Introduction
Mutations in the BRAF oncogene are present in
over 50% of cutaneous melanomas(1), with the vast majority of the
mutations leading to the V600E substitution in the BRAF oncoprotein
and the constitutive activation of the MAPK/ERK signaling pathway,
which is crucial for melanoma growth, survival and invasion
(reviewed in refs. 2 and 3). In patients with BRAF-mutant melanoma,
inhibition of the oncogenic BRAF kinase with vemurafenib or
dabrafenib, or its downstream effector kinases MEK1/2 with
trametinib, yields objective response rates of 50–60 and of 20–30%,
respectively, and significantly prolongs progression-free and
overall survival with respect to dacarbazine chemotherapy (4–6).
Moreover, the combination of dabrafenib and trametinib is well
tolerated and associated with a higher response rate and improved
disease-free and overall survival as compared with single-agent
therapy (7–9).
Despite the impressive initial responses to BRAF
inhibitors (BRAFi), the long-term efficacy of these compounds is
limited by the emergence of drug resistance (reviewed in refs.
10–13). Multiple mechanisms of acquired
resistance to BRAFi have been described, involving both
reactivation of MAPK/ERK signaling and/or activation of alternative
pro-survival pathways. Restoration of MAPK/ERK signaling in the
presence of BRAFi has been shown to occur by activating mutations
in NRAS, MAP2K1 or MAP2K2, loss of NF1,
BRAFV600E amplification, overexpression of
the CRAF or COT1 kinases, as well as by the expression of splice
variants of BRAFV600E (10–13).
Alternative mechanisms of secondary resistance to BRAFi mainly
involve alterations leading to upregulation of the PI3K/AKT/mTOR
signaling axis. In this regard, mutations in AKT1,
AKT3, PIK3CA, PIK3CG, PIK3R2 and
PHLPP1, PTEN loss as well as overexpression and/or
activation of the receptor tyrosine kinases (RTKs) platelet-derived
growth factor receptor α and β, insulin-like growth factor 1
receptor, fibroblast growth factor receptor-3, and epidermal growth
factor receptor (EGFR), have been reported (10–16).
Notably, RTK signaling can also bypass mutant BRAF and activate ERK
via RAS (10,14,15).
Although a large body of experimental data is
available on the effects of BRAFi on melanoma proliferation and
survival, and on the molecular mechanisms involved in acquired
resistance to the growth-suppressive effects of these agents, much
less is known about the impact of BRAFi on melanoma cell migration
and invasion. In this regard, Halaban et al (17) showed that while vemurafenib
inhibited migration of BRAF-mutant melanoma cells, it increased
that of BRAF wild-type cells, as a result of drug-induced CRAF
activation. Girotti et al (18) demonstrated that BRAF-mutant
melanoma cells with acquired resistance to PLX4720, a drug
structurally related to vemurafenib (19), were more invasive, both in
vitro and in vivo, than the corresponding drug-sensitive
counterparts as a consequence of hyperactivation of the EGFR-SRC
family kinase-signal transducers and activator of transcription 3
(STAT3) signaling pathway. Increased invasiveness of BRAF-mutant
melanoma cells that had been rendered resistant to the BRAFi
SB590885 was reported by Vultur et al (20), who also showed activation of the
STAT3 pathway in these cells under basal conditions and upon
exposure to SB590885. Similarly, Zubrilov et al (21) found that in vitro induction
of vemurafenib resistance in BRAF-mutant melanoma cells was
associated with increased potential for metastasis. The
vemurafenib-resistant cells showed, indeed, enhanced migratory and
adherence functions, higher expression levels of several genes
involved in promotion of metastasis and increased capacity to form
lung and liver micro-metastasis in nude mice. Sanchez-Laorden et
al (22) demonstrated that
PLX4720 stimulated the invasive capacity of RAS-mutant melanoma
cells by a mechanism involving activation of the MEK/ERK pathway,
increased expression and secretion of IL-8 and induction of
protease-dependent invasion. Moreover, increased levels of
IL-8 mRNA and similar responses to PLX4720 were observed by
these authors in BRAF-mutant melanoma cells which had developed
resistance to the drug. On the contrary, vemurafenib was reported
to exert a moderate inhibition of the in vitro invasive
activity of a BRAFV600E/NRAS wild-type melanoma cell
line showing primary resistance to the antiproliferative effects of
the drug as a consequence of MET amplification and activation
(23).
To the best of our knowledge, no data are presently
available concerning the effects of dabrafenib on the metastatic
potential of BRAF-mutant melanoma cells. In the present study we,
therefore, evaluated the effect of dabrafenib on the in
vitro invasive capacity of melanoma cells sensitive or with
acquired resistance to the growth suppressive effects of the drug.
We also investigated whether inhibition of the PI3K/AKT/mTOR
signaling pathway could modulate the effects of dabrafenib on the
invasive behavior of the resistant cells.
Materials and methods
Cell cultures
The human melanoma cell line A375 was purchased from
the European Collection of Cell Cultures (Salisbury, UK) and
cultured in BioWhittaker™ RPMI-1640 medium (Lonza, Verviers,
Belgium) supplemented with 10% fetal bovine serum (FBS;
Sigma-Aldrich, St. Louis, MO, USA), 2 mM BioWhittaker™ L-glutamine
(Lonza), and 50 μg/ml BioWhittaker™ gentamicin (Lonza) (hereafter
referred to as complete medium, CM). The A375R subline with
acquired resistance to dabrafenib was generated by growing the
parental A375 cells in gradually increasing concentrations of
dabrafenib (from 1 nM up to 1.5 μM) over a period of 4 months.
After selection, A375R cells were maintained in CM supplemented
with 1.5 μM dabrafenib.
Drugs, chemicals and antibodies
Dabrafenib (GSK2118436A) and GSK2126458A were
dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich) at a final
concentration of 1.92 and 1 mM, respectively. Both drugs were
stored as stock solutions at −80°C and diluted in CM just before
use.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) was purchased from Sigma-Aldrich, dissolved at a
concentration of 5 mg/ml in Gibco™ phosphate-buffered saline (PBS;
Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) and stored
at 4°C.
For western blot analysis, the following rabbit
polyclonal antibodies were used: anti-human ERK1/2 (GTX17942;
GeneTex, Irvine, CA, USA), anti-human phospho-ERK1/2
(Thr185/Tyr187) (44-680G; Invitrogen), anti-human β-tubulin
(sc-9104; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA),
anti-vascular endothelial growth factor receptor-2 (VEGFR-2)
(sc-6251; Santa Cruz Biotechnology), anti-human AKT (9272; Cell
Signaling Technology, Inc., Beverly, MA, USA) and anti-human
phospho-AKT (Ser473) (9271; Cell Signaling Technology).
Reagents for sodium dodecyl sulphate-polyacrylamide
gel electrophoresis (SDS-PAGE) were all purchased from Bio-Rad
Laboratories, Inc. (Hercules, CA, USA).
DNA extraction and whole exome
sequencing
DNA extraction from A375 and A375R cell lines was
performed using the DNeasy Blood & Tissue kit (Qiagen GmbH,
Hilden, Germany) according to the manufacturer’s protocol, and
sheared with a Bioruptor sonicator (Diagenode, Liege, Belgium). DNA
libraries were prepared and captured using the SureSelect Human All
Exon kit (50 Mb; Agilent Technologies, Santa Clara, CA, USA)
according to the manufacturer’s instructions. DNA libraries were
sequenced by Genomix4life S.r.l., (Baronissi, Salerno, Italy) on
Illumina NextSeq 500 platform (Illumina, San Diego, CA, USA) with
100-bp paired-end reads.
Exome sequence analysis
Sequence reads were mapped to the reference human
genome assembly hg19 using Burrows-Wheeler Aligner (BWA-MEM)
version 0.7.12 (24). PCR
duplicates were removed using Picard 1.130 (http://broadinstitute.github.io/picard). The Genome
Analysis Toolkit (GATK) version 3.4 was used for realignment of
insertions and deletions, quality recalibration and variant calling
(25). Then, to create a
sub-selection of high-quality single nucleotide variations (SNVs),
a series of filtering options was applied, including minimum read
depth of 8x, SNV call quality score above 30, and non-reference
allele frequency above 20%. Variants were annotated with
information from RefGenes using wANNOVAR web-based tool (26).
Chemosensitivity assay
A375 and A375R cells were suspended in CM, seeded
(50 μl/well) into BD Falcon™ 96-well plates (BD Biosciences,
Bedford, MA, USA) and allowed to adhere at 37°C in a 5%
CO2 atmosphere for 18 h. Graded amounts of dabrafenib or
GSK2126458A were then added to the cells in 50 μl of CM. As a
control, A375 and A375R cells were treated with DMSO alone. The
plates were incubated at 37°C for five days and cell proliferation
was then evaluated by the MTT assay, as previously described
(27). Three replica wells were
used for each group. Drug concentration producing 50% inhibition of
cell growth (i.e. IC50), was calculated on the
regression line in which absorbance values at 595 nm were plotted
against the logarithm of drug concentration. In a different set of
experiments, A375R cells, plated as described above, were incubated
with DMSO alone, 100 nM dabrafenib, 20 nM GSK2126458A or a
combination of the two drugs for 5 days and then assayed for
proliferation using the MTT assay.
Cell treatment for western blot analysis
and invasion assay
A375 and A375R cells were suspended in CM, seeded
into BD Falcon™ 10-cm-dishes (BD Biosciences) and allowed to adhere
at 37°C in a 5% CO2 atmosphere for 18 h. Thereafter,
dabrafenib (100 nM) or DMSO alone was added to the cultures and the
dishes incubated at 37°C for additional 6 or 48 h. In a different
set of experiments, A375R cells, plated as described above, were
incubated with DMSO alone, 100 nM dabrafenib, 20 nM GSK2126458A or
a combination of the two drugs for 48 h. At the end of the
incubation period, melanoma cells were processed for western blot
analysis or tested for their ability to invade the extracellular
matrix (ECM) in vitro.
Western blot analysis
For analysis of ERK1/2, phosphorylated ERK1/2, AKT
and phosphorylated AKT expression, control and drug-treated cells
were recovered from culture, washed and total cellular extracts
were prepared as described previously (28). For analysis of VEGFR-2 expression,
melanoma cells were left in the culture dishes, washed, lysed
directly in SDS sample buffer (50 mM Tris-HCl pH 6.8, 100 mM
dithiothreitol, 2% SDS, 0.1% bromophenol blue and 10% glycerol) and
then boiled for 5 min. Proteins per sample (15 μg) were run on a 6%
(VEGFR-2) or 12% SDS-polyacrylamide gels, transferred to
nitrocellulose membranes (Amersham Biosciences, Buckinghamshire,
UK) and blocked with 5% non-fat milk in Tris-buffered saline
supplemented with 0.1% Tween-20 (TBST) for 1 h at room temperature.
The membranes were then incubated in the same solution, or in TBST
containing 5% bovine serum albumin (BSA), overnight at 4°C with
primary antibodies at the following dilutions: anti-ERK1/2,
anti-phospho-ERK1/2, anti-AKT and anti-β-tubulin 1:1,000;
anti-phospho-AKT 1:500; anti-VEGFR-2 1:200. The anti-β-tubulin
antibody was used as an internal standard for loading.
Immunodetection was carried out using appropriate horseradish
peroxidase-linked secondary antibodies and enhanced
chemiluminescence (ECL) or ECL Prime (phospho-AKT) detection
reagents (Amersham Biosciences). Where indicated, film was scanned
on a GS-710 Calibrated Imaging Densitometer and analyzed by means
of Quantity One Software Version 4.1.1 (Bio-Rad Laboratories).
Invasion assay in Boyden chambers
This assay was performed as previously described
(29). Briefly, control and
drug-treated melanoma cells were removed from culture, washed,
suspended in invasion medium (1 μg/ml heparin/0.1% BSA in
RPMI-1640) and loaded (2×105 cells) into the upper
compartment of Boyden chambers equipped with 8-μm pore diameter
polycarbonate filters (Nuclepore, Whatman Inc., Clifton, NJ, USA)
coated with 20 μg of BD Matrigel™ Basement Membrane Matrix (BD
Biosciences, hereafter referred to as Matrigel). Invasion medium
or, where indicated, invasion medium containing 20 ng/ml VEGF-A
(ImmunoTools, Friesoythe, Germany) was added to the lower
compartment of the chambers. For each experimental condition, three
Boyden chambers were set up. After incubation of the Boyden
chambers at 37°C in a 5% CO2 atmosphere for 4 h, the
filters were removed from the chambers and the cells were fixed in
ethanol for 5 min and stained in 0.5% crystal violet for 15 min.
The cells from the upper surface of the filter were removed by
wiping with a cotton swab and the migrated cells, attached to the
lower surface of the filters, were counted under the microscope.
Twelve microscopic fields (magnification, x200), randomly selected
on triplicate filters, were scored for each experimental
condition.
In a set of experiments, invasion assays were
performed in the presence of the anti-human VEGF-A monoclonal
antibody bevacizumab (5 μg/ml) (Avastin; Roche, Welwyn Garden City,
UK) or the corresponding control IgG1 immunoglobulins
(MAB002; R&D Systems, Minneapolis, MN, USA). A375R cells were
pre-incubated with the antibodies for 30 min at room temperature in
a rotating wheel. The cells were then loaded in the Boyden chambers
without removing the antibodies.
Three-dimensional spheroid invasion
assay
A375 and A375R spheroids were generated as described
by Naber et al (30) with
minor modifications. Briefly, melanoma cells were suspended
(4×104 cells/ml) in CM containing 0.24% methylcellulose
(Sigma-Aldrich) and seeded (100 μl/well) into non-adhesive,
round-bottom 96-well plates (Corning Inc., Corning, NY, USA).
Plates were centrifuged at 1250 x g for 90 min and then incubated
at 37°C in a 5% CO2 atmosphere for 18 h to allow the
cells to organize into three-dimensional spheroids (one spheroid in
each well). Spheroids were then harvested and individually embedded
between two layers of Matrigel as previously described (31) with minor modifications. Briefly, 50
μl of Matrigel diluted in RPMI-1640 medium (4 μg/ml), were added to
the wells of a flat-bottom 96-well plate, that was then incubated
at 37°C for 15 min to let the Matrigel solidify. Melanoma
spheroids, suspended in 100 μl of Matrigel diluted in invasion
medium (4 μg/ml), were then seeded in the Matrigel-precoated plate
(one spheroid/well, five replicates). After a 30-min incubation at
37°C to allow Matrigel polymerization, spheroids were overlaid with
100 μl of invasion medium and the plate incubated at 37°C for 48 h.
At the end of incubation, images of the invading spheroids were
taken using a Leica inverted microscope and a Canon digital camera
PowerShot G5.
Transient transfection with small
interfering RNA (siRNA) targeting AKT1 and drug treatment of the
transfected cells
Oligonucleotide siRNA targeting AKT1
(CUCACAGCCCUGAAGUACU) (siAKT) and the corresponding scramble
oligonucleotide (GAUCCUAUAUUCGGUUAGU) (siCTRL) to be used as a
control were purchased from Sigma-Proligo (The Woodlands, TX,
USA).
A375R cells were suspended in CM without
antibiotics, seeded into BD Falcon™ 6-well plates (BD Biosciences),
allowed to adhere at 37°C in a 5% CO2 atmosphere for 18
h, and then transfected with 10 nM siAKT or siCTRL using
Lipofectamine™ RNAiMAX reagent (Invitrogen) according to the
manufacturer’s protocol. Twenty-four hours after transfection, the
cells were incubated with 100 nM dabrafenib or DMSO alone. After 48
h of culture, the cells were collected and processed for western
blot analysis or tested for ability to invade the ECM.
Evaluation of VEGF-A and matrix
metalloproteinase-9 (MMP-9) secretion
A375 and A375R cells were suspended in CM, seeded
into BD Falcon 10 cm-dishes (BD Biosciences) and allowed to adhere
at 37°C in a 5% CO2 atmosphere for 18 h. Thereafter,
A375 cells were incubated with DMSO alone or 100 nM dabrafenib,
whereas A375R were incubated with DMSO alone, 100 nM dabrafenib, 20
nM GSK2126458A or a combination of the two drugs. After 48 h of
drug exposure, culture supernatants were collected, centrifuged at
600 x g for 10 min to remove cells in suspension and debris, and
frozen at −20°C until use. Cells were detached with a solution of
1.5 mM EDTA in PBS and counted to determine the total number of
cells in the culture. Quantification of the VEGF-A amount in the
culture supernatants was performed as previously described
(32). For MMP-9 evaluation,
culture supernatants were concentrated at least 5-fold in
Centriplus concentrators (Amicon, Beverly, MA, USA). The amount of
active MMP-9 in the culture supernatants was then determined using
the Quantikine ELISA Human MMP-9 immunoassay (R&D Systems)
according to the manufacturer’s protocol, and calibrated against a
standard curve. The amount of VEGF-A and MMP-9 was normalized to
the number of total cells counted in each culture at the time of
supernatant collection.
Statistical analysis
Statistical significance among different values of
drug IC50, different number of ECM invading cells and
different levels of VEGF-A and MMP-9 secretion, was assessed using
two-sided Student’s t-test analysis.
Results
Generation and characterization of the
A375R subline with acquired resistance to dabrafenib
To generate a cellular model of acquired resistance
to dabrafenib, the human melanoma cell line A375, which harbors the
BRAFV600E mutation and is highly sensitive to the growth
suppressive effects of BRAF inhibitors, was cultured with gradually
increasing concentrations of dabrafenib until a subline (i.e.
A375R) able to growth in the presence of 1.5 μM of the drug was
obtained. This concentration of dabrafenib corresponds to the peak
plasma concentration reported for patients treated with the drug at
the recommended dose of 150 mg BID (33).
A375 and A375R cells were initially analyzed for
sensitivity to the antiproliferative effects of dabrafenib, and for
expression of total and phosphorylated ERK1/2, either under basal
conditions or following a 6-h exposure to 100 nM of the drug. MTT
assays, performed after five days of cell culture with graded
concentrations of dabrafenib, revealed that the A375R subline was
10,000-fold more resistant to the growth inhibitory effect of this
drug than the parental cell line (Fig.
1A). A375R cells also expressed higher basal levels of
phosphorylated ERK1/2 as compared with A375 cells (Fig. 1B). Moreover, while dabrafenib, as
expected, markedly downregulated the level of phosphorylated ERK1/2
in A375 cells, it further increased it in the drug-resistant
subline (Fig. 1B).
Previous studies have shown that melanoma cells with
acquired resistance to the BRAFi vemurafenib, PLX4720 or SB590885
possess a more invasive phenotype (18,20–22),
whereas no data are available regarding the metastatic potential of
dabrafenib-resistant cells. Therefore, we evaluated the ability of
A375 and A375R cells to spontaneously invade the ECM using the
Boyden chamber assay. The results illustrated in Fig. 1C show that the dabrafenib-resistant
A375R cells were ~2-fold more invasive than the
dabrafenib-sensitive A375 cells.
The invasive behavior of A375 and A375R cells was
also investigated using an invasion assay based on
Matrigel-embedded spheroids, that mimics the three-dimensional
tumor architecture and incorporates components of the ECM (34). Consistent with results obtained in
Boyden chamber assays A375R spheroids invaded into Matrigel to a
higher extent as compared with parental A375 cells (Fig. 1D).
Finally, to investigate whether reactivation of the
MAPK/ERK pathway in A375R cells could be dependent on mutations in
HRAS, KRAS, NRAS, MAP2K1, MAP2K2
or on NF1 loss, whole exome sequencing was performed in both
A375 and A375R cell lines. No mutations in the three RAS
genes or in MAP2K1 and MAP2K2, and no loss of
NF1 were detected in A375 and A375R cells.
Dabrafenib impairs invasiveness and
VEGF-A secretion in A375 cells, whereas it stimulates these
functions in A375R cells
We previously showed that an autocrine loop
sustained by the interaction of VEGF-A with VEGFR-2 promoted
melanoma cell ability to migrate and invade the ECM in vitro
(35). Recently, Liu et al
(36) demonstrated that PLX4720
inhibited VEGF production in a melanoma cell line and xenografts,
and that downregulation of VEGF expression occurred in the tumor of
melanoma patients treated with PLX4720. Furthermore, Beazley-Long
et al (37) reported that
vemurafenib decreased VEGF-A expression in BRAF-mutant and
drug-sensitive melanoma cells, whereas it increased this cytokine
in BRAF-wild-type melanoma cells. Therefore, to investigate the
effects of dabrafenib on the metastatic potential of melanoma
cells, A375 and A375R cells were treated with 100 nM of the drug
(or with DMSO as vehicle control) for 48 h and then analyzed for
the ability to invade the ECM under basal conditions and in
response to exogenously added VEGF-A, as well as for VEGF-A
secretion and VEGFR-2 expression.
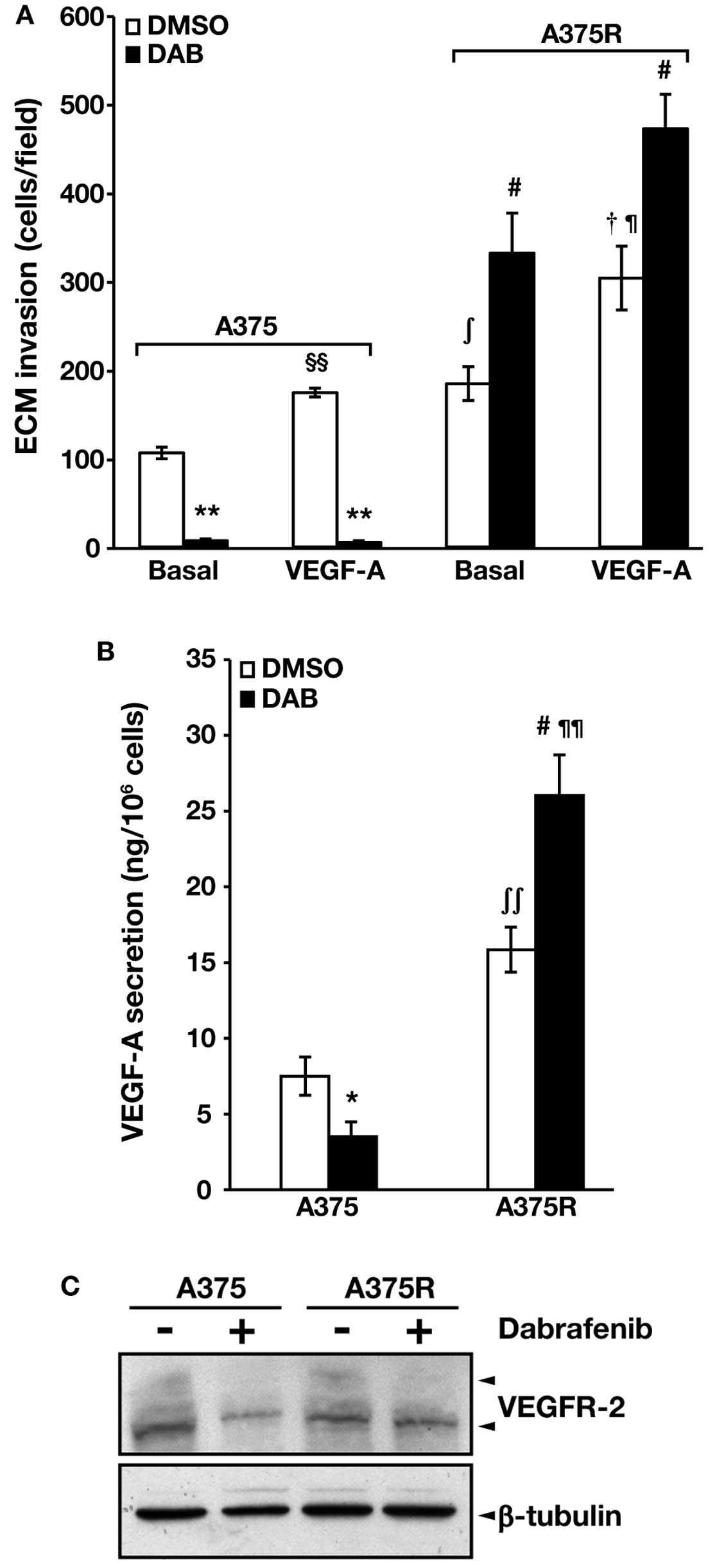
The results illustrated in Fig. 2A show that both cell lines
responded to VEGF-A with an increase of ECM invasion, and that not
only spontaneous but also VEGF-A-induced invasive capacity was
higher in the A375R cell line as compared with the parental
counterpart. Dabrafenib significantly inhibited spontaneous and
VEGF-A-induced ECM invasion in A375 cells. In contrast, A375R cell
invasiveness was markedly stimulated by the drug in both
conditions.
VEGF-A secretion in the culture supernatants of A375
and A375R cells, and VEGFR-2 expression in the two cell lines were
evaluated by specific ELISA and western blot analysis,
respectively. We found that basal secretion of VEGF-A was higher in
A375R cells as compared with parental A375 cells (Fig. 2B). Moreover, VEGF-A secretion was
markedly impaired in dabrafenib-treated A375 cells, whereas it was
significantly increased in drug-treated A375R cells. Exposure to
dabrafenib caused a reduction of VEGFR-2 expression in A375 cells
but it did not substantially affect the receptor levels in A375R
cells (Fig. 2C). Of note, the
effects of 1.5 μM dabrafenib on A375R invasiveness, VEGF-A
secretion and VEGFR-2 expression were comparable to those produced
by 100 nM of the drug (data not shown).
Bevacizumab inhibits dabrafenib-induced
invasion in A375R cells
The findings that A375R cells were more invasive and
secreted higher levels of VEGF-A than A375 cells, and responded to
dabrafenib with a further increase of ECM invasion and VEGF-A
secretion, prompted us to investigate whether inhibition of VEGF-A
binding to its cognate RTKs could impair spontaneous and
dabrafenib-induced invasiveness of A375R cells. To this end, the
cells were treated with 100 nM dabrafenib or DMSO alone for 48 h,
and then assayed for ECM invasion in the absence or in the presence
of the anti-VEGF-A monoclonal antibody bevacizumab (38,39),
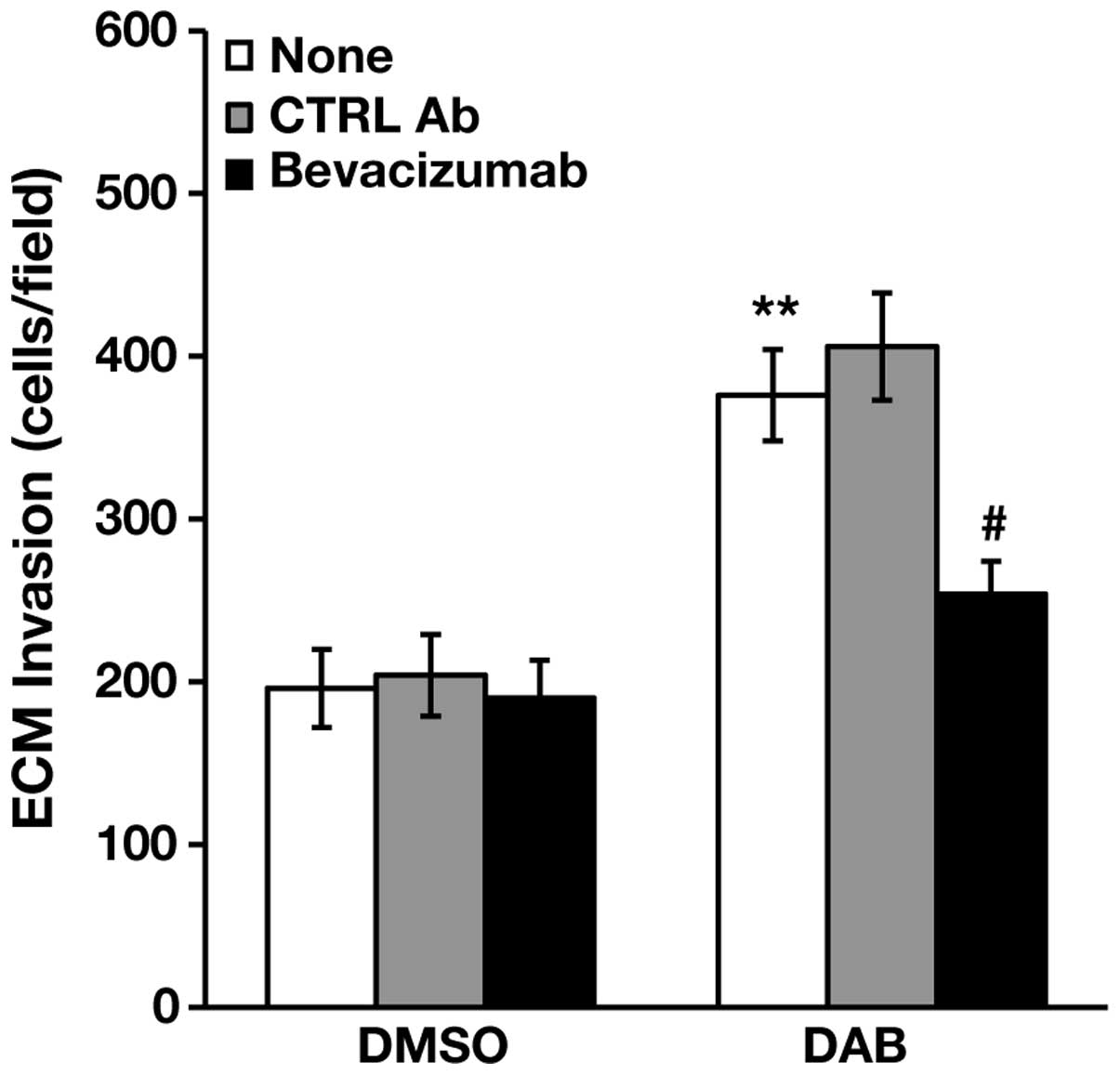
or of a control antibody. As illustrated in Fig. 3, bevacizumab did not affect ECM
invasion of DMSO-treated cells, whereas it abrogated
dabrafenib-induced stimulation of A375R cell invasiveness.
AKT silencing prevents dabrafenib-induced
stimulation of the invasive capacity of A375R cells
VEGF-A binding to VEGFR-2 can activate a number of
downstream signaling pathways, including the MAPK/ERK and PI3K/AKT
cascades (40,41). Moreover, previous studies (42) demonstrated that VEGF-A
overproduction and concomitant expression of its receptors promote
melanoma cell growth and survival through the activation of the
MAPK/ERK and PI3K/AKT signaling pathways. We therefore investigated
whether, in addition to reactivation of the MAKP/ERK signaling in
the presence of dabrafenib, A375R cells also showed hyperactivation
of the PI3K/AKT pathway. Western blot analysis of total and
phosphorylated AKT in A375 and A375R cells, showed that the
drug-resistant subline expressed higher basal levels of
phosphorylated AKT as compared with the parental cell line
(Fig. 4A). Moreover, in both cell
lines exposure to 100 nM dabrafenib for 48 h enhanced the levels of
phosphorylated AKT (Fig. 4A).
Exome sequencing performed in A375 and A375R cells did not reveal
mutations in the AKT1, AKT3, PIK3CA,
PIK3CG, PIK3R2 and PHLPP1 genes, or
PTEN loss.
To establish whether hyperactivation of the PI3K/AKT
pathway could underlie the increased invasive capacity of A375R
cells, AKT1 was silenced in these cells using specific siRNAs. The
results illustrated in Fig. 4B
show that total AKT levels were efficiently reduced in the
siAKT-transfected cells exposed to DMSO or to 100 nM dabrafenib for
48 h. Notably, DMSO-treated and dabrafenib-treated
siAKT-transfected cells also showed a marked reduction of
phospho-AKT levels. This finding indicates that in A375R cells,
basal and dabrafenib-induced phosphorylation of AKT mainly involve
the AKT1 isoform. As observed for bevacizumab, AKT1 silencing
failed to reduce basal invasiveness of A375R cells, whereas it
completely abrogated upregulation of ECM invasion induced by
dabrafenib (Fig. 4C).
The PI3K/mTOR inhibitor GSK2126458A
impairs invasiveness and VEGF-A secretion induced by dabrafenib in
A375R cells
Based on the key role played by MAPK/ERK signaling
reactivation and PI3K/AKT/mTOR signaling upregulation in acquired
resistance to BRAFi, several therapeutic approaches co-targeting
the MAPK/ERK and the PI3K/AKT/mTOR pathways in patients with
BRAF-mutant melanoma are underway to improve the efficacy and
durability of clinical response (12; https://www.clinicaltrials.gov/).
We therefore investigated whether co-treatment of
A375R cells with the dual PI3K/mTOR inhibitor GSK2126458A could
antagonize the stimulating effects exerted by dabrafenib on ECM
invasion and VEGF-A secretion. Preliminary experiments were also
performed to evaluate A375R cell sensitivity to the growth
inhibitory effect of GSK2126458A and to select the drug
concentration to be used in combination with dabrafenib.
In agreement with previous findings (43), MTT proliferation assays, performed
after five days of cell exposure to graded concentrations of
GSK2126458A, revealed that the dabrafenib-resistant subline was
responsive to the PI3K/mTOR inhibitor, displaying a drug
IC50 value comparable to that of the parental cells
(Fig. 5A).
The clinically achievable concentration of 20 nM
GSK2126458A was then selected for the subsequent experiments. This
drug concentration inhibited A375R proliferation by ~70%, either
alone or in combination with dabrafenib (Fig. 5B). Moreover, a 48 h-exposure to 20
nM GSK2126458A completely abrogated AKT phosphorylation in both
DMSO-treated and dabrafenib-treated A375R cells (Fig. 5C) without affecting cell viability
(data not shown).
A375R cells were exposed to DMSO, 20nMGSK2126458A,
100 nM dabrafenib or a combination of the two drugs for 48 h and
then tested for ECM invasion and VEGF-A secretion. Invasiveness of
A375R cells was not substantially affected by GSK2126458A alone
(Fig. 6A). However, the PI3K/mTOR
inhibitor abrogated dabrafenib-induced stimulation of ECM invasion
(Fig. 6A). In agreement with the
invasion assays, VEGF-A secretion was not significantly altered in
A375R exposed to GSK2126458A alone (Fig. 6B). On the other hand, the PI3K/mTOR
inhibitor was able to completely counteract the upregulation of
VEGF-A release caused by the BRAFi (Fig. 6B).
The PI3K/mTOR inhibitor GSK2126458A
impairs MMP-9 secretion induced by dabrafenib in A375R cells
MMPs, a large family of proteolytic enzymes able to
degrade the ECM, play a critical role in many physiological
processes and are also implicated in tumor angiogenesis and
metastatic spreading of cancer cells (reviewed in refs. 44,45).
In melanoma, increased expression of MMP-1, MMP-2 and MMP-9 has
been associated with an invasive phenotype (46). Moreover, VEGF-A has been reported
to upregulate the expression of certain MMPs, including MMP-9, in
endothelial cells and ovarian cancer cells (47,48).
We, therefore, determined MMP-9 levels in culture
supernatants of A375 and A375R cells, treated with DMSO or 100 nM
dabrafenib for 48 h. In A375R cells, MMP-9 secretion was also
evaluated after treatment with 20 nM GSK2126458A, alone or in
combination with dabrafenib.
The results illustrated in Fig. 7 show that A375 cells secreted low
amounts of MMP-9, which were not affected by dabrafenib treatment.
A375R displayed levels of MMP-9 secretion 2-fold higher than those
detected in A375 cells. Moreover, in the resistant subline the
release of MMP-9 was significantly stimulated by dabrafenib.
Similarly to the results obtained with VEGF-A, GSK2126458A alone
did not alter basal MMP-9 secretion in A375R cells, but abrogated
the upregulation caused by dabrafenib treatment (Fig. 7).
Discussion
In the present study we describe for the first time
the effects of dabrafenib on the invasive capacity of BRAF-mutant
melanoma cells sensitive or with acquired resistance to the
antiproliferative activity of this BRAFi, as well as the modulating
effect of GSK2126458A, a dual PI3K/mTOR inhibitor, on the responses
of the resistant cells to dabrafenib.
The invasive behavior of melanoma cells with
acquired resistance to dabrafenib, the only BRAFi presently
approved for melanoma therapy beside vemurafenib, has not been
investigated so far. However, increased invasiveness of melanoma
cells with secondary resistance to the BRAFi PLX4720 (18,22),
SB590885 (20), or vemurafenib
(21) has been previously reported
and linked to hyperactivation of the SRC family kinase-STAT3
signaling pathway (18,20), upregulation of several gene
products involved in the metastatic process (21), or increased expression of IL-8
(22). Our results showing that
the dabrafenib-resistant cell line A375R displayed higher invasive
capacity and higher production of VEGF-A and MMP-9 as compared with
the drug-sensitive parental cell line A375 are consistent with
these previous findings and provide further insight into melanoma
biological characteristics associated with acquired resistance to
BRAFi. Notably, in A375 cells exposure to dabrafenib impaired
invasiveness, VEGFR-2 expression and VEGF-A secretion, and did not
affect MMP-9 release. In contrast, in A375R cells treatment with
dabrafenib caused an increase of the invasive capacity, as well as
of VEGF-A and MMP-9 release, and did not substantially alter
VEGFR-2 expression. These data point out that cells with acquired
resistance to dabrafenib, not only are more invasive than the
sensitive parental cells, but also respond to the drug with a
further increase of their metastatic potential.
In the present study, we focused our investigation
on the possible mechanisms underlying the effects of dabrafenib on
the invasive capacity of the resistant cells, and on the
possibility to modulate those effects. Indeed, there are some
contrasting data regarding the clinical outcome that could be
achieved by continuing BRAFi monotherapy in patients who had
developed secondary resistance. Carlino et al (49) showed that melanoma cells with in
vitro or in vivo acquired resistance to dabrafenib still
responded to the BRAFi with reduced proliferation, even though they
did not undergo apoptosis. These authors also reported a rapid
acceleration of the disease upon dabrafenib suspension in a
melanoma patient who had been treated with the drug for about six
additional months after progression. Accordingly, clinical benefit
of continuing BRAFi beyond RECIST disease progression was
demonstrated in a large group of melanoma patients by Chan et
al (50). In contrast, a
preclinical study conducted with two different primary human
melanoma xenograft models demonstrated that tumors with acquired
resistance to vemurafenib were dependent on this drug for
continuous proliferation, and that withdrawal of vemurafenib caused
regression of established drug-resistant tumors (51). Furthermore, Hartsough et al
(52) showed that melanoma cells
with acquired resistance to PLX4720 due to the expression of mutant
BRAF splice variants grow more efficiently, both in vitro
and in vivo, in the presence of PLX4720 than in the absence
of the inhibitor. Although randomized prospective trials are
necessary to establish the role that prolonged BRAF inhibition may
have in patients undergoing disease progression, the findings that
dabrafenib stimulated ECM invasion and VEGF-A and MMP-9 production
in A375R cells, cast, at least, a note of caution for the
continuation of BRAFi monotherapy beyond progression. In
vivo release of VEGF-A by melanoma cells can promote tumor
growth and metastasis by both paracrine and autocrine mechanisms,
namely supporting angiogenesis and stimulating melanoma cell
proliferation, migration and invasion (35,40–42).
Accordingly, elevated expression of VEGF-A and its receptors has
been associated with melanoma progression (53). Moreover, MMP-9 is an important
regulator of tumor angiogenesis and vasculogenesis, and has also
been implicated in the formation of the metastatic niche (44). Although continuing single agent
dabrafenib beyond progression could suppress the growth of those
cells that within a heterogeneous tumor cell population have
remained sensitive to the drug, it might accelerate the metastatic
spreading of the drug-resistant cells. A better clinical benefit
might be achieved by combining dabrafenib with a drug able to
counteract the stimulating effects of the BRAFi on melanoma cell
invasiveness.
In the present study, we show that
dabrafenib-induced stimulation of A375R invasive capacity was
markedly inhibited in the presence of bevacizumab, a therapeutic
anti-VEGF-A mAb which prevents the interaction of the angiogenic
factor with its receptors VEGFR-1 and VEGFR-2 (38,39).
This finding supports the hypothesis that dabrafenib-induced
stimulation of ECM invasion in A375R cells mainly relies on
drug-mediated upregulation of VEGF-A secretion and the subsequent
engagement of signaling pathways regulating melanoma cell
invasiveness. In this regard, the PI3K/AKT/mTOR signaling cascade,
which is one of the signaling pathways activated upon binding of
VEGF-A to VEGFR-1 and VEGFR-2 (40), appears to play a crucial role.
Indeed, AKT1 silencing by specific siRNAs abrogated the stimulating
effect of dabrafenib on A375R cell invasive capacity. Moreover,
when A375R cells were treated with dabrafenib in combination with
GSK2126458A, phosphorylation of AKT was entirely suppressed and
this molecular event was accompanied by abrogation of
dabrafenib-induced stimulation of ECM invasion and of VEGF-A and
MMP-9 production. Several studies have demonstrated that
co-targeting the PI3K/AKT/mTOR is an effective strategy to reverse
acquired resistance to the growth suppressive effects of BRAFi
(43,54–58).
Our data, showing that A375R cells were highly responsive to
GSK2126458A, as a single agent or in combination with dabrafenib,
are consistent with those studies, and show that inhibition of the
PI3K/AKT/mTOR pathway can also efficiently counteract
dabrafenib-induced stimulation of the invasive capacity of melanoma
cells with required resistance to this agent. Of note, neither
bevacizumab, nor anti-AKT1 siRNA were able to impair basal
invasiveness of A375R. Moreover, GSK2126458A alone, at the
concentration selected for the combination with dabrafenib, did not
alter basal ECM invasion and VEGF-A and MMP-9 secretion in A375R
cells even if efficiently impaired AKT phosphorylation. Taken
together, these data suggest that the higher basal invasive
capacity displayed by A375R cells does not depend on VEGF-A
engagement of its cognate RTKs or on AKT hyperactivation observed
in these cells. It is possible that one or more mechanisms
previously described to be involved in the increased aggressiveness
of melanoma cells resistant to other BRAFi also underlie the
elevated invasive behavior of the dabrafenib-resistant cell line
generated in the present study. However, further investigations are
required to address this issue.
In conclusion, our results demonstrate that
dabrafenib effectively impairs the metastatic potential of
BRAF-mutant melanoma cells sensitive to the antiproliferative
activity of the drug. They also provide evidence that melanoma
cells with acquired resistance to dabrafenib possess a more
invasive phenotype which is further stimulated by exposure to the
inhibitor via upregulation of VEGF-A secretion and activation of
the PI3K/AKT/mTOR signaling pathway. Our data suggest that in
patients progressing on dabrafenib therapy, a regimen combining the
BRAFi with bevacizumab or with an inhibitor of the PI3K/AKT/mTOR
pathway might provide a clinical benefit higher than that of
extended BRAFi monotherapy. Notably, the combination of vemurafenib
and cobimetinib (MEK inhibitor) with or without bevacizumab is
being testing in a randomized phase 2 clinical trial in patients
with unresectable or metastatic BRAF-mutant melanoma (NCT01495988;
https://www.clinicaltrials.gov/). A
phase II clinical study is also ongoing to assess the efficacy and
safety of the combination of LGX818 (BRAFi) with the PI3K inhibitor
BKM120, or other targeted agents, in patients with unresectable or
metastatic BRAF-mutant melanoma after relapse on LGX818
(NCT01820364; https://www.clinicaltrials.gov/).
Acknowledgements
The present study was supported by the Italian
Association for Cancer Research (AIRC, Investigator Grant Project
17585) and by the Italian Ministry of Health (Grant
5PerMille-2010). The authors thank GlaxoSmithKline for the supply
of dabrafenib and GSK2126458A, Maurizio Inzillo (Office of External
Relations and Communication, IDI-IRCCS) for the artwork and Laura
Bonmassar (Laboratory of Molecular Oncology, IDI-IRCCS) for
technical help. The authors wish also to thank Professor Maria
Marino (Department of Science, Biomedical Sciences and Technology
Section, University ‘Roma Tre’, Rome, Italy) for helpful
discussion.
Abbreviations:
|
BRAFi
|
BRAF inhibitors
|
|
BSA
|
bovine serum albumin
|
|
CM
|
complete medium
|
|
DMSO
|
dimethyl sulfoxide
|
|
ECL
|
enhanced chemiluminescence
|
|
ECM
|
extracellular matrix
|
|
EGFR
|
epidermal growth factor receptor
|
|
IC
|
inhibitory concentration
|
|
MMP
|
matrix metalloproteinase
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
siRNA
|
small interfering RNA
|
|
PBS
|
phosphate-buffered saline
|
|
RTK
|
receptor tyrosine kinase
|
|
SDS-PAGE
|
sodium dodecyl sulphate-polyacrylamide
gel electrophoresis
|
|
SEM
|
standard error of the mean
|
|
SNV
|
single nucleotide variation
|
|
STAT3
|
signal transducers and activators of
transcription-3
|
|
TBST
|
Tris-buffered saline/Tween-20
|
|
VEGFR
|
vascular endothelial growth factor
receptor
|
References
|
1
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lopez-Bergami P: The role of mitogen- and
stress-activated protein kinase pathways in melanoma. Pigment Cell
Melanoma Res. 24:902–921. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sullivan RJ and Flaherty K: MAP kinase
signaling and inhibition in melanoma. Oncogene. 32:2373–2379. 2013.
View Article : Google Scholar
|
|
4
|
Chapman PB, Hauschild A, Robert C, Haanen
JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et
al; BRIM-3 Study Group. Improved survival with vemurafenib in
melanoma with BRAF V600E mutation. N Engl J Med. 364:2507–2516.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Flaherty KT, Robert C, Hersey P, Nathan P,
Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, et
al; METRIC Study Group. Improved survival with MEK inhibition in
BRAF-mutated melanoma. N Engl J Med. 367:107–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hauschild A, Grob JJ, Demidov LV, Jouary
T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr,
Kaempgen E, et al: Dabrafenib in BRAF-mutated metastatic melanoma:
A multicentre, open-label, phase 3 randomised controlled trial.
Lancet. 380:358–365. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Flaherty KT, Infante JR, Daud A, Gonzalez
R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N,
et al: Combined BRAF and MEK inhibition in melanoma with BRAF V600
mutations. N Engl J Med. 367:1694–1703. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Robert C, Karaszewska B, Schachter J,
Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R,
Grange F, Mortier L, et al: Improved overall survival in melanoma
with combined dabrafenib and trametinib. N Engl J Med. 372:30–39.
2015. View Article : Google Scholar
|
|
9
|
Long GV, Stroyakovskiy D, Gogas H,
Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A,
Grob JJ, et al: Dabrafenib and trametinib versus dabrafenib and
placebo for Val600 BRAF-mutant melanoma: A multicentre,
double-blind, phase 3 randomised controlled trial. Lancet.
386:444–451. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sullivan RJ and Flaherty KT: Resistance to
BRAF-targeted therapy in melanoma. Eur J Cancer. 49:1297–1304.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Holderfield M, Deuker MM, McCormick F and
McMahon M: Targeting RAF kinases for cancer therapy: BRAF-mutated
melanoma and beyond. Nat Rev Cancer. 14:455–467. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fedorenko IV, Gibney GT, Sondak VK and
Smalley KSM: Beyond BRAF: Where next for melanoma therapy? Br J
Cancer. 112:217–226. 2015. View Article : Google Scholar :
|
|
13
|
Spagnolo F, Ghiorzo P, Orgiano L,
Pastorino L, Picasso V, Tornari E, Ottaviano V and Queirolo P:
BRAF-mutant melanoma: Treatment approaches, resistance mechanisms,
and diagnostic strategies. Onco Targets Ther. 8:157–168. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yadav V, Zhang X, Liu J, Estrem S, Li S,
Gong XQ, Buchanan S, Henry JR, Starling JJ and Peng SB:
Reactivation of mitogen-activated protein kinase (MAPK) pathway by
FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in
human B-RAF V600E mutant melanoma. J Biol Chem. 287:28087–28098.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sabbatino F, Wang Y, Wang X, Flaherty KT,
Yu L, Pepin D, Scognamiglio G, Pepe S, Kirkwood JM, Cooper ZA, et
al: PDGFRα up-regulation mediated by sonic hedgehog pathway
activation leads to BRAF inhibitor resistance in melanoma cells
with BRAF mutation. Oncotarget. 5:1926–1941. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang J, Huang SK, Marzese DM, Hsu SC,
Kawas NP, Chong KK, Long GV, Menzies AM, Scolyer RA, Izraely S, et
al: Epigenetic changes of EGFR have an important role in BRAF
inhibitor-resistant cutaneous melanomas. J Invest Dermatol.
135:532–541. 2015. View Article : Google Scholar
|
|
17
|
Halaban R, Zhang W, Bacchiocchi A, Cheng
E, Parisi F, Ariyan S, Krauthammer M, McCusker JP, Kluger Y and
Sznol M: PLX4032, a selective BRAFV600E kinase
inhibitor, activates the ERK pathway and enhances cell migration
and proliferation of BRAF melanoma cells. Pigment Cell Melanoma
Res. 23:190–200. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Girotti MR, Pedersen M, Sanchez-Laorden B,
Viros A, Turajlic S, Niculescu-Duvaz D, Zambon A, Sinclair J, Hayes
A, Gore M, et al: Inhibiting EGF receptor or SRC family kinase
signaling overcomes BRAF inhibitor resistance in melanoma. Cancer
Discov. 3:158–167. 2013. View Article : Google Scholar
|
|
19
|
Tsai J, Lee JT, Wang W, Zhang J, Cho H,
Mamo S, Bremer R, Gillette S, Kong J, Haass NK, et al: Discovery of
a selective inhibitor of oncogenic B-Raf kinase with potent
antimelanoma activity. Proc Natl Acad Sci USA. 105:3041–3046. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vultur A, Villanueva J, Krepler C, Rajan
G, Chen Q, Xiao M, Li L, Gimotty PA, Wilson M, Hayden J, et al: MEK
inhibition affects STAT3 signaling and invasion in human melanoma
cell lines. Oncogene. 33:1850–1861. 2014. View Article : Google Scholar
|
|
21
|
Zubrilov I, Sagi-Assif O, Izraely S,
Meshel T, Ben-Menahem S, Ginat R, Pasmanik-Chor M, Nahmias C,
Couraud PO, Hoon DSB, et al: Vemurafenib resistance selects for
highly malignant brain and lung-metastasizing melanoma cells.
Cancer Lett. 361:86–96. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sanchez-Laorden B, Viros A, Girotti MR,
Pedersen M, Saturno G, Zambon A, Niculescu-Duvaz D, Turajlic S,
Hayes A, Gore M, et al: BRAF inhibitors induce metastasis in RAS
mutant or inhibitor-resistant melanoma cells by reactivating MEK
and ERK signaling. Sci Signal. 7:ra302014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vergani E, Vallacchi V, Frigerio S, Deho
P, Mondellini P, Perego P, Cassinelli G, Lanzi C, Testi MA,
Rivoltini L, et al: Identification of MET and SRC activation in
melanoma cell lines showing primary resistance to PLX4032.
Neoplasia. 13:1132–1142. 2011. View Article : Google Scholar
|
|
24
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M, et al: The Genome Analysis Toolkit: A MapReduce framework for
analyzing next-generation DNA sequencing data. Genome Res.
20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang H and Wang K: Genomic variant
annotation and prioritization with ANNOVAR and wANNOVAR. Nat
Protoc. 10:1556–1566. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pepponi R, Marra G, Fuggetta MP,
Falcinelli S, Pagani E, Bonmassar E, Jiricny J and D’Atri S: The
effect of O6-alkylguanine-DNA alkyltransferase and
mismatch repair activities on the sensitivity of human melanoma
cells to temozolomide, 1,3-bis(2-chloroethyl)1-nitrosourea, and
cisplatin. J Pharmacol Exp Ther. 304:661–668. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Caporali S, Falcinelli S, Starace G, Russo
MT, Bonmassar E, Jiricny J and D’Atri S: DNA damage induced by
temozolomide signals to both ATM and ATR: Role of the mismatch
repair system. Mol Pharmacol. 66:478–491. 2004.PubMed/NCBI
|
|
29
|
Ruffini F, D’Atri S and Lacal PM:
Neuropilin-1 expression promotes invasiveness of melanoma cells
through vascular endothelial growth factor receptor-2-dependent and
-independent mechanisms. Int J Oncol. 43:297–306. 2013.PubMed/NCBI
|
|
30
|
Naber HP, Wiercinska E, Ten Dijke P and
van Laar T: Spheroid assay to measure TGF-β-induced invasion. J Vis
Exp. 57:e33372011.
|
|
31
|
Merz C, Strecker A, Sykora J, Hill O,
Fricke H, Angel P, Gieffers C and Peterziel H: Neutralization of
the CD95 ligand by APG101 inhibits invasion of glioma cells in
vitro. Anticancer Drugs. 26:716–727. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lacal PM, Failla CM, Pagani E, Odorisio T,
Schietroma C, Falcinelli S, Zambruno G and D’Atri S: Human melanoma
cells secrete and respond to placenta growth factor and vascular
endothelial growth factor. J Invest Dermatol. 115:1000–1007. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Falchook GS, Long GV, Kurzrock R, Kim KB,
Arkenau HT, Brown MP, Hamid O, Infante JR, Millward M, Pavlick A,
et al: Dose selection, pharmacokinetics, and pharmacodynamics of
BRAF inhibitor dabrafenib (GSK2118436). Clin Cancer Res.
20:4449–4458. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Smalley KS, Lioni M and Herlyn M: Life
isn’t flat: Taking cancer biology to the next dimension. In Vitro
Cell Dev Biol Anim. 42:242–247. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lacal PM, Ruffini F, Pagani E and D’Atri
S: An autocrine loop directed by the vascular endothelial growth
factor promotes invasiveness of human melanoma cells. Int J Oncol.
27:1625–1632. 2005.PubMed/NCBI
|
|
36
|
Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo
JA, Chen JQ, Li HS, Watowich SS, Yang Y, et al: BRAF inhibition
increases tumor infiltration by T cells and enhances the antitumor
activity of adoptive immunotherapy in mice. Clin Cancer Res.
19:393–403. 2013. View Article : Google Scholar
|
|
37
|
Beazley-Long N, Gaston K, Harper SJ,
Orlando A and Bates DO: Novel mechanisms of resistance to
vemurafenib in melanoma - V600E B-Raf reversion and switching
VEGF-A splice isoform expression. Am J Cancer Res. 5:433–441.
2014.
|
|
38
|
Ferrara N, Hillan KJ, Gerber HP and
Novotny W: Discovery and development of bevacizumab, an anti-VEGF
antibody for treating cancer. Nat Rev Drug Discov. 3:391–400. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Keating GM: Bevacizumab: A review of its
use in advanced cancer. Drugs. 74:1891–1925. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Koch S, Tugues S, Li X, Gualandi L and
Claesson-Welsh L: Signal transduction by vascular endothelial
growth factor receptors. Biochem J. 437:169–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Goel HL and Mercurio AM: VEGF targets the
tumour cell. Nat Rev Cancer. 13:871–882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Graells J, Vinyals A, Figueras A, Llorens
A, Moreno A, Marcoval J, Gonzalez FJ and Fabra A: Overproduction of
VEGF concomitantly expressed with its receptors promotes growth and
survival of melanoma cells through MAPK and PI3K signaling. J
Invest Dermatol. 123:1151–1161. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Greger JG, Eastman SD, Zhang V, Bleam MR,
Hughes AM, Smitheman KN, Dickerson SH, Laquerre SG, Liu L and
Gilmer TM: Combinations of BRAF, MEK, and PI3K/mTOR inhibitors
overcome acquired resistance to the BRAF inhibitor GSK2118436
dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther.
11:909–920. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kessenbrock K, Plaks V and Werb Z: Matrix
metalloproteinases: Regulators of the tumor microenvironment. Cell.
141:52–67. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gialeli C, Theocharis AD and Karamanos NK:
Roles of matrix metalloproteinases in cancer progression and their
pharmacological targeting. FEBS J. 278:16–27. 2011. View Article : Google Scholar
|
|
46
|
Orgaz JL and Sanz-Moreno V: Emerging
molecular targets in melanoma invasion and metastasis. Pigment Cell
Melanoma Res. 26:39–57. 2013. View Article : Google Scholar
|
|
47
|
Ghosh S, Basu M and Roy SS: ETS-1 protein
regulates vascular endothelial growth factor-induced matrix
metalloproteinase-9 and matrix metalloproteinase-13 expression in
human ovarian carcinoma cell line SKOV-3. J Biol Chem.
287:15001–15015. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Claesson-Welsh L and Welsh M: VEGFA and
tumour angiogenesis. J Intern Med. 273:114–127. 2013. View Article : Google Scholar
|
|
49
|
Carlino MS, Gowrishankar K, Saunders CAB,
Pupo GM, Snoyman S, Zhang XD, Saw R, Becker TM, Kefford RF, Long
GV, et al: Antiproliferative effects of continued mitogen-activated
protein kinase pathway inhibition following acquired resistance to
BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther.
12:1332–1342. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chan MMK, Haydu LE, Menzies AM, Azer MWF,
Klein O, Lyle M, Clements A, Guminski A, Kefford RF and Long GV:
The nature and management of metastatic melanoma after progression
on BRAF inhibitors: Effects of extended BRAF inhibition. Cancer.
120:3142–3153. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Das Thakur M, Salangsang F, Landman AS,
Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M and Stuart
DD: Modelling vemurafenib resistance in melanoma reveals a strategy
to forestall drug resistance. Nature. 494:251–255. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hartsough EJ, Basile KJ and Aplin AE:
Beneficial effects of RAF inhibitor in mutant BRAF splice
variant-expressing melanoma. Mol Cancer Res. 12:795–802. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mehnert JM, McCarthy MM, Jilaveanu L,
Flaherty KT, Aziz S, Camp RL, Rimm DL and Kluger HM: Quantitative
expression of VEGF, VEGF-R1, VEGF-R2, and VEGF-R3 in melanoma
tissue microarrays. Hum Pathol. 41:375–384. 2010. View Article : Google Scholar
|
|
54
|
Jiang CC, Lai F, Thorne RF, Yang F, Liu H,
Hersey P and Zhang XD: MEK-independent survival of B-RAFV600E
melanoma cells selected for resistance to apoptosis induced by the
RAF inhibitor PLX4720. Clin Cancer Res. 17:721–730. 2011.
View Article : Google Scholar
|
|
55
|
Shi H, Kong X, Ribas A and Lo RS:
Combinatorial treatments that overcome PDGFRβ-driven resistance of
melanoma cells to V600EB-RAF inhibition. Cancer Res. 71:5067–5074.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Atefi M, von Euw E, Attar N, Ng C, Chu C,
Guo D, Nazarian R, Chmielowski B, Glaspy JA, Comin-Anduix B, et al:
Reversing melanoma cross-resistance to BRAF and MEK inhibitors by
co-targeting the AKT/mTOR pathway. PLoS One. 6:e289732011.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Su F, Bradley WD, Wang Q, Yang H, Xu L,
Higgins B, Kolinsky K, Packman K, Kim MJ, Trunzer K, et al:
Resistance to selective BRAF inhibition can be mediated by modest
upstream pathway activation. Cancer Res. 72:969–978. 2012.
View Article : Google Scholar
|
|
58
|
Lassen A, Atefi M, Robert L, Wong DJ,
Cerniglia M, Comin-Anduix B and Ribas A: Effects of AKT inhibitor
therapy in response and resistance to BRAF inhibition in melanoma.
Mol Cancer. 13:832014. View Article : Google Scholar : PubMed/NCBI
|