Introduction
Pancreatic ductal adenocarcinoma (PDA) is almost
uniformly lethal with an estimated annual number of 45,220 new
cases approximating 38,460 annual deaths and a 5-year survival rate
of <5% (1–3). Late initial diagnosis, aggressive
metastatic behavior and resistance to chemoradiotherapy render
pancreatic cancer one of the most difficult to treat of all
malignancies. Surgical resection is potentially curative in a
minority of patients; however, >80% of the patients are
diagnosed with locally advanced disease that precludes surgical
intervention. Systemic gemcitabine alone or in combination with
5-FU, irinotecan and oxaliplatin (FOLFIRINOX) is the current
standard of care for advanced pancreatic cancer, providing
short-term symptomatic improvement with minor impact on survival
(4–6). Thus, there is an urgent need for
developing novel agents for the treatment of pancreatic cancer.
Trichothecenes are structurally related low
molecular weight sequiterpenoid metabolites produced by filamentous
fungi, such as Myrothecium, Stachybotrys, Fusarium and
others (7). Trichothecenes are
potent mycotoxins that are believed to be responsible for adverse
respiratory and neurological effects of damp and moldy indoor
environments (8–10). They elicit a wide range of
biological responses in eukaryotic cells, including inhibition of
protein synthesis and cell proliferation; induction of
cytotoxicity, ribotoxic stress responses and modulation of immune
responses (11–16). Verrucarin A (VC-A) is a macrocyclic
trichothecene that evokes strong antiproliferative and proapoptotic
responses in cancer cells (17–20).
There is some evidence that the antiproliferative and
apoptosis-inducing effects of VC-A are due to the inhibition of
protein synthesis through blocking of peptidyl transferase activity
and activation of c-jun N-terminal kinase (c-JNK) and p-38 MAP
kinase (14,17,18).
In a previous report, VC-A was identified as a major
mediator of the anticancer activity in salt water cultures of
Myrothecium verrucaria isolated from a sponge species
collected in Hawaii (21).
However, the mode of cell death or the mechanism by which VC-A
kills tumor cells was not investigated in that study.
In this study, we investigated the anticancer
activity of verrucarin A against PDA cell lines. Data showed that
VC-A inhibits the proliferation and induces cell cycle arrest in S
phase by inhibiting cell cycle-related regulatory proteins. VC-A
induced apoptosis in PDA cells through the activation of
procaspases, induction of mitochondrial depolarization and
inhibition of Bcl-2 and IAP family of proteins that regulate
apoptosis. In addition, VC-A also inhibited prosurvival signaling
proteins (e.g., p-Akt, NF-κB and p-mTOR) and their downstream
mediators.
Materials and methods
Reagents
Verrucarin A isolated from salt water cultures of
Myrothecium verrucaria separated from Spongia Sp. was
obtained from Dr Phillip Crews, University of California, Santa
Cruz. Anti-caspase-3, caspase-8, and caspase-9 antibodies were
purchased from BD Pharmingen (San Diego, CA, USA). Antibodies
against p-Akt (ser473), p-mTOR (Ser2448),
PARP-1, anti-NF-κB (p65), anti-Bcl-2, anti-Bcl-xL, anti-Bax,
anti-Bak, anti-Bad and β-actin were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Anti-cdk2, cdk4, cdk6,
anti-cyclin D, anti-cyclin E and anti-p21 antibodies were from Cell
Signaling Technology (Boston, MA, USA). 96 AQueous One Solution
Proliferation assay system was from Promega (Madison, WI, USA).
Stock solution of VC-A (2 mM) was prepared in DMSO and all test
concentrations were prepared by diluting stock solution in tissue
culture medium.
Cell lines
MiaPaCa-2, Panc-1 and BxPC-3 PDA cell lines were
obtained from the American Type Culture Collection (ATCC,
Rockville, MD, USA). MiaPaCa-2 and Panc-1 cell lines were grown in
DMEM tissue culture medium whereas BxPC-3 cells were cultured in
RPMI-16 (Gibco BRL, Rockville, MD, USA) supplemented with 10% fetal
bovine serum, 1% penicillin/streptomycin, and 25 mM HEPES buffer.
Cells were incubated at 37°C in a humidified atmosphere consisting
of 5% CO2, 95% air and maintained by splitting cultures
twice a week.
MTS assay
Tumor cells (1×104) in 100 μl of tissue
culture medium were seeded into each well of a 96-well plate. After
24-h incubation to allow cells to adhere, cells were treated with
VC-A at concentrations ranging from 0 to 0.625 μM. Cultures were
incubated for additional 72 h and cell viability was then
determined by the colorimetric MTS assay using CellTiter 96 AQueous
One Solution Proliferation assay system from Promega (Madison),
which measures the bioreduction of tetrazolium compound MTS in the
presence of electron-coupling reagent phenazine methosulfate. The
absorbance, which is directly proportional to the number of viable
cells in the cultures, was measured at 490 nm using a microplate
reader.
Cell cycle analysis
The distribution of cells in various cell cycle
phases was analyzed by measuring cellular DNA content. Untreated
(control) (2×106) or VC-A-treated cells were fixed in
70% ethanol overnight at 4°C. Cells were washed twice and
resuspended in 0.8 ml of PBS. To each tube, 100 μl of DNAse free
RNAse (500 μg/ml) and 100 μl of propidium iodide (500 μg/ml) was
added and tubes were incubated at room temperature in the dark for
30 min. Cellular DNA content was determined by flow cytometry using
Accuri C6 flow cytometer (Accuri Cytometers Inc. Ann Arbor, MI,
USA).
Annexin V-FITC binding
Induction of apoptosis was assessed by the binding
of Annexin V to phosphatidylserine, which is externalized to the
outer leaflet of the plasma membrane early during apoptosis.
Briefly, MiaPaCa-2 and BxPC-3 cells treated with VC-A (0-0.625 μM)
for 24 h were resuspended in the binding buffer provided in Annexin
V-FITC apoptosis detection kit II (BD Biosciences, Pharmingen).
Cells were mixed with 5 μl of Annexin V-FITC reagent, 5 μl of PI,
and incubated for 30 min at room temperature in the dark. Stained
cells were analyzed by flow cytometry.
Mitochondrial depolarization assay
Alteration in mitochondrial potential by VC-A was
determined using mitochondrial potential sensor dye JC-1 (Molecular
Probes, Invitrogen, San Diego, CA, USA). Briefly, 1×106
control (untreated) and VC-A- treated cells in 1 ml culture medium
were loaded with JC-1 (10 μg/ml) for 10 min at 22°C and analyzed by
flow cytometry. In normal cells, dye is aggregated in mitochondria,
fluoresces red, and is detected in the FL2 channel. In cells with
altered mitochondrial potential, the dye fails to accumulate in the
mitochondria, remains as monomers in the cytoplasm, fluoresces
green, and is detected in the FL1 channel.
Western blotting
Cell lysates were prepared by detergent lysis [1%
Triton-X 100 (v/v), 10 mM Tris-HCl (pH 7.5), 5 mM EDTA, 150 mM
NaCl, 10% glycerol, 2 mM sodium vanadate, 5 μg/ml leupeptin, 1
μg/ml aprotinin, 1 μg/ml pepstatin A and 10 μg/ml
4-2-aminoethyl-benzenesulfinyl fluoride]. Lysates were clarified by
centrifugation at 14,000 x g for 10 min at 4°C, and protein
concentrations were determined by Bradford assay. Samples (50 μg)
were boiled in an equal volume of sample buffer [20% glycerol, 4%
SDS, 0.2% bromophenol blue, 125 mM Tris-HCl (pH 7.5), and 640 mM
2-mercaptoethanol] and separated on 10% SDS-polyacrilamide gels.
Proteins resolved on the gels were transferred to nitrocellulose
membranes. Membranes were blocked with 5% milk in 10 mM Tris-HCl
(pH 8.0), 150 mM NaCl with 0.05% Tween-20 (TPBS) and incubated with
protein specific antibodies followed by HRP-conjugated secondary
antibody. Immune complexes were visualized with enhanced
chemiluminescence detection system from Amersham Corp. (Arlington
Heights, IL, USA). The immunoblots were imaged and the density of
protein bands was analyzed using Image/J software
(imagej/nih.gov/ij/download/). The protein band densities were
normalized to the corresponding β-actin levels and presented as
fraction of untreated control considered as 1.0.
Statistical analysis
Data are presented as means ± SD. The differences
between control and treatment groups were analyzed using Student’s
t-test and differences with p<0.05 were considered statistically
significant.
Results
VC-A inhibits proliferation and cell
cycle progression in PDA cells
The effect of VC-A on proliferation of MiaPaCa-2,
Panc-1 and BxPC-3 PDA cells was examined using the MTS assay. For
this, cells were treated with VC-A at concentrations of 0–0.625 μM
for 72 h and the viability of cultures was determined. As shown in
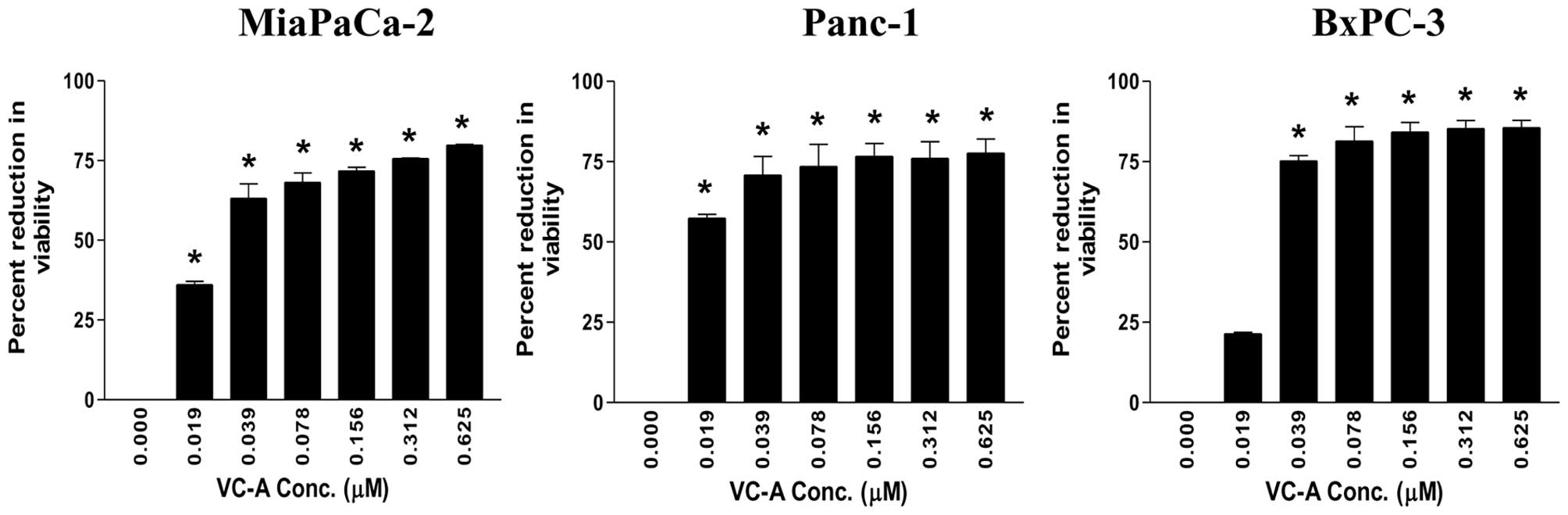
Fig. 1, significant reduction in
viability was observed at the lowest concentration of 0.019 μM VC-A
(MiaPaCa-2 = 36±1.4 SD; Panc-1 = 57±1.8 SD, p<0.05). BxPC-3
cultures also showed measurable reduction in viability at 0.019 μM
VC-A (21±0.6 SD). In all three cell lines, reduction in viability
significantly increased at VC-A concentrations of 0.039–0.625 μM
(MiaPaCa-2 = 63–80% reduction; Panc-1 = 57–78% reduction; BxPC-3 =
75–86% reduction, p<0.01). These data demonstrated potent
antiproliferative activity of VC-A against PDA cells.
In a previous report, VC-A was demonstrated to be
6–10-fold less cytotoxic to normal bone marrow cells compared to
cancer cell lines (21). In
addition, VC-A was also inactive or only weekly active against
non-cancerous Vero cells derived from monkey kidney compared to
cancer cell lines (12).
Verrucarin A induces cell cycle arrest
and inhibits cell cycle-related regulatory proteins in PDA
cells
Since VC-A inhibited the proliferation of PDA cells
we investigated its effect on cell cycle progression and cellular
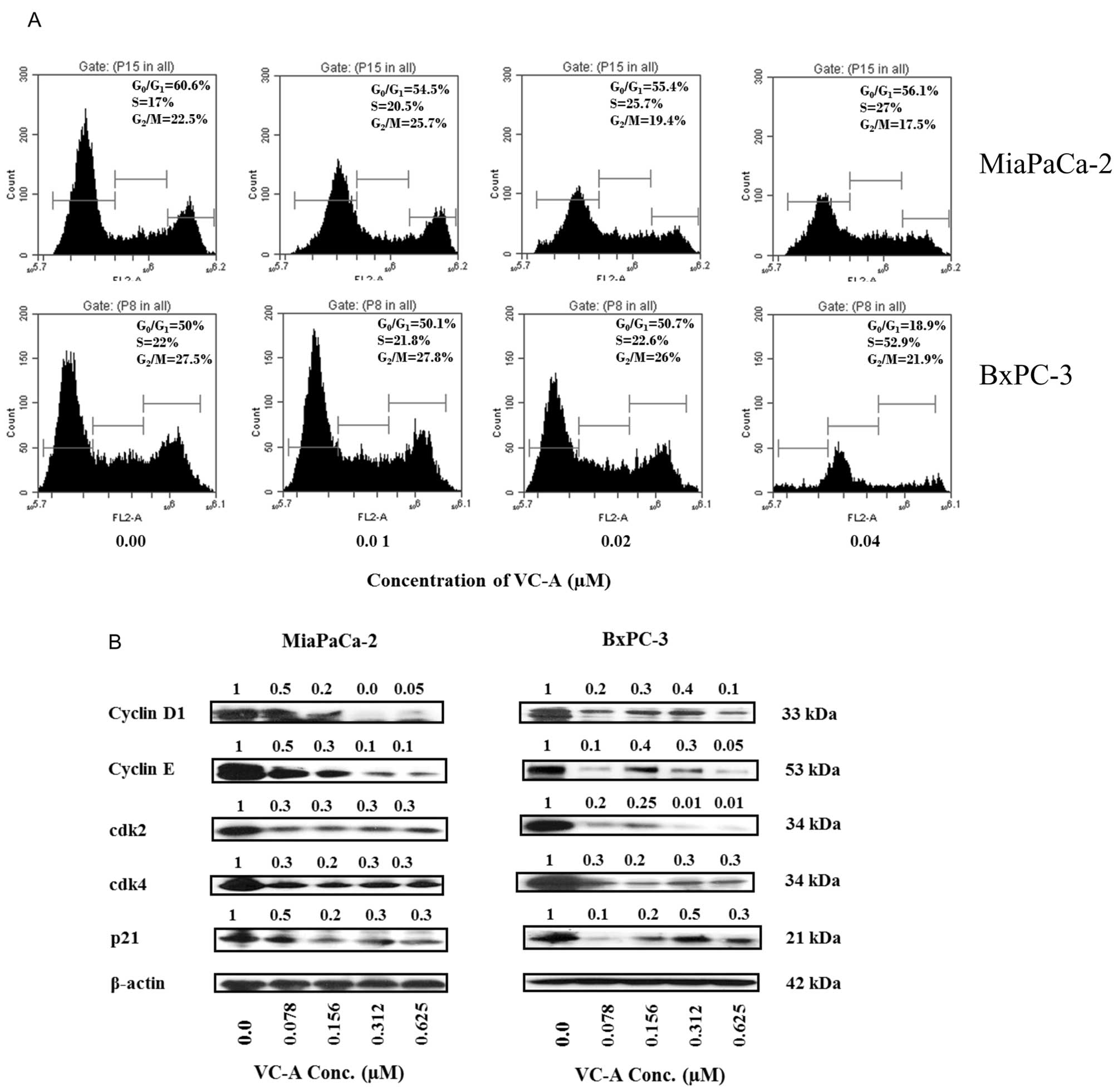
proteins that regulate cell division. For effect on cell cycle
progression, MiaPaCa-2 and BxPC-3 cells were treated with VC-A
(0–0.04 μM) for 24 h, stained with PI and cellular DNA content was
analyzed by flow cytometry. Treatment with VC-A resulted in arrest
of cells in the S cell cycle phase in both cell lines (Fig. 2A). In MiaPaCa-2 cells, the
accumulation of cells in S phase was VC-A concentration-dependent
(e.g., 17–27% at 0–0.04 μM VC-A), whereas in BxPC-3 cells all of
the S-phase arrest occurred at 0.04 μM VC-A. To determine the
effect of VC-A on cell cycle regulatory proteins, cell lysates of
MiaPaCa-2 and BxPC-3 cells treated with VC-A (0.078–0.625 μM) for
24 h were analyzed for the levels of cyclin D1, cyclin E, cdk2,
cdk4 and WAF1/21 (p21) by western blotting. As shown in Fig. 2B, treatment with VC-A reduced the
levels of these proteins mostly in a concentration-dependent
manner. These data suggested that VC-A arrests PDA cells in S cell
cycle phase by inhibiting cell cycle regulatory proteins.
VC-A induces apoptosis in PDA cells
Whether cell cycle arrest leads to the induction of
apoptosis was investigated next. Induction of apoptosis was by
analyzed by the binding of Annexin V-FITC to cells treated with
VC-A. Thus, MiaPaCa-2 and BxPC-3 cells were treated with VC-A
(0.078–0.625 μM) for 24 h and binding of Annexin V-FITC was
determined by flow cytometry. As shown in Fig. 3A, only a small percentage of
untreated MiaPaCa-2 and BxPC-3 cells bound Annexin V-FITC (<5%).
The percentage of Annexin V-FITC binding MiaPaCa-2 increased from
51% at 0.078 μM to 57% at 0.625 VC-A (p<0.01). On the other
hand, the percentage of Annexin V-FITC binding BxPC-3 cells
increased in a dose-dependent manner, e.g., 24, 31, 3 and 43% at
0.078, 0.15, 0.325 and 0.625 μM VC-A.
The induction of apoptosis by VC-A was confirmed by
the cleavage of PARP-1 and procaspases. Tumor cells were treated
with VC-A as described above and the cleavage of PARP-1 and
procaspases was analyzed by western blotting. As shown in Fig. 3B, treatment with VC-A induced the
cleavage of native PARP-1 (110-kDa fragment) as identified by the
emergence of an 89-kDa cleaved PARP-1 fragment in both cell lines.
Treatment with VC-A also induced the processing of procaspases-3,-8
and -9 as determined from a decrease in native proteins
(procaspases-3 and -8) or the emergence of the cleaved fragments
(procaspase-9) (Fig. 3C). However,
the effect of VC-A on the activity of these caspases was not
determined. Together, increase in Annexin V-FITC-binding and the
cleavage of PARP-1 and processing of procaspases-3, -8 and -9
demonstrated induction of apoptosis in PDA cells by VC-A.
VC-A induces mitochondrial depolarization
and release of cytochrome c
Whether mitochondrial pathway of apoptosis was
involved in the apoptotic cell death of PDA cells by VC-A was
examined next. As a measure of mitochondrial involvement in
induction of apoptosis by VC-A, we evaluated mitochondrial
depolarization in cells treated with VC-A. Thus, MiaPaCa-2 and
BxPC-3 cells were treated with VC-A (0–0.625 μM) for 24 h and then
loaded with mitochondrial-potential sensor probe JC-1 and
fluorescence emission was analyzed by flow cytometry. There was
significant change in the mitochondrial potential in both cell
lines after treatment with VC-A. As shown in Fig. 4A, the percentage of MiaPaCa-2 cells
with green fluorescence significantly increased from 3% at 0 μM
VC-A to 29, 42, 44 and 48% at 0.078–0.625 μM VC-A (p<0.01). VC-A
also showed strong mitochondrial depolarizing effect on BxPC-3
cells (e.g., 5, 39, 53, 66 and 62% of cells with green fluorescence
at 0, 0.078, 0.156, 0.312 and 0.625 μM VC-A, p<0.01).
We also analyzed the effect of VC-A on the release
of cytochrome c from mitochondria. Western blot analysis of
mitochondrial fraction of MiaPaCa-2 and BxPC-3 cells treated with
VC-A (0–0.625 μM) demonstrated the release of cytochrome c
in both cell lines in a concentration-dependent manner (Fig. 4B). Taken together, the loss of
mitochondrial membrane potential and release of cytochrome c
from mitochondria indicated that mitochondrial damage plays a
significant role in apoptotic cell death of PDA cells by VC-A.
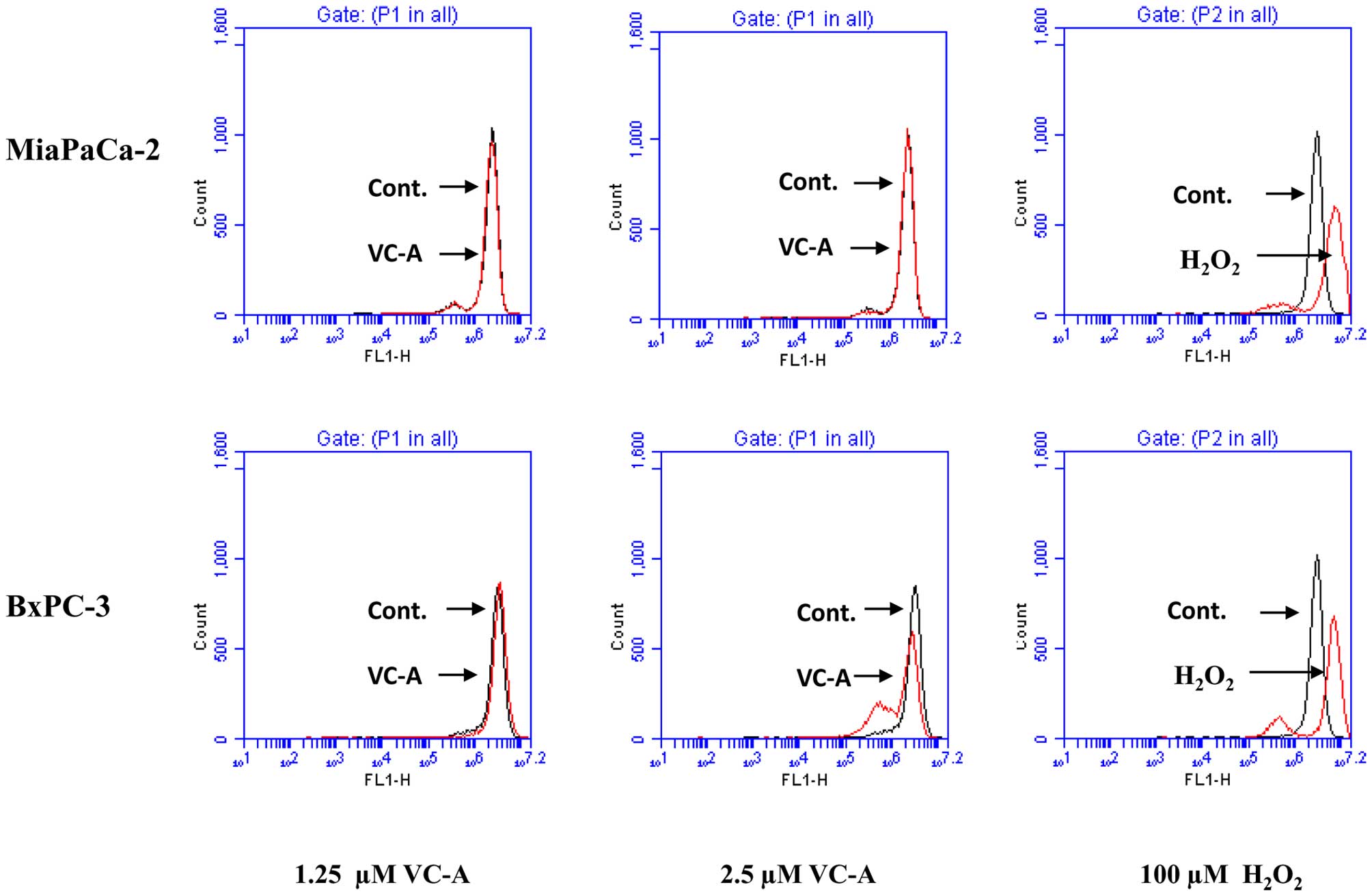
VC-A does not induce ROS generation
Since generation of free radicals is part of the
mechanism by which most chemotherapeutic agents induce apoptosis in
cancer cells we evaluated the generation of ROS by VC-A in PDA
cells. This was accomplished by detecting
H2O2 production by flow cytometry using
H2DCFDA fluorescent probe. As shown in Fig. 5, whereas treatment of MiaPaCa-2 and
BxPC-3 cells with H2O2 resulted in production
of hydroxyl radicals (positive control) treatment with VC-A at 1.25
and 2.5 μM for 2 h failed to induce the generation of these free
radicals. Further, no ROS was detected either when cells were
treated with VC-A for 1 or 4 h (not shown).
VC-A downregulates Bcl-2 and IAP family
proteins
To further characterize apoptotic response of PDA
cells to VC-A, the effect of VC-A on proteins belonging to the
Bcl-2 and IAP families of proteins that regulate apoptosis
positively and negatively was determined. For this, cell lysates
prepared from MiaPaCa-2 and BxPC-3 cells treated or not with VC-A
(0.079–0.625 μM) for 24 h were analyzed for Bcl-2, Bcl-xL, Bax,
p-Bad, survivin and c-IAP-2 by western blotting. As shown in
Fig. 6, treatment with VC-A
significantly to completely inhibited the levels of antiapoptotic
Bcl-2 and Bcl-xL in both cell lines. Proapoptotic Bax was
unaffected in MiaPaCa-2 cells but was reduced in BxPC-3 cells. Even
though Bax levels were somewhat reduced in BxPC-3 cells but the
Bax/Bcl-2 ratio was increased 2–3-fold in both cell lines treated
with VC-A (e.g., MiaPaCa-2: 2.7-, 2.7-, 3.2- and 2.7-fold; BxPC-3:
3-, 2-, 3- and 2-fold at 0.079, 0.158, 0.312 and 0.625 μM,
respectively). Proapototic p-Bad was also reduced but still
detectable in both cell lines. Similarly, antiapoptotic survivin
(IAP family member) was significantly reduced in both cell lines
(60–80%). On the other hand, while cIAP-2 was markedly reduced at
0.312 and 0.625 μM VC-A in MiaPaCa-2 cells, effect on c-IAP-2
expression in BxPC-3 cells was minimal. Overall, these data
suggested that inhibition of antiapoptotic proteins plays a role in
induction of apoptosis by VC-A in PDA cells.
VC-A inhibits Akt, mTOR and NF-κB
signaling proteins and their downstream mediators
Akt/NF-κB/mTOR is a major antiapoptotic
(prosurvival) signaling pathway that confers survival advantage in
cancer cells including PDA cells. We investigated whether induction
of apoptosis in prostate cancer cells by VC-A involved the
inhibition of Akt, mTOR and NF-κB and their downstream mediators.
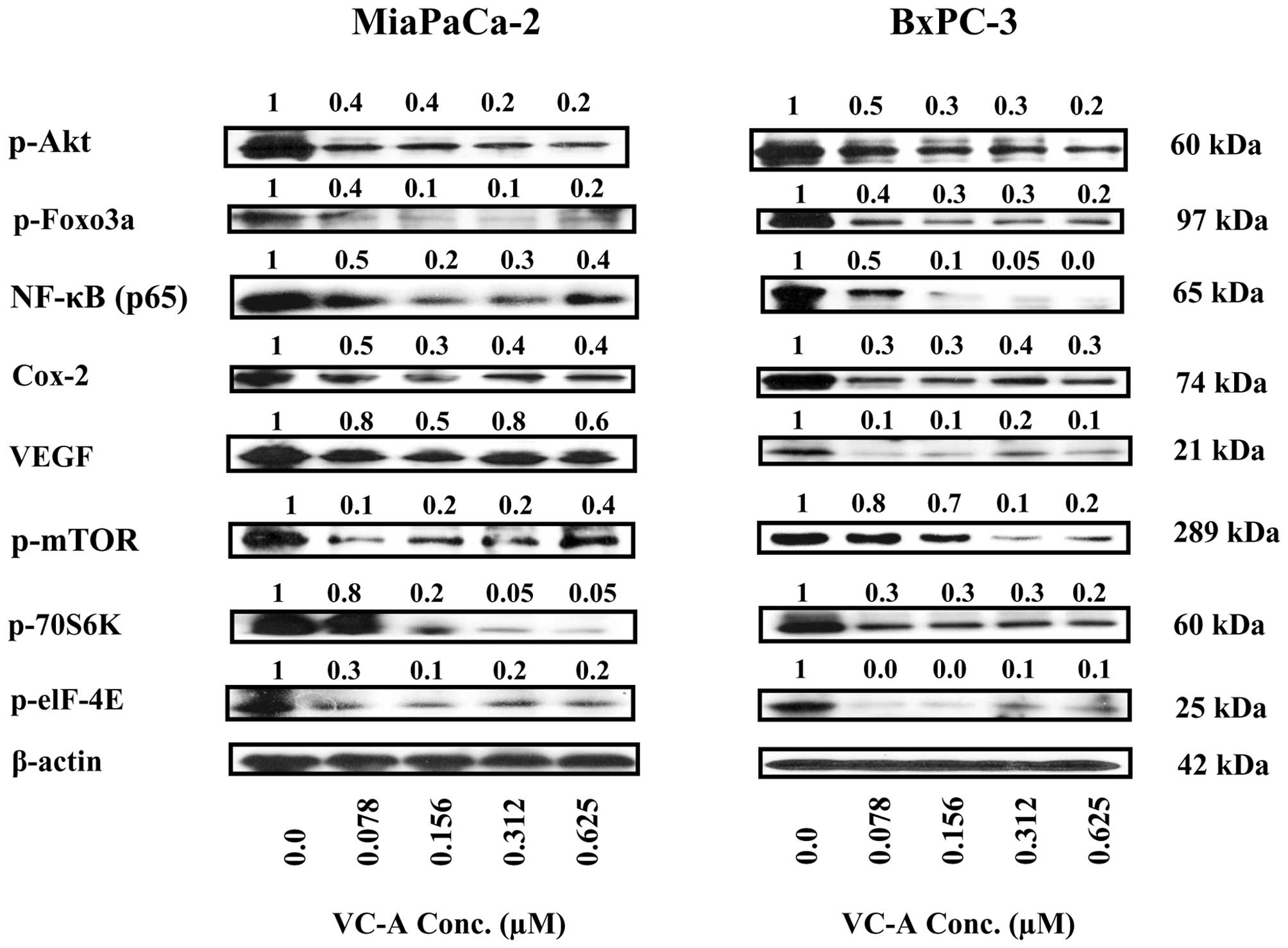
Thus, cell lysates prepared from MiaPaCa-2 and BxPC-3 cells treated
or not with VC-A (0–0.625 μM) for 24 h were analyzed for p-Akt,
mTOR and NF-κB and their effectors by western blotting. Treatment
with VC-A markedly reduced the levels of p-Akt, p-mTOR and NF-κB
(p65) in both cell lines (Fig. 7).
Further analysis of cell lysates showed that VC-A also inhibited
the major downstream intermediary targets of Akt, mTOR and NF-κB.
For instance, VC-A inhibited the expression of p-Foxo-3α, the
downstream target of activated Akt involved in cell proliferation
and apoptosis. Similarly, the levels of mTOR effectors such as
p-S6K1 (p-70S6 kinase) and p-eIF-4e were markedly decreased after
treatment with VC-A. In addition, the levels of angiogenesis
related Cox-2 and VEGF, which are transcriptionally regulated by
NF-κB were also markedly reduced. Collectively, these data
suggested that the inhibition of p-Akt, p-mTOR, NF-κB and their
downstream mediators might be necessary for induction of apoptosis
in PDA tumors by VC-A.
Discussion
There is an urgent need for effective and safe novel
agents with defined therapeutic targets for treating pancreatic
cancer. Natural products have long been recognized as a rich source
of chemical diversity and compounds with a wide spectrum of
anticancer activities (22,23).
Indeed, isolation and identification of bioactive components from
medicinal plants have led to the synthesis of potent anticancer
drugs, including Vinca alkaloids, taxol, camptothecan, etoposide
and retinoids. Verrucarin A (VC-A) is a macrocyclic trichothecene
mycotoxin that has shown strong antiproliferative and proapoptotic
activity in breast cancer cells (17,18),
providing some insights into the mode of action of VC-A. However,
the antitumor activity or the mechanism of action of VC-A in
pancreatic cancer cells has not been investigated. Thus, this study
was undertaken to investigate the antitumor activity and to
identify targets of VC-A in PDA cells. VC-A inhibited the
proliferation of three PDA cell lines (e.g., MiaCa-2, Panc1 and
BxPC-3) at very low concentrations. The antiproliferative effect of
VC-A was attributed to the inhibition of cell cycle progression in
S phase. This finding however differs from a previous report in
which VC-A was shown to arrest cells in G0/G1 phase (17). Furthermore, cell cycle arrest by
VC-A was associated with the inhibition of cyclin D1, cyclin E,
cdk2, cdk-4 and cdk inhibitor p21. Cyclin D and E in conjunction
with cdk2, cdk4 and cdk6 regulate cell cycle progression through G1
and S phases. The inhibition of cyclin E and cdk2 suggests that
inhibition of these proteins would result in decreased formation of
cyclin E-cdk2 complexes and therefore accumulation of cells in S
phase. The cip/kip family member p21 halts cell cycle progression
by inhibiting cyclin-cdk complexes. VC-A only partially reduced p21
in PDA cells, perhaps not enough to hamper the activity of cyclin
E-cdk2 complexes.
The inability to induce apoptosis has been
implicated in cancer development and resistance to cancer therapies
(24). On the other hand,
promotion of apoptosis in cancer cells could potentially lead to
tumor regression and improved prognosis (25). The inhibition of cell proliferation
by anticancer agents normally forces tumor cells to undergo
apoptosis. Indeed, VC-A induced apoptosis in PDA cells as
demonstrated by the increased binding of Annexin V and cleavage of
PARP-1 and procaspases-3, -8 and -9. These results corroborate the
findings of another study that also reported cell cycle arrest and
induction of apoptosis in breast cancer cells by VC-A (17). The cleavage of procaspases-8 and -9
suggested that VC-A induces both the extrinsic and intrinsic
pathways of apoptosis. However, additional studies, such as an
increase in the expression of death receptors DR4 and DR5, are
required to confirm the activation of extrinsic pathway of
apoptosis by VC-A. Mitochondria plays a critical role in the
intrinsic pathway of apoptosis. Breakdown in mitochondrial
integrity and the release of cytochrome c and other
apoptogenic mediators are the best indicators of apoptotic cell
death via the intrinsic pathway of apoptosis. VC-A induced
mitochondrial depolarization and release of cytochrome c
indicating the involvement of mitochondrial or intrinsic pathway of
apoptosis in death of PDA cells by VC-A.
The generation of free radicals facilitates cancer
cell death by most chemotherapeutic agents and ionizing radiation
(26,27). Our findings demonstrated that
production of hydrogen peroxide is not involved in death of PDA
cells by VC-A. This finding contradicts a previous report in which
ROS was shown to be involved in killing of breast cancer by VC-A
(17). Our results, however, do
not rule out participation of other reactive oxygen species such as
superoxide anion in VC-A induced death of PDA cells.
Apoptosis is regulated by members of the Bcl-2
family of proteins that includes both pro- and anti-apoptotic
molecules (28,29). Members of the IAP family are also
potent inhibitors of apoptosis (30). Bcl-2 and Bcl-xL are two major
antiapoptotic Bcl-2 family members that reside in the mitochondrial
membrane and inhibit apoptosis by preventing the activation of
inner mitochondrial permeability transition pore and release of
proapoptogenic mitochondrial contents including cytochrome c
(29). VC-A inhibited Bcl-2 and
Bcl-xL in PDA cells. Bax and p-Bad which counteract antiapoptotic
Bcl-2 and Bcl-xL were either not affected or only partially reduced
by VC-A. Since both anti- and proapoptotic Bcl-2 family members
were inhibited or reduced the exact nature of the interplay between
antiapoptotic and proapoptotic members of Bcl-2 family in induction
of apoptosis in PDA cells by VC-A remains unresolved. On the
contrary, however, increase in Bax:Bcl-2 ratio after treatment with
VC-A suggests that VC-A may exert proapoptotic effect by displacing
Bcl-2 from mitochondria through Bax.
Prosurvival phosphatidylinositol-3
kinase/Akt/NF-κB/mTOR (PI3K/Akt/NF-κB/mTOR) is a major signaling
axis which controls cell proliferation, survival, apoptosis, and
malignant transformation (31).
PI3K/Akt signaling is frequently activated in a variety of
malignancies (32). p-Akt promotes
cell growth and survival by inactivating downstream substrates such
as Bad, procaspase-9, and Forkhead transcription factors (33,34).
Antiapoptotic NF-κB and progrowth mTOR signaling pathways are
downstream targets of activated Akt. NF-κB family of transcription
factors controls the expression of genes involved in immune and
inflammatory responses, cell proliferation, oncogenesis,
angiogenesis, and several Bcl-2 family members (35). NF-κB also plays a critical role in
resistance of cancer cells to anticancer therapies by protecting
them from apoptosis. mTOR, a 290-kDa serine-threonine kinase, which
controls cell growth, survival and division is activated in a
variety of human tumors (36). Our
results demonstrated that Akt, NF-κB and mTOR are constitutively
active in PDA cells and their expression was inhibited by VC-A.
VC-A also reduced the expression of several downstream effectors of
Akt, NF-κB and mTOR that regulate cell proliferation, cell cycle,
apoptosis and ribogenesis. These findings suggest that the
inhibition of Akt/NF-κB/mTOR signal transduction pathway plays a
critical role in induction of apoptosis in pancreatic cancer cells
by VC-A.
Collectively, results of this study indicate that
VC-A is a promising agent worthy of further development for the
treatment of pancreatic cancer.
Acknowledgements
This study was supported by an institutional grant
A10176 to S.C.G.
References
|
1
|
National Cancer Institute. Pancreatic
Cancer-National Cancer Institute, U.S. National Institutes of
Health. Cancer. https://www.cancer.gov/types/pancreatic.
Accessed 06-04-2010
|
|
2
|
Maitra A and Hruban RH: Pancreatic cancer.
Annu Rev Pathol. 3:157–188. 2008. View Article : Google Scholar
|
|
3
|
Li D, Xie K, Wolff R and Abbruzzese JL:
Pancreatic cancer. Lancet. 363:1049–1057. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mulcahy MF, Wahl AO and Small W Jr: The
current status of combined radiotherapy and chemotherapy for
locally advanced or resected pancreas cancer. J Natl Compr Canc
Netw. 3:637–642. 2005.PubMed/NCBI
|
|
5
|
Pino SM, Xiong HQ, McConkey D and
Abbruzzese JL: Novel therapies for pancreatic adenocarcinoma. Curr
Oncol Rep. 6:199–206. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vaccaro V, Sperduti I and Milella M:
FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N
Engl J Med. 365:768–769; author reply 769. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pestka JJ, Yike I, Dearborn DG, Ward MD
and Harkema JR: Stachybotrys chartarum, trichothecene mycotoxins,
and damp building-related illness: New insights into a public
health enigma. Toxicol Sci. 104:4–26. 2008. View Article : Google Scholar
|
|
8
|
Hossain MA, Ahmed MS and Ghannoum MA:
Attributes of Stachybotrys chartarum and its association with human
disease. J Allergy Clin Immunol. 113:200–208; quiz 209. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kilburn KH: Role of molds and mycotoxins
in being sick in buildings: Neurobehavioral and pulmonary
impairment. Adv Appl Microbiol. 55:339–359. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hardin BD, Kelman BJ and Saxon A: Adverse
human health effects associated with molds in the indoor
environment. J Occup Environ Med. 45:470–478. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Leatherman DL and Middlebrook JL: Effect
of emetine on T-2 toxin-induced inhibition of protein synthesis in
mammalian cells. J Pharmacol Exp Ther. 266:741–748. 1993.PubMed/NCBI
|
|
12
|
Ge HM, Jiao RH, Zhang YF, Zhang J, Wang YR
and Tan RX: Cytotoxicity and phytotoxicity of trichothecene
macrolides from Myrothecium gramminum. Planta Med. 75:227–229.
2009. View Article : Google Scholar
|
|
13
|
Abbas HK, Johnson BB, Shier WT, Tak H,
Jarvis BB and Boyette CD: Phytotoxicity and mammalian cytotoxicity
of macrocyclic trichothecene mycotoxins from Myrothecium
verrucaria. Phytochemistry. 59:309–313. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shifrin VI and Anderson P: Trichothecene
mycotoxins trigger a ribotoxic stress response that activates c-Jun
N-terminal kinase and p38 mitogen-activated protein kinase and
induces apoptosis. J Biol Chem. 274:13985–13992. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chung YJ, Jarvis B and Pestka J:
Modulation of lipopolysaccharide-induced proinflammatory cytokine
production by satratoxins and other macrocyclic trichothecenes in
the murine macrophage. J Toxicol Environ Health A. 66:379–391.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hughes BJ, Taylor MJ and Sharma RP:
Effects of verrucarin A and roridin A, macrocyclic trichothecene
mycotoxins, on the murine immune system. Immunopharmacology.
16:79–87. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Palanivel K, Kanimozhi V and Kadalmani B:
Verrucarin A alters cell-cycle regulatory proteins and induces
apoptosis through reactive oxygen species-dependent p38MAPK
activation in the human breast cancer cell line MCF-7. Tumour Biol.
35:10159–10167. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Palanivel K, Kanimozhi V, Kadalmani B and
Akbarsha MA: Verrucarin A induces apoptosis through ROS-mediated
EGFR/MAPK/Akt signaling pathways in MDA-MB-231 breast cancer cells.
J Cell Biochem. 115:2022–2032. 2014.PubMed/NCBI
|
|
19
|
Woldemichael GM, Turbyville TJ, Vasselli
JR, Linehan WM and McMahon JB: Lack of a functional VHL gene
product sensitizes renal cell carcinoma cells to the apoptotic
effects of the protein synthesis inhibitor verrucarin A. Neoplasia.
14:771–777. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang GH, Jarvis BB, Chung YJ and Pestka
JJ: Apoptosis induction by the satratoxins and other trichothecene
mycotoxins: Relationship to ERK, p38 MAPK, and SAPK/JNK activation.
Toxicol Appl Pharmacol. 164:149–160. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Amagata T, Rath C, Rigot JF, Tarlov N,
Tenney K, Valeriote FA and Crews P: Structures and cytotoxic
properties of trichoverroids and their macrolide analogues produced
by saltwater culture of Myrothecium verrucaria. J Med Chem.
46:4342–4350. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chin YW, Balunas MJ, Chai HB and Kinghorn
AD: Drug discovery from natural sources. AAPS J. 8:E239–E253. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Itokawa H, Morris-Natschke SL, Akiyama T
and Lee KH: Plant-derived natural product research aimed at new
drug discovery. J Nat Med. 62:263–280. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kerr JF, Winterford CM and Harmon BV:
Apoptosis. Its significance in cancer and cancer therapy. Cancer.
73:2013–2026. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cusack JC Jr: Overcoming antiapoptotic
responses to promote chemosensitivity in metastatic colorectal
cancer to the liver. Ann Surg Oncol. 10:852–862. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ramanathan B, Jan KY, Chen CH, Hour TC, Yu
HJ and Pu YS: Resistance to paclitaxel is proportional to cellular
total antioxidant capacity. Cancer Res. 65:8455–8460. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun Y and Rigas B: The thioredoxin system
mediates redox-induced cell death in human colon cancer cells:
Implications for the mechanism of action of anticancer agents.
Cancer Res. 68:8269–8277. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chao DT and Korsmeyer SJ: BCL-2 family:
Regulators of cell death. Annu Rev Immunol. 16:395–419. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Deveraux QL and Reed JC: IAP family
proteins - suppressors of apoptosis. Genes Dev. 13:239–252. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Altomare DA and Testa JR: Perturbations of
the AKT signaling pathway in human cancer. Oncogene. 24:7455–7464.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Datta SR, Dudek H, Tao X, Masters S, Fu H,
Gotoh Y and Greenberg ME: Akt phosphorylation of BAD couples
survival signals to the cell-intrinsic death machinery. Cell.
91:231–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo
P, Hu LS, Anderson MJ, Arden KC, Blenis J and Greenberg ME: Akt
promotes cell survival by phosphorylating and inhibiting a Forkhead
transcription factor. Cell. 96:857–868. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mayo MW and Baldwin AS: The transcription
factor NF-kappaB: Control of oncogenesis and cancer therapy
resistance. Biochim Biophys Acta. 1470:M55–M62. 2000.PubMed/NCBI
|
|
36
|
Bjornsti MA and Houghton PJ: The TOR
pathway: A target for cancer therapy. Nat Rev Cancer. 4:335–348.
2004. View
Article : Google Scholar : PubMed/NCBI
|