Introduction
Despite multimodal treatment with surgery,
radiotherapy and chemotherapy, glioblastoma is still associated
with poor prognosis (1). Reasons
why glioma are refractory to current conventional therapeutic
approaches involve their infiltrative growth patterns preventing
radical resection and cell autonomous anti-apoptotic mechanisms.
These properties could be mediated at least partly by the epidermal
growth factor receptor (EGFR) which is among the most overexpressed
and/or mutated protein in gliomas (2). Therefore, targeting the EGFR pathway
could be a promising therapeutic approach. However, clinical
results exploring EGFR inhibitors against glioma have been
disappointing thus far (3), and
the causes for the lack of clinical activity of EGFR inhibitors are
only incompletely understood.
A factor which might influence the efficacy of EGFR
inhibitors could be the tumor microenvironment characterised by
areas of nutrient and oxygen deprivation (4). We therefore previously investigated
the effects of EGFR inhibition under severe hypoxia and found that
hypoxia-induced cell death in glioma cells was reduced by an EGFR
inhibitor, at least partly by reduced energy consumption (5). This energy conserving mechanism
included reduced glucose consumption, resulting in slower ATP
depletion and decreased cytochrome c release (5). Further analyses showed that
inhibition of mammalian target of rapamycin (mTOR), a downstream
effector of growth factor receptors, similarly reduced energy
demands and cell death under hypoxia (6), underlining the importance of mTOR as
a central regulator of cellular energetics (7). As hypoxia is present in large areas
of glioma before treatment and probably is even more severe after
anti-angiogenic treatments (8),
these observations might be an explanation for the low efficacy of
EGFR inhibitors in glioma patients. On the contrary, EGFR
inhibition sensitised glioma cells to death ligands under normoxic
conditions (9) indicating that
glioma cells are not completely refractory against anti-apoptotic
effects of EGFR inhibition. However, how exactly metabolic
conditions mediate these different effects is unclear, but
understanding the mechanism that determine the outcome of EGFR
inhibition is important to improve the efficacy of growth factor
receptor-inhibiting therapies. In this study, we therefore
investigated in more detail how glucose and oxygen influence the
effect of EGFR blockade in glioma cells and found that the
AMP-activated kinase (AMPK) increases resistance against EGFR
inhibition.
Materials and methods
Cell culture
LNT-229 glioma cells were described previously
(10). U87MG, the leukemia cell
line K562 as well as the breast cancer cell line MDA-MB-435 were
obtained from the ATCC (Wesel, Germany). Cells were cultured in
Dulbecco's modified Eagle's medium (DMEM, Sigma-Aldrich,
Taufkirchen, Germany) containing 10% fetal calf serum (FCS), 2 mM
glutamine, 100 IU/ml penicillin, and 100 mg/ml streptomycin. For
experimental procedures involving limited glucose concentrations,
we used Dulbecco's modified Eagle's glucose-free medium (Life
Technologies, Darmstadt, Germany) without FCS, and glucose was
added to obtain the required concentration. Cells were seeded at a
density of 80,000 cells/well in 24-well plates for FACS analysis
and 1,500,000 cells/well in 10-cm dishes for protein extraction if
not otherwise specified. LNT-229 cells stably transfected with
wild-type-EGFR (pLERNL) were described previously (9).
Primary cell culture
The glioblastoma derived cell line MNOF132 was
kindly provided by Stefan Momma (Edinger Institute,
Goethe-University Frankfurt). Cells were cultured in DMEM-F12
Medium containing 20 ng/ml of epidermal growth factor (EGF,
ReliaTech, Wolfenbüttel, Germany) and human basic fibroblast growth
factor-2 (bFGF-2, ReliaTech, Wolfenbüttel, Germany) as well as 20%
BIT admixture 100 supplement (Pelo Biotech, Planegg/Martinsried,
Germany). During cell culture EGF and bFGF-2 were added to the
cells twice per week.
Reagents and treatments
The following reagents were used: 2-deoxy-D-glucose
(2DG) (Sigma-Aldrich), PD153035 hydrochloride (Biozol, Eching,
Germany), A769662 (RnD Systems, Minneapolis, MN, USA), zVAD-fmk
(Bachem, Weil am Rhein, Germany) and compound C (Sigma-Aldrich).
Cells were seeded and, after 24 h, treated with either PD153035 (10
μM), A769662 (100 μM), 2DG (10 mM), compound C (5 μM) or zVAD (100
μM) for the indicated time-points in Dulbecco's modified Eagle's
glucose-free medium (Life Technologies) in which glucose was added
as required. For treatment of MDA-MB-435 and of K562 cells,
lapatinib (10 μM) was obtained from Sequoia laboratory (Berkshire,
UK), and imatinib (10 μM) was obtained from Enzo Life Sciences
(Lörrach, Germany).
Lentiviral transduction
For AMPK double knockout LNT229 cells were
transfected with pLKO.1 short hairpin RNA (shRNA) plasmids from
Sigma-Aldrich (TRCN0000196482, TRCN0000355741). The non-targeting
plamids were from Addgene (#1864, #10905). Production of lentivirus
and transfection of the cells were done as previously described
(11). For selection 2 μg/ml
puromycin and 800 μg/ml hygromycin was used.
Induction of hypoxia
To induce 0.1% O2, cells were incubated
in GasPak pouches for anaerobic culture (Becton-Dickinson,
Heidelberg, Germany) (10).
Moderate hypoxia (1% O2) was induced in a
CO2-incubator (Binder, Germany) (12).
Cell death analysis
Cells were seeded at 80,000 cells per well in
24-well-plates. After 24 h, medium was removed, cells were washed,
and incubated with the appropriate reagents. For the detection of
cell death, cells were stained with propidium iodide (PI) and
analysed by FACS as previously described (12). Experiments were performed in
triplicates and presented as mean ± SD.
Cell cycle analysis
To assess cell cycle distribution, cells were
treated as indicated, fixed in 70% ice-cold ethanol and incubated
gently vortexing for 45 min. After centrifugation, the pellet was
resuspended in 0.1 ml RNaseA (20 μg/ml) and incubated for 5 min
before 400 μl PI (50 μg/ml) was added. After a second incubation
step for 30 min, cells were filtered through 50 μl filcons before
starting FACS analysis. Cells with a sub-G1 DNA content were
considered as dead cells.
Caspase activation assay
Caspase activation was assessed with caspase
activity assay (Roche Diagnostic, Mannheim, Germany) according to
the manufacturer's protocol.
Flow cytometry analysis
Cells were harvested with accutase, and 500,000
cells per sample were washed twice with FACS buffer (PBS with 2%
FCS) and centrifuged by 1200 × g for 3 min. Thereafter, the isotype
(M5534, Sigma-Aldrich, Steinheim, Germany) or the anti-EGFR
antibody (Alexa Fluor 647 (sc-101 AF647), R-1, Santa Cruz, USA) was
added, the pellet was resuspended, and the cells were incubated on
ice for 1 h. Subsequently, cells were washed twice and kept on ice
until the measurement started. Cells were analysed by flow
cytometry in a BD Canto II using the PE-channel. Specific
fluorescence index (SFI) was calculated as median fluorescence
intensity of specific antibody/median fluorescence index of isotype
antibody.
Immunoblot analysis
Cells were seeded in 10-cm plates and treated as
indicated. Thereafter, the cells were washed with ice cold
phosphate-buffered saline (PBS) and lysed in lysis buffer (50 mM
Tris-HCl pH 8.0, 120 mM NaCl, 5 mM EDTA, 0.5% Nonidet P-40)
containing protease inhibitors (Roche Applied Science, Mannheim,
Germany). Cellular lysates were prepared as described (13) and subjected to sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Membranes
were probed with antibodies as listed below. The chemiluminescence
solution used for detection was composed of 1 ml of solution A (200
ml of 0.1 M Tris-HCl pH 8.6, 50 mg of luminol), 100 μl of solution
B [11 mg of p-hydroxycoumarin acid, 10 ml dimethyl sulfoxide
(DMSO)], and 0.3 μl of H2O2 (30%). Antibodies
against the following antigens were used: rabbit anti-phospho AMPKα
Thr172, rabbit anti-AMPKα, rabbit anti-pACC Ser79 (D7D11), rabbit
anti-ACC and rabbit Pathscan Muliplex Western Cocktail I
(phospho-p90RSK, phospho-Akt, phospho-Erk1/2, phospho-S6 ribosomal
protein) were obtained from Cell Signaling Technologies (Danvers,
MA, USA), and mouse anti-GAPDH (MAB374) from Chemicon (Nuernberg,
Germany). Secondary antibodies and rabbit anti-EGFR were purchased
from Santa Cruz Biotechnology.
RNA extraction and quantitative RT-PCR
(qRT-PCR)
Total RNA was extracted using TRIzol and
PureLink® RNA Mini kit (Life Technologies
Ambion®). The Vilo cDNA synthesis kit (Life Technologies
Invitrogen™) was used for the synthesis of first strand cDNA for 10
min at 25°C and 2 h at 42°C. Following this, the enzyme was
inactivated at 85°C for 10 min. PCR was performed using 15 μg RNA
and absolute Blue Q-PCR master mix with SYBR Green + fluorescein
(Thermo Fisher Scientific, Hamburg, Germany). The reactions were
cycled 30 times [50°C for 2 min and 95°C for 10 min (94°C for 15
sec, 58–60°C for 1 min, and 72°C for 1 min) × 30 cycles]. The
following primer pairs were used: AMPKα1 forward
5′-AGAAGCAGAAACACGACGGG-3′, AMPKα1 reverse
5′-GCGGATTTTTCCTACCACATCA-3′, AMPKα2 forward
5′-CGGCTCTTTCAGCAGATTCTGT-3′, AMPKα2 reverse
5′-ATCGGCTATCTTGGCATTCATG-3′, SDHA forward
5′-TGGGAACAAGAGGGCATCTG-3′ and SDHA reverse
5′-CCACCACTGCATCAAATTCATG-3′. Cycle threshold (Ct) values were
normalized for amplification of the SDHA RNA, and the data were
analysed using the Vandesompele method as described (13).
Glucose/lactate measurements
To determine glucose consumption and lactate
production, the supernatant was collected and cells were removed by
centrifugation. Glucose and lactate concentrations were measured
using the biochemistry analyser Hitachi 917.
Ethics statement
The use of the primary glioma cell line was approved
by the ethics committee of the University Hospital Frankfurt
(reference number 4/09).
Results
Metabolic conditions influence the effect
of EGFR inhibition
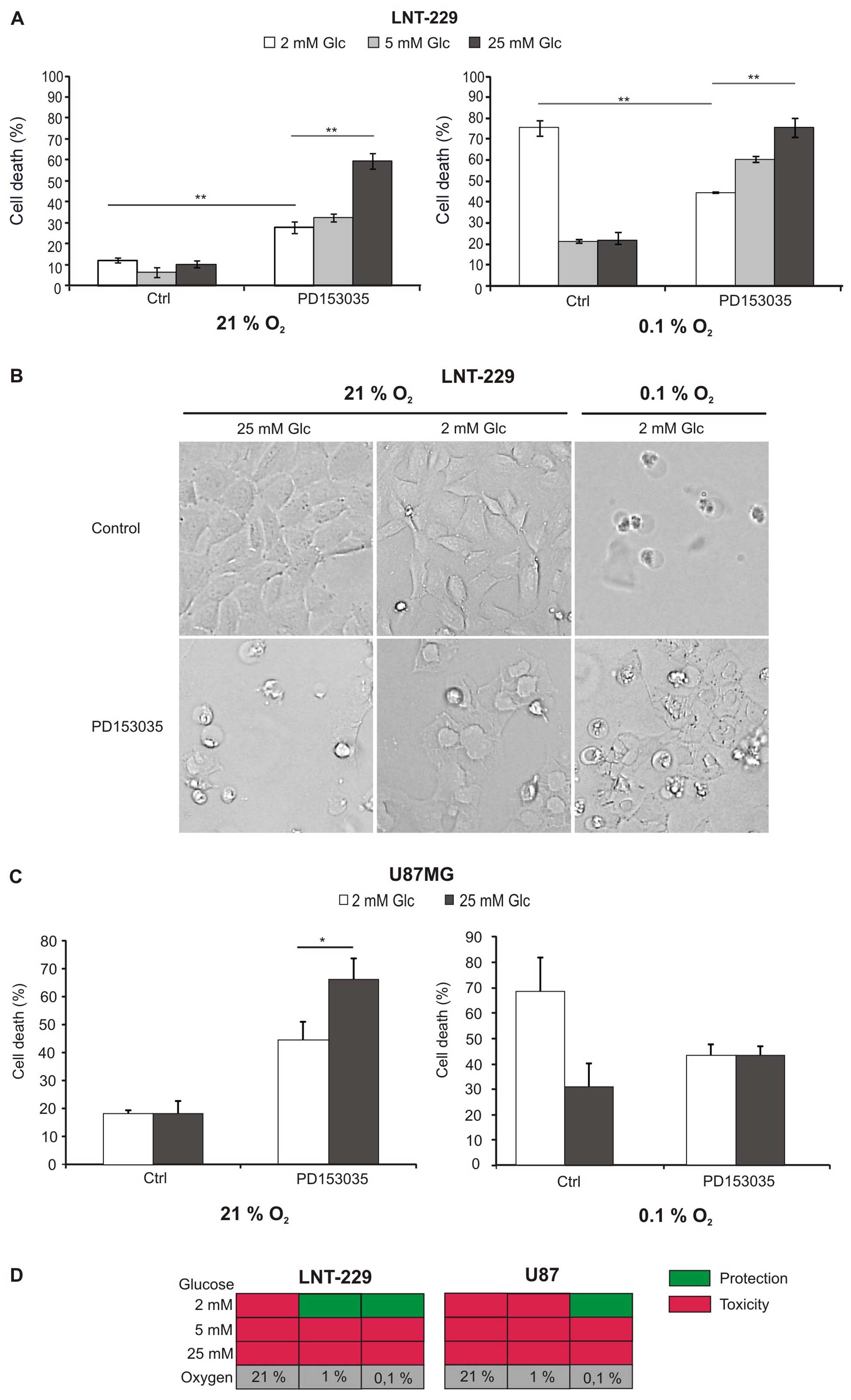
To investigate whether two important metabolic
determinants of the tumor microenvironment, glucose and oxygen,
modulate the toxicity of EGFR inhibition, glioma cells were exposed
to different concentrations of glucose and oxygen in the absence or
presence of an EGFR inhibitor (PD153035), and cell death was
analysed (Fig. 1A and C).
Hypoxia-induced cell death was enhanced at low glucose
concentrations, suggesting that glucose availability is a limiting
factor for survival of glioma cells under hypoxia (Fig. 1A and C right panel; Fig. 1B). As previously shown, PD153035
inhibited glioma cell death at hypoxia and low glucose levels
(5). Under normoxia, however,
PD153035 induced cell death, and, surprisingly, this effect was
more pronounced if large amounts of glucose were available
(Fig. 1A and C left panel;
Fig. 1B). Increased toxicity under
abundant glucose was also present under hypoxia in LNT-229 cells,
but less pronounced (Fig. 1A).
Fig. 1D summarises the effect of
EGFR inhibition under the different metabolic conditions.
EGFR inhibition induces caspase-dependent
cell death
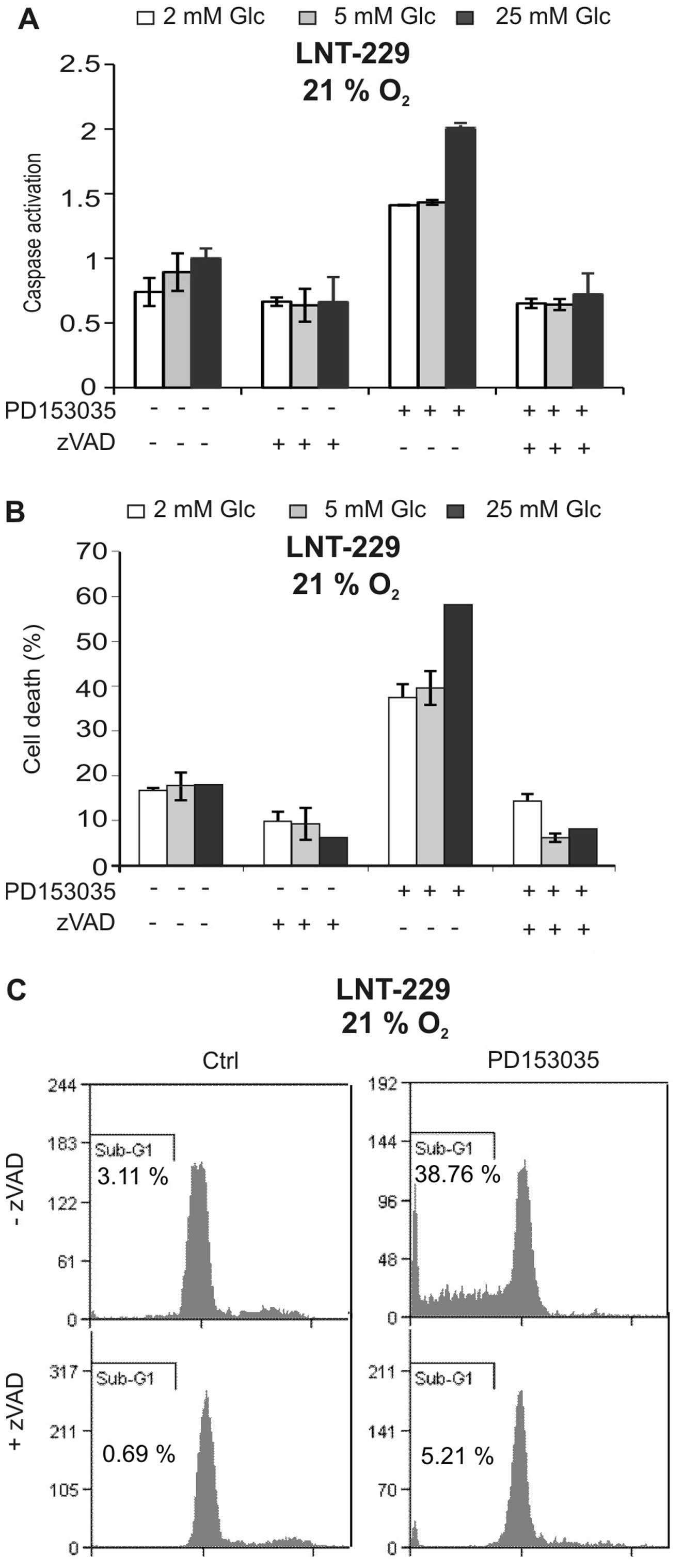
To characterise cell death induced by PD153035, we
asked whether caspases were involved. As shown in Fig. 2A, the EGFR inhibitor induced
caspase activation, and this effect again was suppressed by low
glucose concentrations. The pan-caspase inhibitor zVAD-FMK strongly
suppressed caspase activation and blocked PD153035-mediated cell
death, suggesting that cytotoxicity of the EGFR inhibitor was
caspase-dependent (Fig. 2A and B).
Additionally, the PD153035-induced increase in sub-G1 fraction
indicative of nuclear fragmentation during apoptosis was inhibited
by zVAD-FMK (Fig. 2C). Together,
these experiments suggest that the EGFR inhibitor PD153035 induced
caspase-dependent cell death.
EGF receptor expression and downstream
signaling
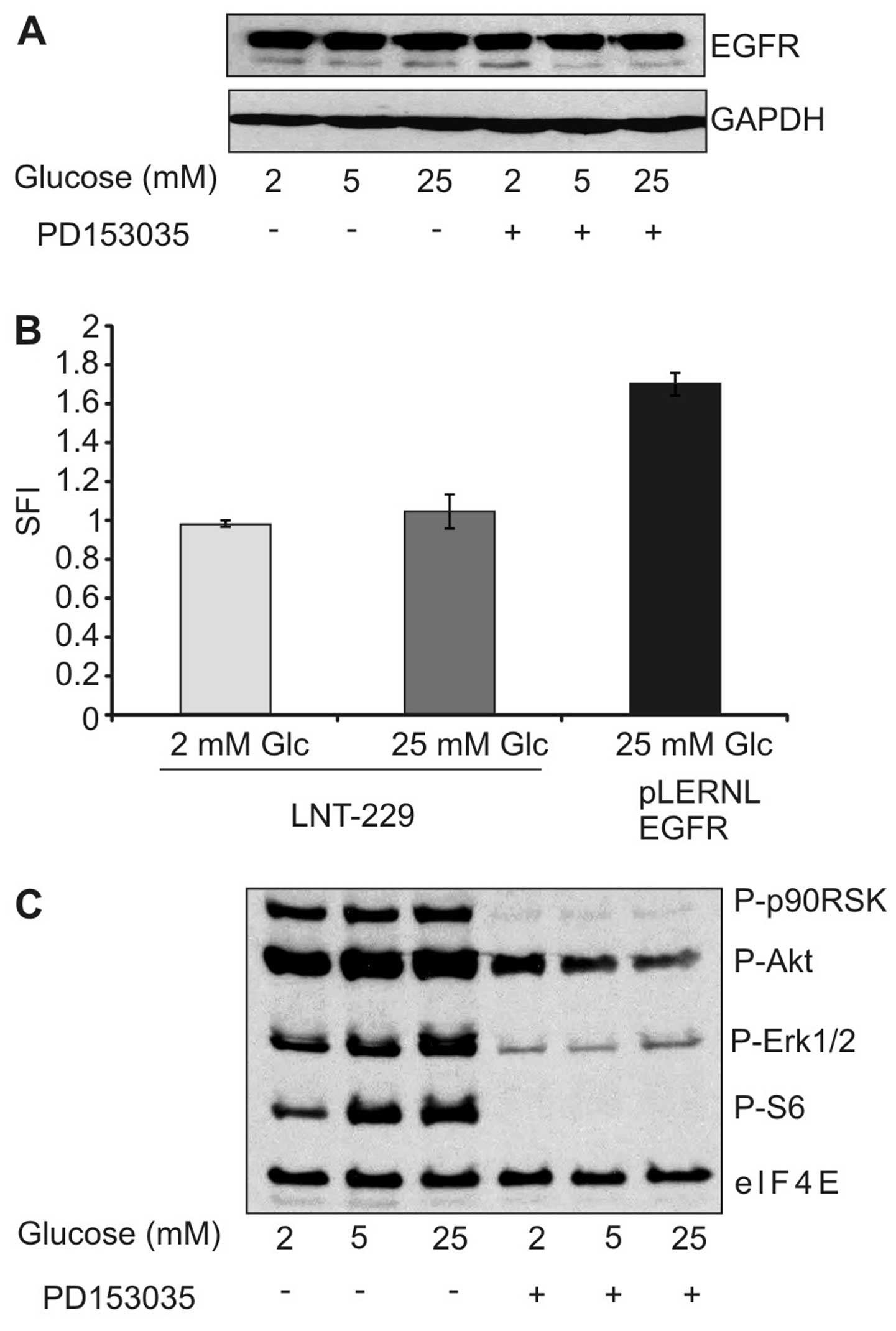
Having established that low glucose levels inhibit
cytotoxicity of the EGFR inhibitor, we asked whether reduced
expression of EGFR under low glucose conditions might be
responsible for the observed effect. However, immunoblot analysis
did not reveal any modulation in EGFR expression under different
glucose concentrations (Fig. 3A).
Similarly, surface expression of EGFR remained unaltered by
different glucose concentrations (Fig.
3B). We further investigated whether different glucose
concentrations modulated response of downstream effectors to the
EGFR inhibitor. As expected, PD153035 diminished the
phosphorylation of extracellular signal-regulated kinase 1/2
(ERK1/2), protein kinase B (PKB)/Akt and S6 kinase, but this effect
was unaffected by the different experimental conditions (Fig. 3C). Together, the reduced toxicity
of EGFR inhibition under low glucose concentration could not be
explained by reduced expression of EGFR or diminished pathway
inhibition.
Activation of AMPK protects against EGFR
inhibition
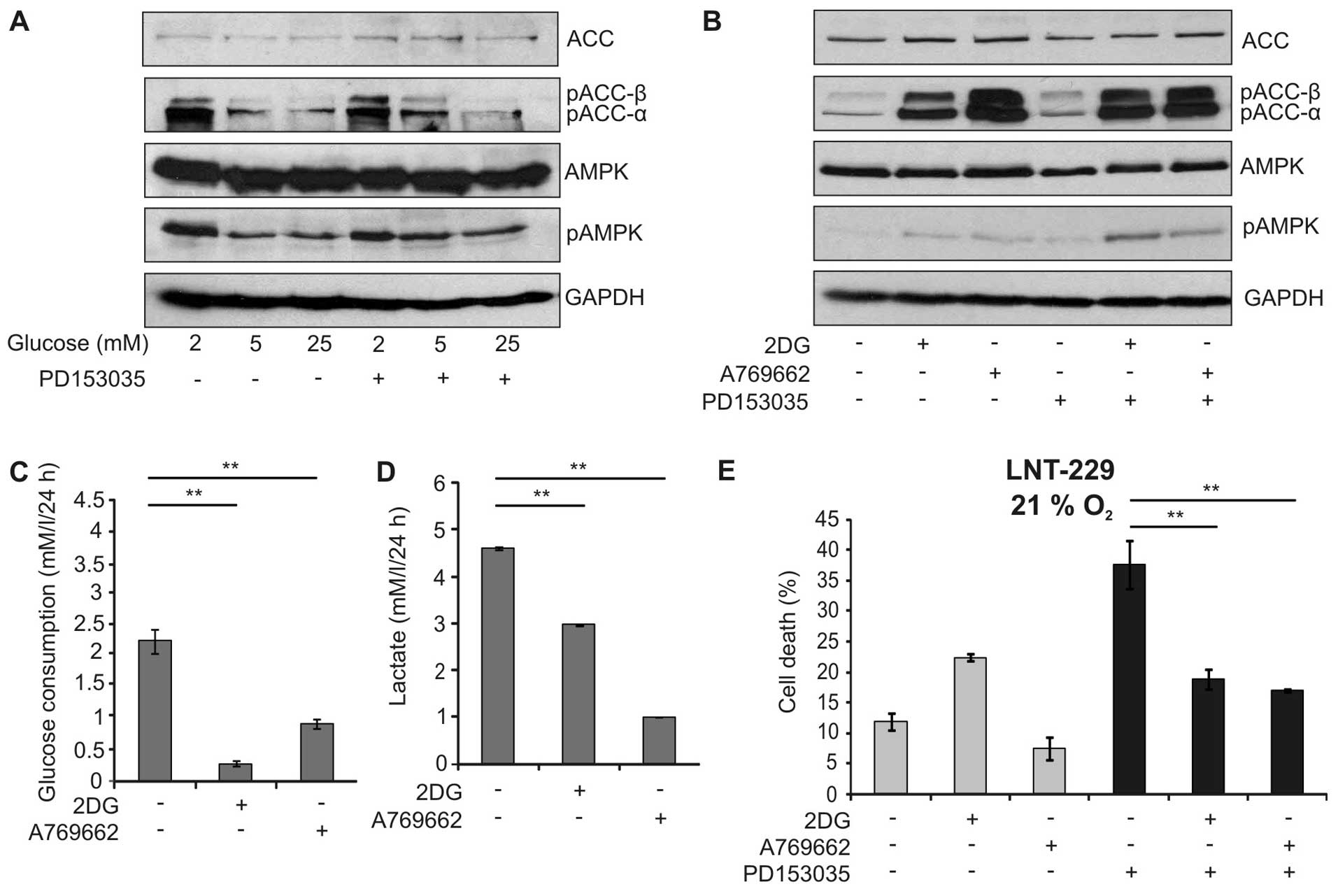
The suppression of PD153035-mediated toxicity by
limited glucose availability suggested that AMP-activated protein
kinase (AMPK), a major sensor for cellular energy status, might
influence the outcome of EGFR inhibition. Indeed, low glucose
concentrations led to phosphorylation of AMPK and its downstream
target acetyl-CoA-carboxylase (ACC) (Fig. 4A). Therefore, we investigated
whether activation of AMPK by two chemically unrelated activators,
the glucose analogue 2-deoxy-D-glucose (2DG) and A769662, a direct
activator of AMPK, modulated EGFR-inhibitor dependent cytotoxicity.
As expected, these activators induced phosphorylation of AMPK and
ACC in a similar extent (Fig. 4B).
In line with the AMPK function in suppressing glycolysis, the
activators significantly reduced glucose consumption and lactate
production (Fig. 4C and D).
Interestingly, 2DG and A769662 significantly diminished the
toxicity of PD153035 suggesting that activation of AMPK indeed
protected against EGFR inhibitor-induced cell death (Fig. 4E). Similar results could be
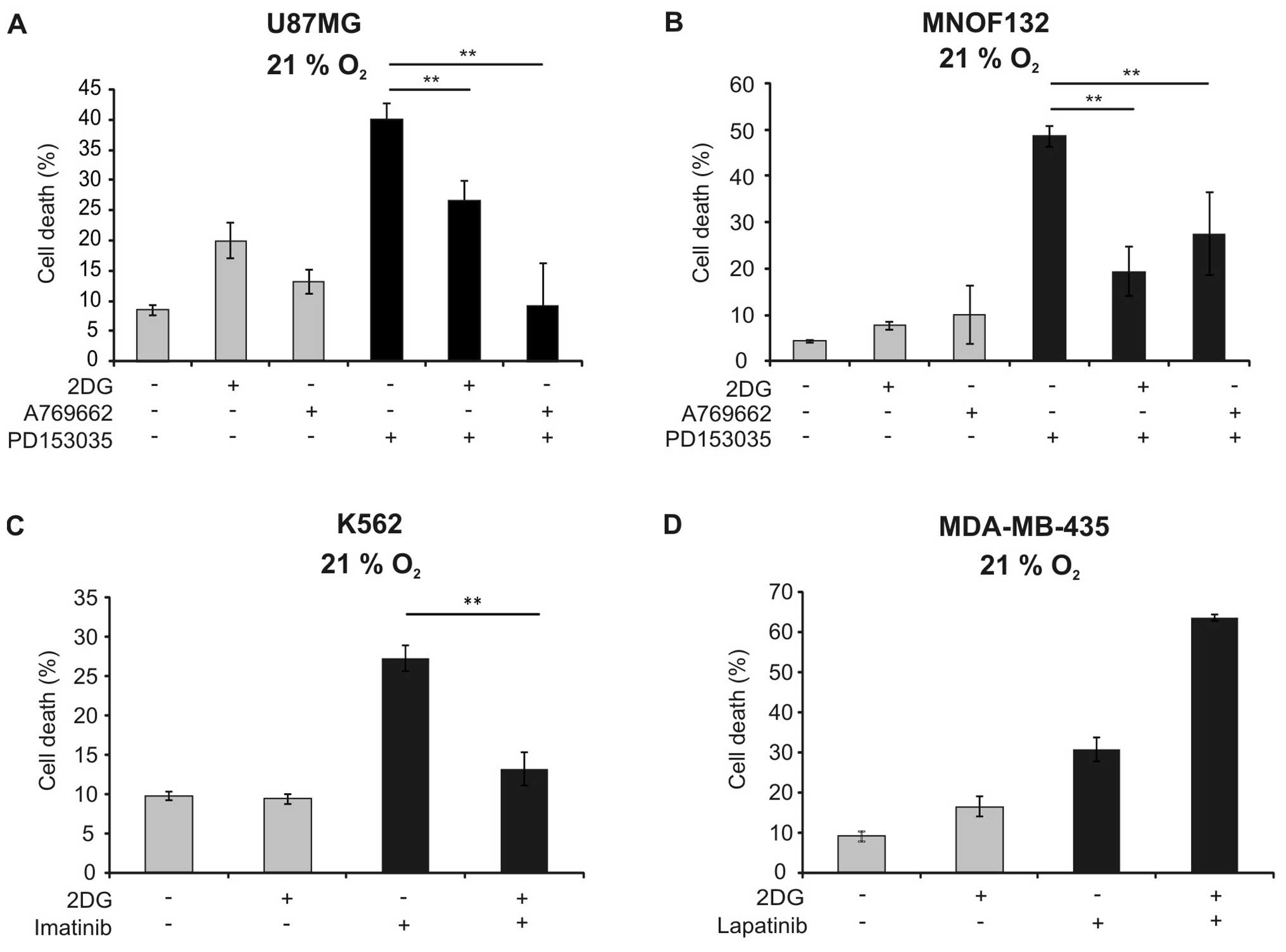
observed in the U87MG cell line and in a primary glioma cell line
(Fig. 5A and B). To analyse
whether the antagonistic effects of AMPK activators towards
tyrosine kinase inhibitors are specific for glioma cells,
additional cellular models of tyrosine-kinase-inhibitor-induced
cell death were studied. 2DG similarly reduced cytotoxicity of
imatinib against K562 cells (Fig.
5C). In contrast, it sensitised MDA-MB-435 cells against cell
death induction by lapatinib (Fig.
5D).
Knockdown or inhibition of AMPK
sensitises against EGFR inhibition
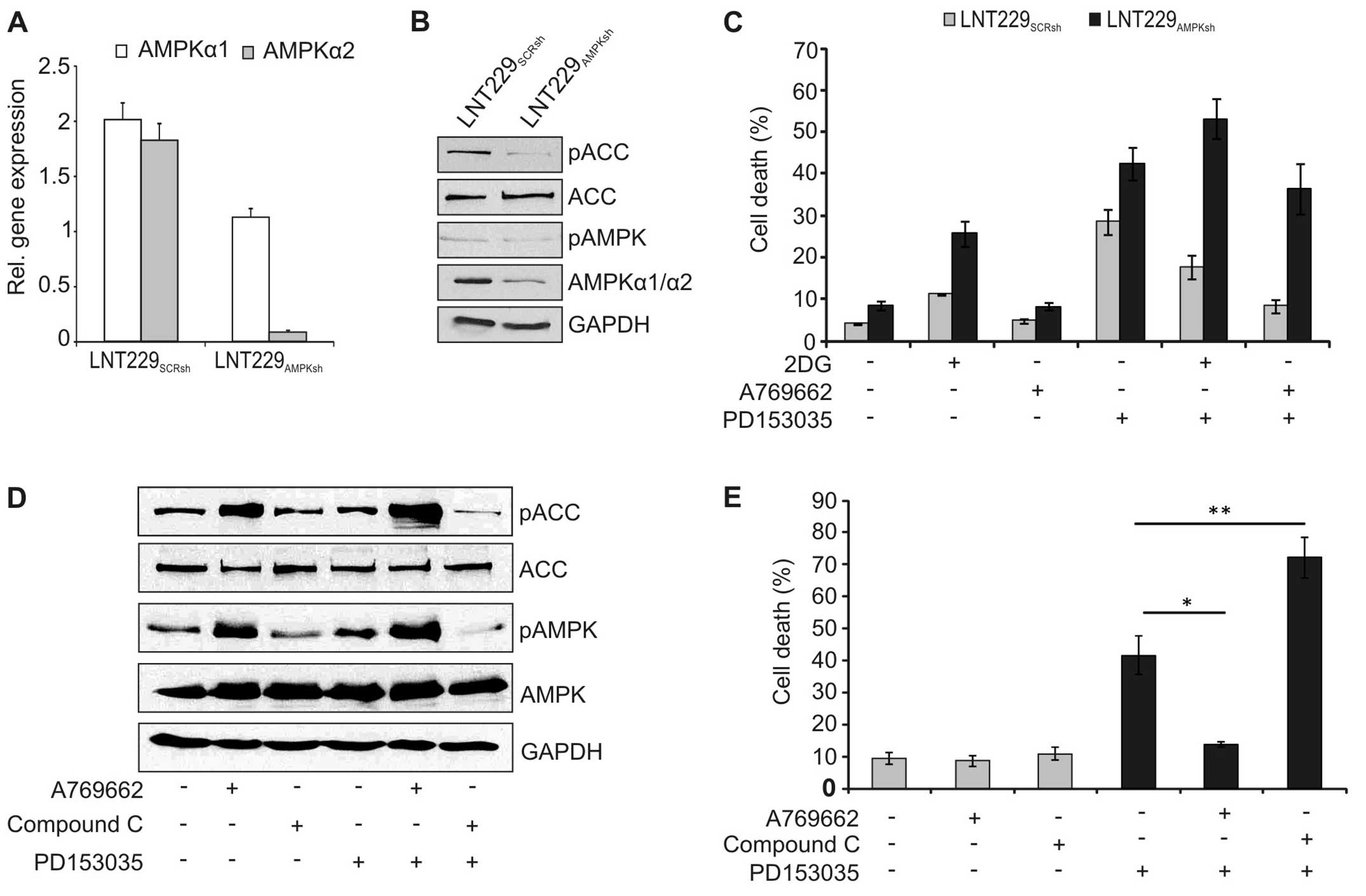
To further confirm the role of AMPK, expression of
AMPK was inhibited by lentiviral knockdown. Stable expression of
the AMPK-specific shRNA significantly reduced expression of AMPK on
the RNA (Fig. 6A) and protein
level (Fig. 6B). Accordingly,
phosphorylation of the downstream effector ACC was inhibited in
these cells (Fig. 6B).
Additionally, the cytotoxicity of PD153035 was higher in the
AMPK-knockdown cells, and 2DG and A769662 could no longer block
cell death in these cells (Fig.
6C). Similarly, pharmacological blockade of AMPK reduced
phosphorylation of AMPK and ACC (Fig.
6D) and increased cell death induction by PD153035 in wild-type
LNT-229 cells (Fig. 6E).
Discussion
Although EGFR is often activated in glioma, and
inhibition of EGFR leads to cell death in vitro, EGFR
inhibitors did not show substantial efficacy in clinical trials in
glioma patients (3,14). Because the microenvironment of
glioma is characterised by glucose deprivation comprising levels of
~0.5 and 2 mM (15) and severe
hypoxia (16), we speculated
whether these conditions might be responsible for the observed
failure of tyrosine kinase inhibitors.
Whereas inhibition of EGFR led to caspase-dependent
apoptosis in the presence of excess glucose (Figs. 2 and 3), cell death induction was indeed
impaired by hypoxia and low glucose availability (Fig. 1). Under low glucose,
phosphorylation of the cellular energy sensor AMPK was stimulated,
and activation of AMPK by 2DG and A769662 reduced toxicity of EGFR
inhibition (Fig. 4). In contrast,
inhibition of AMPK by sh-mediated knockdown or pharmacological
blockade reverted the effects of 2DG and A769662 (Fig. 5), confirming that their effect was
dependent on AMPK. These results indicate that, in glioma cells,
activation of AMPK might limit cytotoxicity of tyrosine-kinase
inhibition. The phenomenon was not restricted to glioma cells as
2DG reduced toxicity also of imatinib in K562 cells (Fig. 5C). In contrast, 2DG did not block,
but rather increased cell death induction by lapatinib in the
breast cancer cell line MDA-MB-435 (Fig. 5D). The latter results are in
agreement to findings in non-small cell lung cancer cells where 2DG
increased cell death induction by the EGFR inhibitor afatinib
(17). Therefore, the observed
cytoprotective effect of AMPK activation against tyrosine-kinase
inhibitors seems to be cell-type specific and might be an
explanation why EGFR inhibitors proved to be effective in breast
and lung cancer, but not in glioma trials.
Interestingly, 2DG was more toxic in the
AMPK-suppressed cells (Fig. 6C),
consistent with findings in lymphoma cells (18). Similarly, synergistic cell death
induction between 2DG and the AMPK inhibitor compound C was
described in leukemia cells (19).
These observations support the hypothesis that AMPK is necessary to
preserve energy homeostasis during nutrient starvation in glioma.
This is in agreement with the recent observation that AMPK is
highly activated (20) in glioma
tissue. By dissociating different molecular effects of metformin,
AICAR and A769662, these authors similarly speculated that AMPK
activation has pro-survival functions in nutrient starved tumors.
Supporting this assumption, the eukaryotic translation elongation
factor 2 kinase (eEF2K), which is activated by AMPK, has been shown
to be important for resistance of tumor cells against nutrient
deprivation (21). Interestingly,
these authors found that expression of eEF2K is increased in glioma
and associated with a worse prognosis. The finding that AMPK
activators reduced glucose consumption and lactate production
(Fig. 4) is congruent to the
energy saving function of AMPK and a recently proposed model where
AMPK is a central regulator of aerobic glycolysis (the ‘Warburg
effect’) (18). The exact
molecular mechanism how AMPK activation blocks EGFR
inhibitor-induced toxicity remains unclear. Possible mechanisms
might include the suppression of reactive oxygen species (ROS) by
AMPK (22) as ROS have been
proposed to be involved in the toxicity of tyrosine-kinase
inhibitors (23). Additionally,
metabolic alterations occurring as a result of AMPK inhibition
might modulate sensitivity against tyrosine kinase inhibitors,
offering novel opportunities how the efficacy of these drugs could
be enhanced (24,25). Together, these results suggest that
activation of AMPK limits the toxicity of EGFR inhibitors in glioma
cells. Determining which molecular and metabolic mechanisms mediate
these cytoprotective effects of AMPK could result in strategies
improving efficacy of tyrosine-kinase inhibitors.
Acknowledgements
This study was supported by grant RI2175/1-1 from
the ‘Deutsche Forschungsgemeinschaft’ (DFG) to J.P.S. and J.R. The
Dr. Senckenberg Institute of Neurooncology is supported by the
Hertie foundation and the Dr. Senckenberg foundation. J.P.S. is
‘Hertie Professor for Neurooncology’. S.W. was supported by grant
1748-0-0 from the interdisciplinary center for clinical research of
the University of Tübingen (IZKF). M.R. received funding from the
Medical Faculty, University Hospital Frankfurt (Program
‘Nachwuchsforscher 2012’). J.P.S. and J.R. have served as
consultants for Roche, the European distributor of bevacizumab.
Abbreviations:
|
2DG
|
2-deoxy-D-glucose
|
|
ACC
|
acetyl-CoA-carboxylase
|
|
AMPK
|
AMP-activated kinase
|
|
EGFR
|
epidermal growth factor receptor
|
|
ERK1/2
|
extracellular signal-regulated kinase
1/2
|
|
mTOR
|
mammalian target of rapamycin
|
|
PI
|
propidium iodide
|
|
PKB
|
protein kinase B
|
|
ROS
|
reactive oxygen species
|
|
shRNA
|
short-hairpin RNA
|
References
|
1
|
Ohgaki H and Kleihues P: Population-based
studies on incidence, survival rates, and genetic alterations in
astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol.
64:479–489. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fleming TP, Saxena A, Clark WC, Robertson
JT, Oldfield EH, Aaronson SA and Ali IU: Amplification and/or
overexpression of platelet-derived growth factor receptors and
epidermal growth factor receptor in human glial tumors. Cancer Res.
52:4550–4553. 1992.PubMed/NCBI
|
|
3
|
van den Bent MJ, Brandes AA, Rampling R,
Kouwenhoven MC, Kros JM, Carpentier AF, Clement PM, Frenay M,
Campone M, Baurain JF, et al: Randomized phase II trial of
erlotinib versus temozolomide or carmustine in recurrent
glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol.
27:1268–1274. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Harris AL: Hypoxia - a key regulatory
factor in tumour growth. Nat Rev Cancer. 2:38–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Steinbach JP, Klumpp A, Wolburg H and
Weller M: Inhibition of epidermal growth factor receptor signaling
protects human malignant glioma cells from hypoxia-induced cell
death. Cancer Res. 64:1575–1578. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ronellenfitsch MW, Brucker DP, Burger MC,
Wolking S, Tritschler F, Rieger J, Wick W, Weller M and Steinbach
JP: Antagonism of the mammalian target of rapamycin selectively
mediates metabolic effects of epidermal growth factor receptor
inhibition and protects human malignant glioma cells from
hypoxia-induced cell death. Brain. 132:1509–1522. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Düvel K, Yecies JL, Menon S, Raman P,
Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S,
et al: Activation of a metabolic gene regulatory network downstream
of mTOR complex 1. Mol Cell. 39:171–183. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
de Groot JF, Fuller G, Kumar AJ, Piao Y,
Eterovic K, Ji Y and Conrad CA: Tumor invasion after treatment of
glioblastoma with bevacizumab: Radiographic and pathologic
correlation in humans and mice. Neuro-oncol. 12:233–242. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Steinbach JP, Supra P, Huang H-JS, Cavenee
WK and Weller M: CD95-mediated apoptosis of human glioma cells:
Modulation by epidermal growth factor receptor activity. Brain
Pathol. 12:12–20. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Steinbach JP, Wolburg H, Klumpp A, Probst
H and Weller M: Hypoxia-induced cell death in human malignant
glioma cells: Energy deprivation promotes decoupling of
mitochondrial cytochrome c release from caspase processing and
necrotic cell death. Cell Death Differ. 10:823–832. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fischer S, Ronellenfitsch MW, Thiepold
A-L, Harter PN, Reichert S, Kögel D, Paschke R, Mittelbronn M,
Weller M, Steinbach JP, et al: Hypoxia enhances the antiglioma
cytotoxicity of B10, a glycosylated derivative of betulinic acid.
PLoS One. 9:e949212014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wanka C, Brucker DP, Bähr O,
Ronellenfitsch M, Weller M, Steinbach JP and Rieger J: Synthesis of
cytochrome C oxidase 2: A p53-dependent metabolic regulator that
promotes respiratory function and protects glioma and colon cancer
cells from hypoxia-induced cell death. Oncogene. 31:3764–3776.
2012. View Article : Google Scholar
|
|
13
|
Wanka C, Steinbach JP and Rieger J:
Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects
glioma cells from starvation-induced cell death by up-regulating
respiration and improving cellular redox homeostasis. J Biol Chem.
287:33436–33446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Peereboom DM, Shepard DR, Ahluwalia MS,
Brewer CJ, Agarwal N, Stevens GH, Suh JH, Toms SA, Vogelbaum MA,
Weil RJ, et al: Phase II trial of erlotinib with temozolomide and
radiation in patients with newly diagnosed glioblastoma multiforme.
J Neurooncol. 98:93–99. 2010. View Article : Google Scholar
|
|
15
|
Marcus HJ, Carpenter KLH, Price SJ and
Hutchinson PJ: In vivo assessment of high-grade glioma biochemistry
using microdialysis: A study of energy-related molecules, growth
factors and cytokines. J Neurooncol. 97:11–23. 2010. View Article : Google Scholar
|
|
16
|
Collingridge DR, Piepmeier JM, Rockwell S
and Knisely JP: Polarographic measurements of oxygen tension in
human glioma and surrounding peritumoural brain tissue. Radiother
Oncol. 53:127–131. 1999. View Article : Google Scholar
|
|
17
|
Kim SM, Yun MR, Hong YK, Solca F, Kim J-H,
Kim H-J and Cho BC: Glycolysis inhibition sensitizes non-small cell
lung cancer with T790M mutation to irreversible EGFR inhibitors via
translational suppression of Mcl-1 by AMPK activation. Mol Cancer
Ther. 12:2145–2156. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Faubert B, Boily G, Izreig S, Griss T,
Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et
al: AMPK is a negative regulator of the Warburg effect and
suppresses tumor growth in vivo. Cell Metab. 17:113–124. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miwa H, Shikami M, Goto M, Mizuno S,
Takahashi M, Tsunekawa-Imai N, Ishikawa T, Mizutani M, Horio T,
Gotou M, et al: Leukemia cells demonstrate a different metabolic
perturbation provoked by 2-deoxyglucose. Oncol Rep. 29:2053–2057.
2013.PubMed/NCBI
|
|
20
|
Liu X, Chhipa RR, Pooya S, Wortman M,
Yachyshin S, Chow LM, Kumar A, Zhou X, Sun Y, Quinn B, et al:
Discrete mechanisms of mTOR and cell cycle regulation by AMPK
agonists independent of AMPK. Proc Natl Acad Sci USA.
111:E435–E444. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Leprivier G, Remke M, Rotblat B, Dubuc A,
Mateo AR, Kool M, Agnihotri S, El-Naggar A, Yu B, Somasekharan SP,
et al: The eEF2 kinase confers resistance to nutrient deprivation
by blocking translation elongation. Cell. 153:1064–1079. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jeon S-M, Chandel NS and Hay N: AMPK
regulates NADPH homeostasis to promote tumour cell survival during
energy stress. Nature. 485:661–665. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qian X, Li J, Ding J, Wang Z, Zhang W and
Hu G: Erlotinib activates mitochondrial death pathways related to
the production of reactive oxygen species in the human non-small
cell lung cancer cell line A549. Clin Exp Pharmacol Physiol.
36:487–494. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao F, Mancuso A, Bui TV, Tong X, Gruber
JJ, Swider CR, Sanchez PV, Lum JJ, Sayed N, Melo JV, et al:
Imatinib resistance associated with BCR-ABL upregulation is
dependent on HIF-1alpha-induced metabolic reprograming. Oncogene.
29:2962–2972. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
De Rosa V, Iommelli F, Monti M, Fonti R,
Votta G, Stoppelli MP and Del Vecchio S: Reversal of Warburg effect
and reactivation of oxidative phosphorylation by differential
inhibition of EGFR signaling pathways in non-small cell lung
cancer. Clin Cancer Res. 21:5110–5120. 2015. View Article : Google Scholar : PubMed/NCBI
|