834–838. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun T, Wang Q, Balk S, Brown M, Lee GS and
Kantoff P: The role of microRNA-221 and microRNA-222 in
androgen-independent prostate cancer cell lines. Cancer Res.
69:3356–3363. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
He L, Thomson JM, Hemann MT,
Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe
SW, Hannon GJ, et al: A microRNA polycistron as a potential human
oncogene. Nature. 435:828–833. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ota A, Tagawa H, Karnan S, Tsuzuki S,
Karpas A, Kira S, Yoshida Y and Seto M: Identification and
characterization of a novel gene, C13orf25, as a target for
13q31-q32 amplification in malignant lymphoma. Cancer Res.
64:3087–3095. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Volinia S, Calin GA, Liu CG, Ambs S,
Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et
al: A microRNA expression signature of human solid tumors defines
cancer gene targets. Proc Natl Acad Sci USA. 103:2257–2261. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mogilyansky E and Rigoutsos I: The
miR-17/92 cluster: A comprehensive update on its genomics,
genetics, functions and increasingly important and numerous roles
in health and disease. Cell Death Differ. 20:1603–1614. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mu P, Han YC, Betel D, Yao E, Squatrito M,
Ogrodowski P, de Stanchina E, D'Andrea A, Sander C and Ventura A:
Genetic dissection of the miR-17~92 cluster of microRNAs in
Myc-induced B-cell lymphomas. Genes Dev. 23:2806–2811. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
van Haaften G and Agami R: Tumorigenicity
of the miR-17-92 cluster distilled. Genes Dev. 24:1–4. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Conkrite K, Sundby M, Mukai S, Thomson JM,
Mu D, Hammond SM and MacPherson D: miR-17-92 cooperates with RB
pathway mutations to promote retinoblastoma. Genes Dev.
25:1734–1745. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsuchida A, Ohno S, Wu W, Borjigin N,
Fujita K, Aoki T, Ueda S, Takanashi M and Kuroda M: miR-92 is a key
oncogenic component of the miR-17-92 cluster in colon cancer.
Cancer Sci. 102:2264–2271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Osada H and Takahashi T: let-7 and
miR-17-92: Small-sized major players in lung cancer development.
Cancer Sci. 102:9–17. 2011. View Article : Google Scholar
|
|
14
|
Cho WC: OncomiRs: The discovery and
progress of microRNAs in cancers. Mol Cancer. 6:602007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pesta M, Klecka J, Kulda V, Topolcan O,
Hora M, Eret V, Ludvikova M, Babjuk M, Novak K, Stolz J, et al:
Importance of miR-20a expression in prostate cancer tissue.
Anticancer Res. 30:3579–3583. 2010.PubMed/NCBI
|
|
16
|
Sikand K, Slane SD and Shukla GC:
Intrinsic expression of host genes and intronic miRNAs in prostate
carcinoma cells. Cancer Cell Int. 9:212009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sylvestre Y, De Guire V, Querido E,
Mukhopadhyay UK, Bourdeau V, Major F, Ferbeyre G and Chartrand P:
An E2F/miR-20a autoregulatory feedback loop. J Biol Chem.
282:2135–2143. 2007. View Article : Google Scholar
|
|
18
|
Watahiki A and Wang Y, Morris J, Dennis K,
O'Dwyer HM, Gleave M, Gout PW and Wang Y: MicroRNAs associated with
metastatic prostate cancer. PLoS One. 6:e249502011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo F, Kang S, Zhou P, Guo L, Ma L and Hou
J: Maspin expression is regulated by the non-canonical NF-κB
subunit in androgen-insensitive prostate cancer cell lines. Mol
Immunol. 49:8–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu F, Zhou J, Zhou P, Chen W and Guo F:
The ubiquitin ligase CHIP inactivates NF-κB signaling and impairs
the ability of migration and invasion in gastric cancer cells. Int
J Oncol. 46:2096–2106. 2015.PubMed/NCBI
|
|
21
|
Olive V, Li Q and He L: mir-17-92: A
polycistronic oncomir with pleiotropic functions. Immunol Rev.
253:158–166. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiao C, Srinivasan L, Calado DP, Patterson
HC, Zhang B, Wang J, Henderson JM, Kutok JL and Rajewsky K:
Lymphoproliferative disease and autoimmunity in mice with increased
miR-17-92 expression in lymphocytes. Nat Immunol. 9:405–414. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wong P, Iwasaki M, Somervaille TC, Ficara
F, Carico C, Arnold C, Chen CZ and Cleary ML: The miR-17-92
microRNA polycistron regulates MLL leukemia stem cell potential by
modulating p21 expression. Cancer Res. 70:3833–3842. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nittner D, Lambertz I, Clermont F,
Mestdagh P, Köhler C, Nielsen SJ, Jochemsen A, Speleman F,
Vandesompele J, Dyer MA, et al: Synthetic lethality between Rb, p53
and Dicer or miR-17-92 in retinal progenitors suppresses
retinoblastoma formation. Nat Cell Biol. 14:958–965. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hayashita Y, Osada H, Tatematsu Y, Yamada
H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y and
Takahashi T: A polycistronic microRNA cluster, miR-17-92, is
overexpressed in human lung cancers and enhances cell
proliferation. Cancer Res. 65:9628–9632. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhu H, Han C and Wu T: MiR-17-92 cluster
promotes hepatocarcinogenesis. Carcinogenesis. 36:1213–1222. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhu H, Han C, Lu D and Wu T: miR-17-92
cluster promotes cholangiocarcinoma growth: Evidence for PTEN as
downstream target and IL-6/Stat3 as upstream activator. Am J
Pathol. 184:2828–2839. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu Q, Luo G, Yang Z, Zhu F, An Y, Shi Y
and Fan D: miR-17-5p promotes proliferation by targeting SOCS6 in
gastric cancer cells. FEBS Lett. 588:2055–2062. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park D, Lee SC, Park JW, Cho SY and Kim
HK: Overexpression of miR-17 in gastric cancer is correlated with
proliferation-associated oncogene amplification. Pathol Int.
64:309–314. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Grillari J, Hackl M and Grillari-Voglauer
R: miR-17-92 cluster: Ups and downs in cancer and aging.
Biogerontology. 11:501–506. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Krysan K, Kusko R, Grogan T, O'Hearn J,
Reckamp KL, Walser TC, Garon EB, Lenburg ME, Sharma S, Spira AE, et
al: PGE2-driven expression of c-Myc and oncomiR-17-92 contributes
to apoptosis resistance in NSCLC. Mol Cancer Res. 12:765–774. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Matsubara H, Takeuchi T, Nishikawa E,
Yanagisawa K, Hayashita Y, Ebi H, Yamada H, Suzuki M, Nagino M,
Nimura Y, et al: Apoptosis induction by antisense oligonucleotides
against miR-17-5p and miR-20a in lung cancers overexpressing
miR-17-92. Oncogene. 26:6099–6105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Takakura S, Mitsutake N, Nakashima M,
Namba H, Saenko VA, Rogounovitch TI, Nakazawa Y, Hayashi T, Ohtsuru
A and Yamashita S: Oncogenic role of miR-17-92 cluster in
anaplastic thyroid cancer cells. Cancer Sci. 99:1147–1154. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ernst A, Campos B, Meier J, Devens F,
Liesenberg F, Wolter M, Reifenberger G, Herold-Mende C, Lichter P
and Radlwimmer B: De-repression of CTGF via the miR-17-92 cluster
upon differentiation of human glioblastoma spheroid cultures.
Oncogene. 29:3411–3422. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Olive V, Bennett MJ, Walker JC, Ma C,
Jiang I, Cordon-Cardo C, Li QJ, Lowe SW, Hannon GJ and He L: miR-19
is a key oncogenic component of mir-17-92. Genes Dev. 23:2839–2849.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Uziel T, Karginov FV, Xie S, Parker JS,
Wang YD, Gajjar A, He L, Ellison D, Gilbertson RJ, Hannon G, et al:
The miR-17-92 cluster collaborates with the Sonic Hedgehog pathway
in medulloblastoma. Proc Natl Acad Sci USA. 106:2812–2817. 2009.
View Article : Google Scholar
|
|
37
|
Gupta S, Read DE, Deepti A, Cawley K,
Gupta A, Oommen D, Verfaillie T, Matus S, Smith MA, Mott JL, et al:
Perk-dependent repression of miR-106b-25 cluster is required for ER
stress-induced apoptosis. Cell Death Dis. 3:e3332012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gantier MP, Stunden HJ, McCoy CE, Behlke
MA, Wang D, Kaparakis-Liaskos M, Sarvestani ST, Yang YH, Xu D, Corr
SC, et al: A miR-19 regulon that controls NF-κB signaling. Nucleic
Acids Res. 40:8048–8058. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Trenkmann M, Brock M, Gay RE, Michel BA,
Gay S and Huber LC: Tumor necrosis factor α-induced microRNA-18a
activates rheumatoid arthritis synovial fibroblasts through a
feedback loop in NF-κB signaling. Arthritis Rheum. 65:916–927.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li Y, Choi PS, Casey SC, Dill DL and
Felsher DW: MYC through miR-17-92 suppresses specific target genes
to maintain survival, autonomous proliferation, and a neoplastic
state. Cancer Cell. 26:262–272. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li H, Bian C, Liao L, Li J and Zhao RC:
miR-17-5p promotes human breast cancer cell migration and invasion
through suppression of HBP1. Breast Cancer Res Treat. 126:565–575.
2011. View Article : Google Scholar
|
|
42
|
Cohen R, Greenberg E, Nemlich Y, Schachter
J and Markel G: miR-17 regulates melanoma cell motility by
inhibiting the translation of ETV1. Oncotarget. 6:19006–19016.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wu Q, Yang Z, An Y, Hu H, Yin J, Zhang P,
Nie Y, Wu K, Shi Y and Fan D: MiR-19a/b modulate the metastasis of
gastric cancer cells by targeting the tumour suppressor MXD1. Cell
Death Dis. 5:e11442014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ohyagi-Hara C, Sawada K, Kamiura S, Tomita
Y, Isobe A, Hashimoto K, Kinose Y, Mabuchi S, Hisamatsu T,
Takahashi T, et al: miR-92a inhibits peritoneal dissemination of
ovarian cancer cells by inhibiting integrin α5 expression. Am J
Pathol. 182:1876–1889. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Barkan D and Chambers AF: β1-integrin: A
potential therapeutic target in the battle against cancer
recurrence. Clin Cancer Res. 17:7219–7223. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kato H, Liao Z, Mitsios JV, Wang HY,
Deryugina EI, Varner JA, Quigley JP and Shattil SJ: The primacy
ofβ1 integrin activation in the metastatic cascade. PLoS One.
7:e465762012. View Article : Google Scholar
|
|
47
|
Lee YC, Jin JK, Cheng CJ, Huang CF, Song
JH, Huang M, Brown WS, Zhang S, Yu-Lee LY, Yeh ET, et al: Targeting
constitutively activated β1 integrins inhibits prostate cancer
metastasis. Mol Cancer Res. 11:405–417. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Somanath PR, Kandel ES, Hay N and Byzova
TV: Akt1 signaling regulates integrin activation, matrix
recognition, and fibronectin assembly. J Biol Chem.
282:22964–22976. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dhar S, Gu FX, Langer R, Farokhzad OC and
Lippard SJ: Targeted delivery of cisplatin to prostate cancer cells
by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles.
Proc Natl Acad Sci USA. 105:17356–17361. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dhar S, Kolishetti N, Lippard SJ and
Farokhzad OC: Targeted delivery of a cisplatin prodrug for safer
and more effective prostate cancer therapy in vivo. Proc Natl Acad
Sci USA. 108:1850–1855. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jacobsen C and Honecker F: Cisplatin
resistance in germ cell tumours: Models and mechanisms. Andrology.
3:111–121. 2015. View Article : Google Scholar
|
|
52
|
Jiang Z, Yin J, Fu W, Mo Y, Pan Y, Dai L,
Huang H, Li S and Zhao J: MiRNA 17 family regulates
cisplatin-resistant and metastasis by targeting TGFbetaR2 in NSCLC.
PLoS One. 9:e946392014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang X, Martindale JL and Holbrook NJ:
Requirement for ERK activation in cisplatin-induced apoptosis. J
Biol Chem. 275:39435–39443. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Persons DL, Yazlovitskaya EM and Pelling
JC: Effect of extra-cellular signal-regulated kinase on p53
accumulation in response to cisplatin. J Biol Chem.
275:35778–35785. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mendoza J, Martínez J, Hernández C,
Pérez-Montiel D, Castro C, Fabián-Morales E, Santibáñez M,
González-Barrios R, Díaz-Chávez J, Andonegui MA, et al: Association
between ERCC1 and XPA expression and polymorphisms and the response
to cisplatin in testicular germ cell tumours. Br J Cancer.
109:68–75. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kirschner K and Melton DW: Multiple roles
of the ERCC1-XPF endonuclease in DNA repair and resistance to
anticancer drugs. Anticancer Res. 30:3223–3232. 2010.PubMed/NCBI
|
|
57
|
Li W and Melton DW: Cisplatin regulates
the MAPK kinase pathway to induce increased expression of DNA
repair gene ERCC1 and increase melanoma chemoresistance. Oncogene.
31:2412–2422. 2012. View Article : Google Scholar
|
|
58
|
Andrieux LO, Fautrel A, Bessard A,
Guillouzo A, Baffet G and Langouët S: GATA-1 is essential in
EGF-mediated induction of nucleotide excision repair activity and
ERCC1 expression through ERK2 in human hepatoma cells. Cancer Res.
67:2114–2123. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Cummings M, Higginbottom K, McGurk CJ,
Wong OG, Köberle B, Oliver RT and Masters JR: XPA versus ERCC1 as
chemosensitising agents to cisplatin and mitomycin C in prostate
cancer cells: Role of ERCC1 in homologous recombination repair.
Biochem Pharmacol. 72:166–175. 2006. View Article : Google Scholar : PubMed/NCBI
|