Introduction
Autophagy is a highly conserved self-degradation
process of cellular constituents using the lysosomal machinery
(1). It is physiologically induced
by cellular stress or nutrient deprivation for maintenance of cell
homeostasis and recycling of cellular debris. In human
malignancies, including pancreatic ductal adenocarcinoma (PDAC),
autophagy plays a dual, but contrary role and promotes both, tumor
cell survival and death, probably depending on the tumor type and
stage of tumor progress (2,3). In
addition to direct mechanisms of tumor growth promotion, autophagy
counteracts chemotherapy-induced cell death, and autophagy
inhibition sensitizes PDAC to standard chemotherapeutics such as
gemcitabine (1,4–7). The
effectiveness of currently available chemotherapeutics against PDAC
is disappointing; even in patients with resectable PDAC, the median
survival is still limited to 21–24 months after curative surgery
and adjuvant standard chemotherapy with gemcitabine or
5-fluorouracil (8). Therefore, a
combination of chemotherapy and autophagy inhibition is intriguing,
and has entered the clinical phase (7,9).
Clinical trials of autophagy inhibition in PDAC are further
supported by the intense crosstalk of oncogenic Ras and autophagy
pathways (10). Oncogenic
KRAS mutations are characteristic for >90% of all PDAC,
and the link between mutation status and response to autophagy
inhibition is controversially discussed (11,12).
Autophagy and lysosomal gene expression are
centrally orchestrated by the transcription factor EB (TFEB)
(13,14). Nuclear translocation and activity
of TFEB is sensed and controlled by the mammalian target of
rapamycin complex (mTORC1) at the cytosolic lysosome (15). Inhibition of mTORC1 or nutrient
starvation abrogate TFEB phosphorylation and trigger its nuclear
shift and transcription of the autophagic/lysosomal gene cascade.
Interestingly, TFEB was found to be responsible for the development
of a rare malignant tumor: Aberrant expression of TFEB through a
specific gene translocation (Alpha-TFEB fusion) causes a distinct
subtype of pediatric renal cell carcinoma (16).
This study aims to investigate the expression of the
master autophagy regulator TFEB in PDAC, and its response to
standard chemotherapy, to elucidate its role as a potential target
for autophagy inhibition in pancreatic cancer patients.
Materials and methods
Human pancreas samples
Specimen collection was approved by the ethics
committee of the University of Heidelberg (Decision no.
S-134-2010), and written informed consent was obtained from the
patients. The study was performed with tissue samples obtained from
patients admitted to the Department of General, Visceral and
Transplantation Surgery, University Hospital Heidelberg, for
surgical treatment of PDAC or benign disease. Diagnoses were
established by a pathologist according to the World Health
Organization (WHO) classification.
Cell lines, reagents, and antibodies
The human pancreatic cancer cell lines PANC-1, MIA
PaCa-2, and BxPC-3 were purchased from ATCC (LGC Standards, Wesel,
Germany). They were cultured in a humidified atmosphere containing
5% CO2 at 37°C. RPMI-1640 supplemented with 10% fetal
calf serum (FCS) was used as the medium for PANC-1. MIA PaCa-2
cells were cultured in DMEM, with 10% FCS and 2.5% horse serum
added.
The following antibodies were used: mouse anti-KRAS
(sc-30, Santa Cruz Biotechnology, Heidelberg, Germany), rabbit
anti-LC3A/B (#4108, Cell Signaling, Danvers, MA, USA), rabbit
anti-Akt (#9272, Cell Signaling), rabbit anti-phospho-AKT (Ser473,
#9271, Cell Signaling), rabbit anti-TFEB (#4240, Cell Signaling),
anti-TFEB (V-17, Santa Cruz Biotechnology), mouse monoclonal
anti-TFEB (clone 3E1-G6, Sigma-Aldrich, Taufkirchen, Germany),
anti-PARP (#9542, Cell Signaling), and rabbit anti-GAPDH (#2118,
Cell Signaling).
The chemotherapeutic agent gemcitabine (Hexal,
Holzkirchen, Germany) was added to the cells after their treatment
with siRNAs in indicated concentrations, and incubated for 48–72 h.
Choloroquine (chloroquine disphophate salt, Sigma-Aldrich) was
diluted in H2O to the indicated concentrations.
Transfection and siRNA
The plasmid ptfLC3 was purchased from Addgene
(#21074, Cambridge, MA, USA). It encodes for LC3 tagged with mRFP
and EGFP to distinguish between autophagosomes and
autophagolysosomes based on its loss of GFP in lysosomal acidic
environment (17). For the
transfection, Lipofectamine 2000 (Life Technologies, Darmstadt,
Germany) was used according to the manufacturer's protocol.
Transfection of siRNAs was performed using HiPerfect
Transfection reagent (Qiagen, Hilden, Germany) according to the
manufacturer's protocol. The following sequences were targeted by
siRNAs: KRAS 5′-GGCTATATTTACATGCTACTA-3′ [Eurofins MWG
Operon, Ebersberg, Germany (18)],
TFEB 5′-CACAACTTAATTGAAAGGAGA-3′ (Qiagen), and KIF11
(Eg5) 5′-AACTGAAGACCTGAAGACAAT-3′ as the positive control (19); and nonsense siRNA (Allstars,
Qiagen) served as the negative control. All transfections were
performed with a concentration of 30 nM per target gene and an
incubation time of 48–72 h.
Quantitative RT-PCR
RNA was isolated with the Nucleo Spin II kit
(Macherey-Nagel, Düren, Germany) and converted to cDNA with the
High Capacity cDNA Reverse Transcription kit (Applied Biosystems,
Foster City, CA, USA) according to the manufacturers' protocols.
The quantitative RT-PCR was performed using the Power SYBR Green
Master Mix (Life Technologies). ACTB served as a housekeeping gene
for normalization of Ct-values. The
Delta-Delta-Ct-method was used to quantify gene
expression levels. The specific TFEB primers were obtained from
Search-LC (Heidelberg, Germany). RT-PCR results in human tissues
are presented as TFEB transcripts per 10,000 cyclophilin B copies
(10 kCPB).
Western blotting
Cells were washed and lysed on ice by RIPA buffer
containing protease and phosphatase inhibitors (Thermo Fisher
Scientific, Rockford, IL, USA). For extracting proteins, cell
lysates were centrifuged for 15 min at 11,000 × g followed by
concentration measurement of the supernatant with the Pierce BCA
Protein assay kit (Thermo Fisher Scientific). A total of 10–15 μg
protein per sample was then mixed with 4X LDS sample buffer and
Sample Reducing Agent (Thermo Fisher Scientific) and heated to 90°C
for 10 min with gentle agitation according to the manufacturer's
protocol of the NuPAGE SDS-PAGE Gel system (Thermo Fisher
Scientific). Probes were separated electrophoretically in a 4–12%
Bis-Tris NuPAGE Gel (Thermo Fisher Scientific) and blotted onto a
nitrocellulose membrane using the semidry blot NuPAGE system. After
blotting, the membrane was blocked for 1 h at room temperature in a
blocking solution, containing 5% Slim Fast powder (Allpharm
Vertriebs GmbH, Messel, Germany) dissolved in TBS-Tween 0.1%
buffer. The blots were then incubated with primary antibodies
(anti-Akt 1:1,000, anti-GAPDH 1:5,000, anti-KRAS 1:100, anti-LC3A/B
1:500, anti-phospho-Akt 1:1,000, anti-TFEB 1:500) in a blocking
solution overnight at 4°C. After three washing steps with TBS-Tween
0.1% buffer, the membrane was incubated with HRP-tagged,
host-specific secondary antibodies (Cell Signaling) for 1 h at room
temperature. After three additional washing steps,
chemiluminescence solution (Immobilon Western Chemi, Millipore,
Billerica, MA, USA) was added for detection of protein bands using
G:Box Chemi XT4 (Syngene, Cambridge, UK). GAPDH served as the
loading control. Quantification of protein intensity was performed
using ImageJ (Version 2.0.0; http://imagej.net/).
Immunhistochemistry
The paraffin-embedded tissue sections were
immunostained with horseradish peroxidase (HRP) labeled polymer
(Dako Deuschland GmbH, Hamburg, Germany). Shortly, consecutive
tissue sections were deparaffinized and rehydrated in progressively
decreasing concentrations of ethanol. After the antigens were
retrieved by boiling the tissue sections in 10 mM citrate buffer
for 3×5 min in the microwave oven, endogenous peroxidase activity
was blocked with 3% hydrogen peroxide in methanol for 10 min. The
slides were then placed in a washing buffer (10 mM Tris-HCl, 0.85%
NaCl, 0.1% bovine serum albumin, pH 7.4), and the sections were
incubated overnight at 4°C with the rabbit anti-TFEB antibody
(#4240, Cell Signaling) diluted in antibody diluent (Dako) or
rabbit immunoglobulin as negative control (Dako). Then, the slides
were incubated with a secondary anti-rabbit-labeled polymer (Dako)
for 45 min at room temperature. The immunoreactivity was visualized
using a 3,3′-diaminobenzidine (DAB)
chromogen/H2O2 system (Dako), and the
sections were counterstained with Mayer's hematoxylin. Finally, the
slides were viewed with an Axiophot microscope (Carl Zeiss,
Göttingen, Germany) coupled with a charge-coupled device (CCD)
camera (AxioVision, Zeiss, Jena, Germany).
Immunofluorescence staining and confocal
imaging
Cells were grown on collagen-A (0.1 mg/ml) coated
glass cover slips and treated with serum starvation or siRNA
transfection. Immunostaining was performed as described previously
(20). Briefly, cells were fixed
with 2% paraformaldehyde, permeabilized by 0.3% Triton X-100 for 5
min, and incubated in a blocking solution (2% fetal calf serum, 2%
bovine serum albumin, 0.2% fish gelatine resolved in PBS). Samples
were then incubated with the primary antibody for 1 h. After
several washing steps with PBS, cover slips were treated with Cy3-
or Alexa Fluor 488-tagged secondary antibodies. Cell nuclei were
stained with DAPI (1:200). Confocal imaging was performed on a
Leica SP5 laser-scanning microscope using the 63× oil-immersion
objectives (Leica Microsystems, Heidelberg, Germany). The
fluorescence intensity of TFEB was quantified by calculating the
ratio of the mean intensity in the nucleus and cytoplasm of the
respective cells using ImageJ (Version 2.0.0; http://imagej.net/).
Apoptosis assay and cell count
Cells including the supernatant were collected in a
FACS tube. After centrifugation for 5 min at 4°C and 1,500 rpm, the
supernatant was discarded and pellets were used for propidium
iodide (PI)/Annexin V apoptosis assays. For the apoptosis assay,
the samples were washed twice with FACS binding buffer (10 mM HEPES
pH 7.4, 2.5 mM CaCl2, 140 nM NaCl), and PI (50 μg/ml)
(Sigma-Aldrich) and Annexin V (BD Biosciences, Heidelberg, Germany)
were added. Measurements were performed with FACSCalibur (BD
Biosciences) and analyzed using WinMDI (Scripps Research Institute,
La Jolla, CA, USA). Apoptotic cells were defined as the sum of
PI+/Annexin V+ (late apoptosis) and
PI−/Annexin V+ (early apoptosis) cells.
The relative cell count was determined, which
reflects proliferation inhibition and apoptosis. The treated cells
were harvested by trypsinization and counted with the TC-20
Automated Cell Counter (Bio-Rad, München, Germany). Cell numbers
were normalized to the media controls.
Statistical analysis
If not indicated otherwise, data are presented as
mean ± standard error of the mean (SEM) and the two-sided, unpaired
t-test was used as the statistical test. Results were analyzed and
plotted using the Prism 5 software (GraphPad Software, La Jolla,
CA, USA).
Results
Expression analysis of TFEB in human
pancreas and PDAC
The expression of TFEB transcription was analyzed in
human normal pancreas tissue, in PDAC samples and in liver
metastases. The expression levels in primary tumor samples (n=45)
were significantly elevated compared with normal pancreas tissue
(P<0.001), and PDAC liver metastasis had also higher TFEB
expression compared with normal pancreas (P=0.02, Fig. 1A). Immunohistochemistry revealed a
weak to moderate cytoplasmic TFEB expression in normal ductal
cells, but no nuclear staining; moderate or high TFEB staining and
nuclear localization was found in 11 of 15 cancer specimens
(P=0.02; Fisher's exact test) (Fig.
1B).
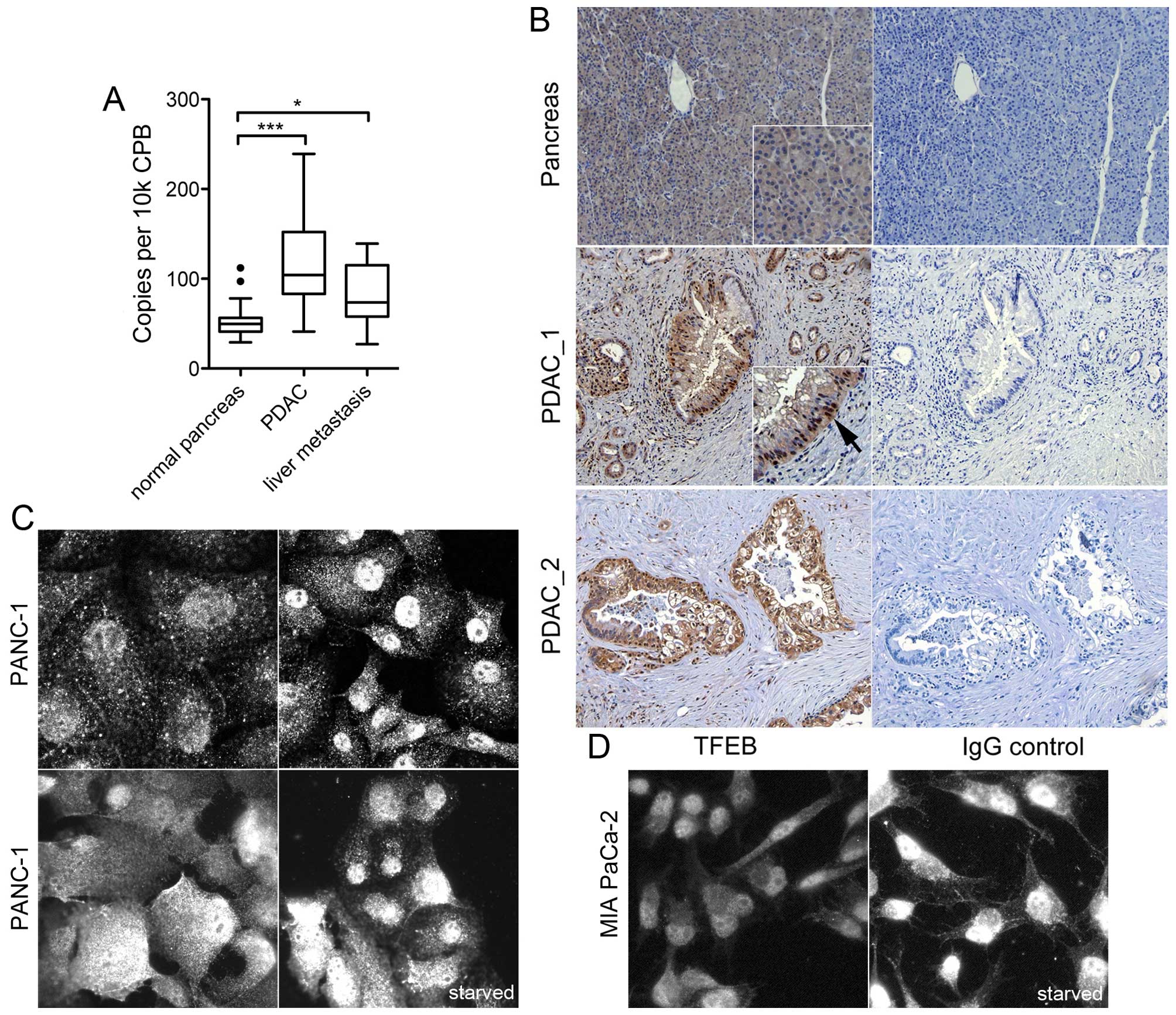 | Figure 1Expression analysis of TFEB in human
PDAC samples and cell lines. (A) Quantitative mRNA expression in
human normal pancreas (n=18), PDAC (n=45), and PDAC liver
metastasis (n=8). Data were normalized to cyclophilin B (CPB) and
presented as box plot (median, interquartile range).
***P<0.0001; **P<0.01;
*P<0.05. (B) Representative immunohistochemistry
analysis of TFEB in normal pancreas, and in two PDAC samples
(PDAC_1 and PDAC_2) next to the respective immunoglobulin G control
(right). The arrow points to the nuclear staining. Magnification
was ×200. (C and D) Immunofluorescence analysis of TFEB in PANC-1
(C) and MIA PaCa-2 cells (D) under normal serum conditions, or
under serum deprivation for 24 h (starved). Immunoreactivity was
tested with three different antibodies: goat polyclonal anti-TFEB
V-17 [(C), upper left)], mouse monoclonal anti-TFEB clone 3E1-G6
[(C), upper right], and rabbit anti-TFEB #4240 [(C), lower row;
(D)]. The image width represents 100 μm except the upper left image
in (C) (80 μm). |
Likewise, TFEB expression was investigated in human
PDAC cell lines. Immunoreactivity was compared using three
different, specific antibodies directed against TFEB. The primary
PDAC cell lines PANC-1 and MIA PaCa-2 showed both cytoplasmic and
nuclear TFEB expression under normal culture conditions. Serum
starvation led to minor enhancement of the nuclear staining
(Fig. 1C and D). The ratio of
nucleus and cytoplasmic fluorescence intensity was significantly
increased by starvation (P=0.01, n=35 cells). These results
indicate TFEB activation in PDAC cells even under basal conditions
and functional sensing of the available nutrient supply.
Autophagy dependence on TFEB in PDAC
cells
To assess whether TFEB expression affected autophagy
in PDAC cells, a transient TFEB RNA interference (siRNA) was
established, which achieved a mean protein downregulation to 23 and
11% in PANC-1 and MIA PaCa-2 cells, respectively (Fig. 2A and B). Both PDAC cell lines
responded to starvation with increased expression of TFEB and
microtubule-associated protein 1A/1B-light chain 3 (LC3)-II. The
starvation-induced upregulation of LC3-II was even maintained
during TFEB silencing (Fig. 2C and
D). We further explored whether TFEB silencing affected the
autophagy route from autophago-to autophagolysosomes. To this end,
a red fluorescent protein-green fluorescent protein (RFP-GFP)
tandem tagged LC3 protein was exogenously expressed to
differentiate autophagosomes (GFP and RFP-positive) from
autophagolysosomes (GFP-negative, but RFP-positive) (17). However, there was neither a
measurable difference in the LC3-positive vesicle number, nor in
the ratio of autophago-to autophagolysosomes after TFEB silencing
(Fig. 2E). Consequently, TFEB is
an indicator of autophagy in PDAC cells, but low expression levels
are sufficient to run the autophago-to-lysosome machinery. This was
further supported by chemotherapeutic treatment of the cancer cells
with gemcitabine, which caused a nearly dose-dependent LC3-II
accumulation using gemcitabine at concentrations ≤100 μM,
indicating increased autophagy, regardless of the amount of TFEB
protein (Fig. 2F and G). Thus, the
autophagy response of PDAC cells to gemcitabine did not solely
depend on the TFEB expression levels.
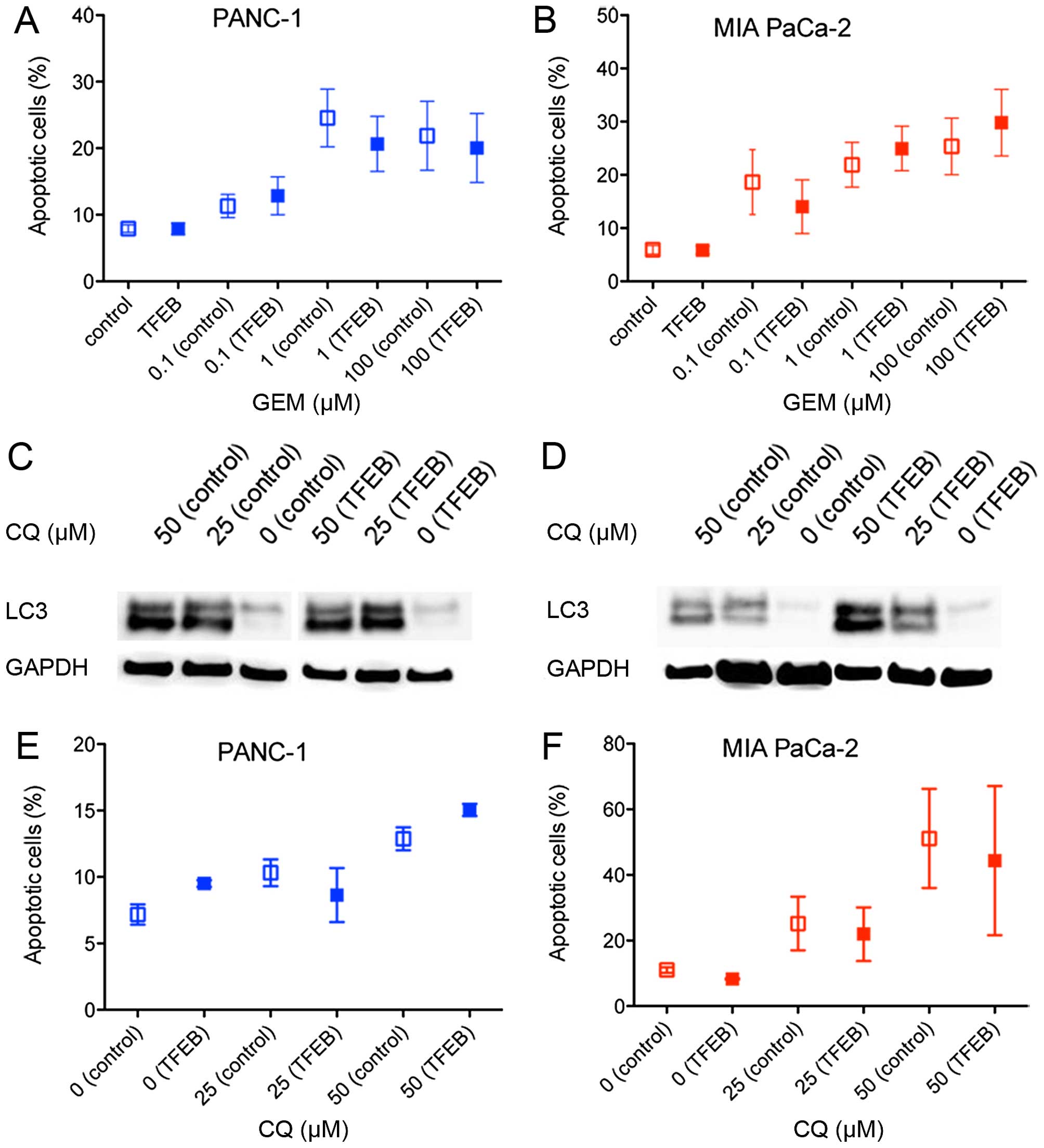
Chemotherapy-induced cytotoxicity
Previous data have demonstrated that autophagy
sustained PDAC cell survival and counteracted chemotherapeutic
cytotoxicity, but it is unknown whether these effects are mediated
by TFEB (5,7). Therefore, cell death (apoptosis)
induced by gemcitabine treatment was examined after TFEB siRNA
depletion. Only 20–30% of the cancer cells went into apoptosis at
concentrations >1 μM gemcitabine, and there was no significant
difference depending on the TFEB expression level (Fig. 3A and B). Because it has been shown
that the known inhibitor of autophagosome degradation, chloroquine
(CQ), can suppress PDAC growth, we further explored whether the
cytotoxic effect of CQ depends on TFEB activity. Autophagy
inhibition by CQ was seen in both cell lines (Fig. 3C and D). Induction of apoptosis was
found in 15–50% of the cells using established concentrations of
CQ, but there was no difference in the apoptosis rate when TFEB
expression was silenced (Fig. 3E and
F).
KRAS and TFEB expression - a link between
driver mutation and autophagy regulation
Because the results indicated that autophagy
sustained tumor cell survival in PDAC, we further investigated the
crosstalk between the PDAC driver oncogene KRAS and
autophagy. Ras signaling can promote, but also inhibit, autophagy
through use of different cascades including Rac1, Raf1, and
phosphoinositide-3-kinase (PI3K) signaling (10). The latter PI3K downsignaling
involves the Akt/mTOR1 pathway, and can negatively regulate
autophagy activity (21).
Silencing of KRAS expression increased cell death and apoptosis of
the cancer cells independently of TFEB expression, and TFEB
knockdown had no significant effect on cell death (Fig. 4A–C). Blockage of KRAS expression
led to an unchanged or minor increase in the LC3-II fraction, which
was not accompanied by changes in Akt phosphorylation (Fig. 4D). However, KRAS silencing resulted
in reciprocally augmented levels of TFEB protein and transcripts
(Fig. 4E and F), whereas TFEB
knockdown did not enhance KRAS expression. If oncogenic KRAS
activity instead of KRAS transcription levels would determine TFEB
expression, wild-type PDAC cells (e.g., BxPC-3) were expected to
harbor higher TFEB concentrations. However, TFEB expression was
comparably low in the KRAS wild-type BxPC-3 cells (data not
shown). These findings were supported by analysis of published data
of an inducible KRAS mouse model; oncogenic KRAS
activity status (on/off) did not influence TFEB expression, but
KRAS expression levels negatively and significantly correlated with
TFEB expression (Fig. 4G and H).
Moreover, the inverse relationship of KRAS and TFEB expression
levels could be confirmed in human PDAC samples, and again, there
was no TFEB dependence on the KRAS mutation status (Fig. 4I).
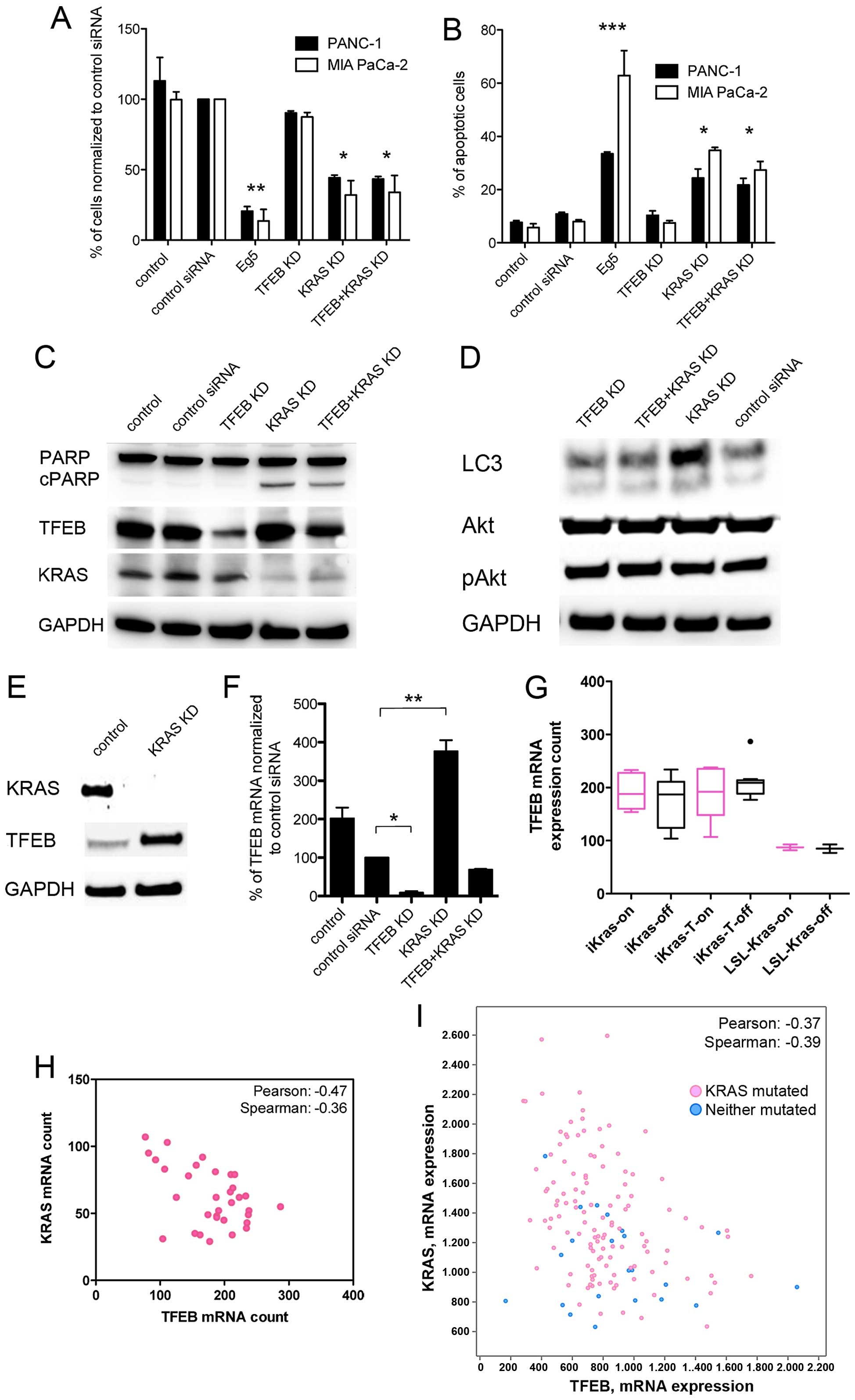 | Figure 4Correlation of KRAS and TFEB
expression in pancreatic cancer cells. (A and B) PDAC cell count
(A) and apoptosis (B) after siRNA knockdown (KD) of Eg5, TFEB, KRAS
or simultaneous TFEB, and KRAS KD for 72 h. Experiments were
performed as triplicates (mean ± SEM). (C) Western blotting of
indicated proteins in PANC-1 cells after TFEB and KRAS KD for 72 h.
Cleaved PARP (cPARP) indicates apoptosis after KRAS KD. (D) Western
blotting of LC3-I and -II after TFEB and KRAS KD. KRAS KD was not
accompanied by increased phospho-AKT (pAkt) in PANC-1 cells. (E)
KRAS KD for 48 h led to increased TFEB protein expression (PANC-1
cells). (F) Quantitative mRNA analysis after TFEB and KRAS KD in
PANC-1 cells (mean ± SEM). (G) TFEB expression analysis using the
data set record GDS4343 (NCBI Gene Expression Omnibus Data Set).
Cultured parenteral iKras p53L/+ PDAC cells (iKras, n=10), the
respective xenograft tumors (iKras-T, n=19), and control cells
(LSL-iKras, n=4) expressed doxycycline-dependent oncogenic KRAS
G12D (on). KRAS expression was significantly elevated in LSL-iKras
cells compared with the iKras and iKras-T cells (26). Data are presented as a box plot.
(H) Correlation of TFEB and KRAS mRNA expression in the iKras
parenteral, xenograft, and control cells (n=33). (I) Correlation of
TFEB and KRAS expression in human PDAC samples using the cBioPortal
database (http://www.cbioportal.org/;
pancreatic adenocarcinoma, 145 samples included). Pink dots
represent tumor samples with KRAS mutations.
***P<0.0001; **P<0.01;
*P<0.05. |
Discussion
Although autophagy can synergize with chemotherapy
in other types of cancer, accumulating evidence points to the fact
that autophagy is required for or supports PDAC growth (5,7,22).
In our previous studies, we could demonstrate large lysosomal
compartments and oncogene-associated, autophagy-mediated pathways
in metastasized PDAC cells (20).
For these reasons, targeting autophagy has grown to an important
field in PDAC research. This study explored the central autophagy
regulator TFEB in PDAC, and its role for autophagy and tumor cell
survival. TFEB belongs to the microphtalmia MiT/TFE family of
transcription factors (MITF, TFE3, TFEB and TFEC) (23), and this study demonstrated its
elevated expression and nuclear localization in human PDAC cells.
While this report was under preparation, Perera et al
published a comprehensive analysis of the MiT/TFE proteins in PDAC
(24). These data support both,
TFEB expression in PDAC, and the constitutive nuclear translocation
in these cells. The authors further found that the MiT/TFE proteins
are decoupled from regulatory control mechanisms and are required
to maintain the intracellular amino acid pool (24). In contrast to this study, they
found the MiT/TFE proteins to be critical for autophagy-lysosome
function, and that knockdown impaired primary PDAC cell growth.
Moreover, knockdown of TFE3 and MITF abolished xenograft tumor
growth of PANC-1 cells. These discrepant results might be explained
by the relative high expression of TFE3 in PDAC and PANC-1 cells
compared with TFEB (24), which
may compensate for autophagy biogenesis after TFEB silencing. Both
studies strongly indicate nuclear expression of TFE3 (24) or TFEB (this study) in PDAC, and
therefore conflict with an immunohistochemistry analysis of a
subset of 56 PDAC samples, which lacked significant nuclear
staining (16). Hence, one can
speculate whether nuclear TFE3/TFEB positivity depends on a
different antibody affinity in the studies or on the biological
tumor stage.
These data also show that chemotherapeutic treatment
with gemcitabine induces autophagy, and that autophagy inhibition
with chloroquine increases tumor cell apoptosis; both results are
in line with a potentiated PDAC cell response to simultaneous
chemotherapy and chloroquine treatment (5). However, PDAC cells may use other
central autophagy regulators than TFEB for chemotherapy-induced
autophagy, as TFEB knockdown did not impair autophagy
induction.
In fact, promotion of tumorigenesis by autophagy
appears to be an outstanding hallmark of PDAC, and was linked to
oncogenic KRAS activity (3,12).
Oncogenic KRAS is the signature mutation of PDAC and orchestrates
not only altered metabolism of glucose and glutamine (25,26),
but also promotes macropinocytosis and makes the tumor cells
(namely, their mitochondrial respiration) dependent on autophagic
flux; defective autophagosome formation causes accumulation of
abnormal mitochondria and reduced oxygen consumption (7,27–30).
In contrast, oncogenic KRAS activity can impair autophagy through
the PI3K/Akt/mTORC1 signaling pathway (10). Activated mTORC1 retains the
cytoplasmic TFEB pool and inhibits its nuclear activity (15), but it had been unknown whether TFEB
is critical for KRAS-mediated autophagy regulation. These results
demonstrate that PANC-1 and MIA PaCa-2 cell growth is sustained on
KRAS expression, and KRAS inhibition, but not TFEB inhibition,
increased apoptosis. This is in good agreement with our published
results of simultaneous gene silencing of KRAS and apoptotic
genes (18). Both primary PDAC
cell lines (PANC-1, MIA PaCa-2) express activating mutations of
KRAS and inactivating mutations of the P53 gene
(31). Although under debate,
recent experiments provided evidence that autophagy fuels PDAC
growth regardless of a simultaneous P53 inactivation
(3), which is supported by the
data that demonstrated impaired growth of both PDAC cell lines with
inactivated P53 status after autophagy inhibition. Because of the
high basal autophagic flux in PDAC cells, pro-autophagy signaling
by KRAS should outweigh its inhibitory effect on autophagy (e.g.,
through the PI3K/Akt/mTORC1 pathway). Thus, KRAS knockdown did not
significantly augment autophagy, and the phosphorylation status of
Akt remained unchanged. One can hypothesize that increased TFEB
expression compensates for depleted KRAS, to maintain autophagy in
PDAC cells. The inverse correlation of KRAS and TFEB expression in
PDAC cells was a novel finding and was confirmed in a mouse model
and in human PDAC samples. The diverging effects of KRAS
gene silencing and oncogenic KRAS activity in PDAC may suggest that
KRAS and TFEB are interrelated in a way that loss of oncogenic KRAS
levels elicits enhanced TFEB transcription for promotion of
pro-tumorigenic, autophagic flux and amino acid supply (24).
This study has some drawbacks and limitations. For
example, TFEB expression was not completely depleted in the cell
lines, leaving a basal gene expression, and the presented results
were not confirmed by additional siRNA sets; other members of the
MiT/TFE protein family were not examined, and the effect of
increased TFEB expression after KRAS knockdown on the tumor cells
was not further elucidated. However, the results strongly point to
the fact that PDAC cells require TFEB, but can compensate for TFEB
depletion to maintain tumorigenic autophagy. Although PDAC growth
and metabolism is tailored to and depends on autophagy, and TFEB
centrally orchestrates cellular lysosomal and autophagic programs,
targeting TFEB appears not to be a promising anticancer strategy
for PDAC. Based on these findings, further studies will focus on
how TFEB and the associated MiT/TFE proteins are regulated by the
key oncogene KRAS.
Acknowledgements
This study was supported by a German Research
Foundation (DFG) research grant to T.W. (WE 4816/1–2). Pancreatic
samples were obtained from Pancobank (Professor Büchler, Dr Giese,
E. Soyka, M. Stauch) supported by the BMBF (BMBF grant 01EY1101 to
Professor Schirmacher), Heidelberger Stiftung Chirurgie, and the
BMBF grants 01GS08114 and 01ZX1305C.
References
|
1
|
Galluzzi L, Pietrocola F, Bravo-San Pedro
JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J,
Gewirtz DA, Karantza V, et al: Autophagy in malignant
transformation and cancer progression. EMBO J. 34:856–880. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ropolo A, Bagnes CI, Molejon MI, Lo Re A,
Boggio V, Gonzalez CD and Vaccaro MI: Chemotherapy and
autophagy-mediated cell death in pancreatic cancer cells.
Pancreatology. 12:1–7. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang A and Kimmelman AC: Inhibition of
autophagy attenuates pancreatic cancer growth independent of
TP53/TRP53 status. Autophagy. 10:1683–1684. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Donohue E, Thomas A, Maurer N, Manisali I,
Zeisser-Labouebe M, Zisman N, Anderson HJ, Ng SS, Webb M, Bally M,
et al: The autophagy inhibitor verteporfin moderately enhances the
antitumor activity of gemcitabine in a pancreatic ductal
adenocarcinoma model. J Cancer. 4:585–596. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hashimoto D, Bläuer M, Hirota M, Ikonen
NH, Sand J and Laukkarinen J: Autophagy is needed for the growth of
pancreatic adenocarcinoma and has a cytoprotective effect against
anticancer drugs. Eur J Cancer. 50:1382–1390. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Papademetrio DL, Cavaliere V, Simunovich
T, Costantino S, Campos MD, Lombardo T, Kaiser CM and Alvarez E:
Interplay between autophagy and apoptosis in pancreatic tumors in
response to gemcitabine. Target Oncol. 9:123–134. 2014. View Article : Google Scholar
|
|
7
|
Yang S, Wang X, Contino G, Liesa M, Sahin
E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al:
Pancreatic cancers require autophagy for tumor growth. Genes Dev.
25:717–729. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Neoptolemos JP, Stocken DD, Bassi C,
Ghaneh P, Cunningham D, Goldstein D, Padbury R, Moore MJ, Gallinger
S, Mariette C, et al; European Study Group for Pancreatic Cancer.
Adjuvant chemotherapy with fluorouracil plus folinic acid vs
gemcitabine following pancreatic cancer resection: A randomized
controlled trial. JAMA. 304:1073–1081. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wolpin BM, Rubinson DA, Wang X, Chan JA,
Cleary JM, Enzinger PC, Fuchs CS, McCleary NJ, Meyerhardt JA, Ng K,
et al: Phase II and pharmacodynamic study of autophagy inhibition
using hydroxychloroquine in patients with metastatic pancreatic
adenocarcinoma. Oncologist. 19:637–638. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schmukler E, Kloog Y and Pinkas-Kramarski
R: Ras and autophagy in cancer development and therapy. Oncotarget.
5:577–586. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eser S, Schnieke A, Schneider G and Saur
D: Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer.
111:817–822. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Morgan MJ, Gamez G, Menke C, Hernandez A,
Thorburn J, Gidan F, Staskiewicz L, Morgan S, Cummings C, Maycotte
P, et al: Regulation of autophagy and chloroquine sensitivity by
oncogenic RAS in vitro is context-dependent. Autophagy.
10:1814–1826. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Settembre C and Ballabio A: TFEB regulates
autophagy: An integrated coordination of cellular degradation and
recycling processes. Autophagy. 7:1379–1381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Settembre C, Di Malta C, Polito VA, Garcia
Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D,
Colella P, et al: TFEB links autophagy to lysosomal biogenesis.
Science. 332:1429–1433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Settembre C, Zoncu R, Medina DL, Vetrini
F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et
al: A lysosome-to-nucleus signalling mechanism senses and regulates
the lysosome via mTOR and TFEB. EMBO J. 31:1095–1108. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Argani P, Laé M, Hutchinson B, Reuter VE,
Collins MH, Perentesis J, Tomaszewski JE, Brooks JS, Acs G, Bridge
JA, et al: Renal carcinomas with the t(6;11)(p21;q12):
Clinicopathologic features and demonstration of the specific
alpha-TFEB gene fusion by immunohistochemistry, RT-PCR, and DNA
PCR. Am J Surg Pathol. 29:230–240. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kimura S, Noda T and Yoshimori T:
Dissection of the autophagosome maturation process by a novel
reporter protein, tandem fluorescent-tagged LC3. Autophagy.
3:452–460. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Werner K, Lademann F, Thepkaysone ML,
Jahnke B, Aust DE, Kahlert C, Weber G, Weitz J, Grützmann R and
Pilarsky C: Simultaneous gene silencing of KRAS and anti-apoptotic
genes as a multitarget therapy. Oncotarget. 26:3984–3992. 2016.
|
|
19
|
Rückert F, Samm N, Lehner AK, Saeger HD,
Grützmann R and Pilarsky C: Simultaneous gene silencing of Bcl-2,
XIAP and Survivin re-sensitizes pancreatic cancer cells towards
apoptosis. BMC Cancer. 10:3792010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Welsch T, Younsi A, Disanza A, Rodriguez
JA, Cuervo AM, Scita G and Schmidt J: Eps8 is recruited to
lysosomes and subjected to chaperone-mediated autophagy in cancer
cells. Exp Cell Res. 316:1914–1924. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Furuta S, Hidaka E, Ogata A, Yokota S and
Kamata T: Ras is involved in the negative control of autophagy
through the class I PI3-kinase. Oncogene. 23:3898–3904. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim SE, Park HJ, Jeong HK, Kim MJ, Kim M,
Bae ON and Baek SH: Autophagy sustains the survival of human
pancreatic cancer PANC-1 cells under extreme nutrient deprivation
conditions. Biochem Biophys Res Commun. 463:205–210. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Haq R and Fisher DE: Biology and clinical
relevance of the micropthalmia family of transcription factors in
human cancer. J Clin Oncol. 29:3474–3482. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Perera RM, Stoykova S, Nicolay BN, Ross
KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK,
Ferrone CR, et al: Transcriptional control of autophagy-lysosome
function drives pancreatic cancer metabolism. Nature. 524:361–365.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Son J, Lyssiotis CA, Ying H, Wang X, Hua
S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et
al: Glutamine supports pancreatic cancer growth through a
KRAS-regulated metabolic pathway. Nature. 496:101–105. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ying H, Kimmelman AC, Lyssiotis CA, Hua S,
Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff
JL, et al: Oncogenic Kras maintains pancreatic tumors through
regulation of anabolic glucose metabolism. Cell. 149:656–670. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bryant KL, Mancias JD, Kimmelman AC and
Der CJ: KRAS: Feeding pancreatic cancer proliferation. Trends
Biochem Sci. 39:91–100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Commisso C, Davidson SM, Soydaner-Azeloglu
RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin
JA, Thompson CB, et al: Macropinocytosis of protein is an amino
acid supply route in Ras-transformed cells. Nature. 497:633–637.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guo JY, Chen HY, Mathew R, Fan J,
Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM,
Karantza V, et al: Activated Ras requires autophagy to maintain
oxidative metabolism and tumorigenesis. Genes Dev. 25:460–470.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo JY, Xia B and White E:
Autophagy-mediated tumor promotion. Cell. 155:1216–1219. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Moore PS, Sipos B, Orlandini S, Sorio C,
Real FX, Lemoine NR, Gress T, Bassi C, Klöppel G, Kalthoff H, et
al: Genetic profile of 22 pancreatic carcinoma cell lines. Analysis
of K-ras, p53, p16 and DPC4/Smad4. Virchows Arch. 439:798–802.
2001. View Article : Google Scholar
|