Introduction
Lung carcinoma, the most common type of tumor, is
currently the leading cause of tumor-related deaths world-wide.
Lung cancer is a bronchogenic carcinoma and histologically
subdivided into small cell lung cancer (SCLC) and non-small cell
lung cancers (NSCLC). Lung adenocarcinoma, the most common subtype
of NSCLC, is the most prevalent pathological form of lung cancer
(1–3). Although surgical therapy,
chemotherapy, radiation therapy and targeted therapy have made
significant progress in recent years, the prognosis of lung
adenocarcinoma is still poor (4).
Thus, a more detailed understanding of the key biomarkers and
molecular mechanisms of initiation, development and progression of
lung adenocarcinoma is extremely important for improving the
diagnosis, prevention and treatment of this disease (5,6).
The Aurora kinases family is comprised of three
members: AURK-A, -B and -C. These kinase members are key regulators
of mitosis and multiple signaling pathways. Human AURKA gene maps
to chromosome 20q13.2, and is more extensively studied, especially
in tumor fields (7). AURKA
functions as an oncogene and overexpressed in several kinds of
cancer including malignancies breast, and colon cancers, as well as
in neuroblastoma (7,8). Due to essential roles of AURKA in
mitotic entry, DNA damage checkpoint recovery and centrosome and
spindle maturation, the inhibition of AURKA expression is a
promising therapeutic for multiple cancers. With the development of
AURKA inhibitors, several clinical trials using AURKA inhibitors in
multiple tumor types have been applied, especially Alisertib, a
potent and selective inhibitor currently in phase III. The results
of AURKA inhibitors from clinical trials indicated the complexity
of such treatment in cancers, which depends on many factors. To
select those patients that better react to AURKA inhibitors, and
test the cooperative effect of AURKA inhibitors with different
antitumoral drugs should be explored in future studies (7,9,10).
RNA interference (RNAi), the process of sequence-specific
post-transcriptional gene silencing, is a revolutionary tool for
the analysis of gene function and gene therapy for cancer and other
diseases (11).
To our knowledge, this is the first study associated
with the relationship between AURKA and lung adenocarcinoma. We
confirmed that AURKA is highly expressed in lung adenocarcinoma
tissues and human lung adenocarcinoma cell lines. Furthermore, we
found that knockdown of AURKA in human lung adenocarcinoma could
inhibit cell growth and proliferation in vitro. Importantly,
AURKA has cooperative effects with VCR on suppressing human lung
adenocarcinoma proliferation. Therefore, our results provide novel
insights into AURKA as a therapeutic target for lung
adenocarcinoma.
Materials and methods
Main reagents
Mouse anti-human AURKA polyclonal antibody was
purchased from ProteinTech Group (Wuhan, Hubei, China); mouse
anti-human AURKA monoclonal antibody was purchased from Abcam
(Abcam, Cambridge, UK); Taq DNA polymerase was obtained from
Fermentas, Inc. (Waltham, MA, USA); Lipofectamine 2000, Opti-MEM
and the SuperScript III reverse transcriptase (RT) kit were
purchased from Invitrogen (Carlsbad, CA, USA); Glyceraldehyde
3-phosphate dehydrogenase (GAPDH) and bovine serum albumin were
purchased from Sigma Chemical Co. (St. Louis, MO, USA); VCR were
purchased from Shenzhen Wan Le Pharmaceutical Co., Ltd. (Shenzhen,
Guangdong, China); cell culture media, fetal bovine serum (FBS) and
other supplementary materials were purchased from Gibco Co. (Grand
Island, NY, USA).
Cell culture
Human lung adenocarcinoma H1299 cells and A549 cells
were obtained from the Cell Bank of Chinese Academy of Sciences
(Shanghai, China) in 2013 and were identified by STR (short tandem
repeat) method in 2014. All cells were cultured in DMEM (Dulbecco’s
modified Eagle’s medium) (Gibco Co.) supplemented with 10% fetal
calf serum (FCS) and grown in a humidified incubator at 37°C and 5%
CO2.
Tissue collection
From January 2005 to January 2010, 101 patients with
lung adenocarcinoma underwent resection in the Kunshan First
People’s Hospital affiliated to Jiangsu University. All cases of
lung adenocarcinoma were clinically and pathologically proven. This
study was approved by the medical ethics committee of the Kunshan
First People’s Hospital with the reference number: 10. All
participants have provided verbal informed consent to participate
in this study. We recorded participant consents through the
telephone communication. This consent procedure was approved by the
ethics committees.
Immunohistochemical detection of AURKA in
lung adenocarcinoma tissues
Immunohistochemistry (IHC) studies were performed
according to the manufacturer’s instructions. Briefly, 3 μm-thick
sections was deparaffinized and rehydrated, then incubated in 3%
hydrogen peroxide for 15 min to block endogenous peroxidase
activity. These tissue slides were boiled in EDTA buffer (pH 9.0)
for 10 min for antigen retrieval. At room temperature, 10% normal
rabbit serum was introduced for blocking non-specific binding. At
4°C refrigerator, the slides was incubated with polyclonal antibody
against AURKA at a dilution of 1:100 in PBS for 1 h, rinsed five
times with PBS, they were incubated with goat anti-mouse IgG
conjugated with horseradish peroxidase for 30 min at room
temperature. The histologic sections were developed with DAB
(3,3′-diaminobenzidine-tetrahydrochloride-dihydrate) and lightly
counterstained with haematoxylin. The histologic sections were read
using light microscopy.
Each case was scored according to the percentage of
positive cells to total cancer cells and the staining intensity of
the positive cells. Regarding cell counting under microscope, at
least 10 high-power fields were randomly selected. The area of
staining was divided into four levels as follows: no staining of
cells in any microscopic fields was scored 0; <30% of tissue
stained positive was scored 1; between 30 and 60% stained positive
was scored 2; >60% stained positive was scored 3. In each slice,
no staining, weak staining, moderate staining and strong staining
were scored as 0, 1, 2, and 3, respectively.
AURKA expression and prognosis
A total of 443 samples from 4 institutions [Moffitt
Cancer Center (HLM), University of Michigan Cancer Center (UM), the
Dana-Farber Cancer Institute (DFCI) and Memorial Sloan-Kettering
Cancer Center (MSK)] were used to investigate the association of
the AURKA expression and prognosis in lung adenocarcinoma, as
previously published (12).
Patients were separated into high or low AURKA expression groups
based on the first quantile (25%) of the AURKA expression values of
total samples. Kaplan-Meier survival analysis was used to estimate
survival curves and difference between curves was evaluated by
log-rank test. Multivariate Cox proportional hazards regression
with covariate age, gender, and stage was carried out to measure
the independent prognostic factors. All tests were two-tailed and
p<0.05 were considered significant.
RT-PCR (Reverse transcription PCR)
analysis
The TRIzol reagent (Invitrogen) following the
protocol of the manufacturer (Invitrogen). RNA (1 μg) was subjected
to reverse transcription. The PCR primers used were as follows: for
AURKA: forward 5′-GCCCTGTCTTACTGTCATTCG-3′ and reverse
5′-AGGTCTCTTGGTATGTGTTTGC-3′; for GAPDH: forward
5-TGACTTCAACAGCGACACCCA-3′ and reverse 5′-CACCCTGTTGCTGTAGCCAAA-3′;
PCR products were separated by electrophoresis in 1% agarose gel,
visualized by staining with ethidium bromide and photographed under
ultraviolet light.
Recombination lentivirus generation and
cell infection
The human AURKA-specific small interfering RNA
(siRNA) sequence is 5′-GAAAGCTCCACATCAATAA-3′, designed with an
online software of Invitrogen using AURKA sequence (GeneBank code:
NM_003600) as a reference. The non-silencing (NS) sequence
(5′-TTCTCCGAACGTGTCACGT-3′) was used as scrambled control that has
been widely used (13). The short
hairpin RNA (shRNA) cassette against AURKA is
5′-CCGGCAGAAAGCTCCACATCAATAATTCAAGAGA
TTATTGATGTGGAGCTTTCTGTTTTTG-3′, with two cohesive ends for ligation
into the pGCSIL-GFP vector. The double stranded shRNA
oligonucleotide were ligated into pGCSIL-GFP vector linearized by
restriction enzyme EcoRI and AgeI.
Next, lentiviral vector that expressed the
AURKA-specific siRNA or negative control siRNA, together with
pHelper 1.0 and pHelper 2.0 plasmids were co-transfected into
HEK293T cells with Lipofectamine 2000 for lentivirus generation,
according to the manufacturer’s instructions (Invitrogen). After 48
h of transfection, the lentiviral particles were harvested and
purified with ultracentrifugation. Due to the produced lentiviruses
carrying green fluorescence protein (GFP), the viral titer was
determined by counting green cells with serial dilutions under
fluorescence microscopy at 5 days after infection. For lentivirus
infection, H1299 and A549 cells were grown in 6-well plates at
70–80% confluence and infected with AURKA-specific siRNA lentivirus
or control lentivirus at MOI of 20. Five days after infection,
cells expressing GFP protein were observed using fluorescence
microscopy to determine the infection efficiency. There were two
experimental groups for each cell line: LV-AURKA infected cells
(AURKA-siRNA) and LV-NS infected cells (scr-siRNA).
Real-time PCR analysis
Total RNA was initially extracted from cultured lung
adenocarcinoma cells using TRIzol (Invitrogen) and treated with
RNase-free DNase I. Standard reverse transcription reaction was
performed using a Promega M-MLV cDNA synthesis kit following the
manufacturer’s instructions. The real-time reverse transcription
polymerase chain reaction was performed using the SYBR Green
One-Step qRT-PCR kit (Invitrogen) according to the kit’s procedure
manual. GAPDH was used as an internal control. The PCR primers used
were: AURKA: forward 5′-GCCCTGTCTTACT GTCATTCG-3′, AURKA: reverse
5′-AGGTCTCTTGGTAT GTGTTTGC-3′; GAPDH: forward 5′-TGACTTCAACAGC
GACACCCA-3′, and GAPDH: reverse 5′-CACCCTGTTGCT GTAGCCAAA-3. The
relative gene expression levels were calculated using the
2−ΔΔCT algorithm.
Cellomics to test inhibition of lung
adenocarcinoma cell proliferation following knockdown of AURKA
The monolayer culture growth rate was determined by
using a Cellomics Arrayscan (Thermo Fisher Scientific Inc.,
Waltham, MA, USA). Briefly, after the lung adenocarcinoma H1299
cells were infected with virus for 3 days, cells were seeded into
96-well plates and cultured in a humidified atmosphere of 5%
CO2 at 37°C. The Cell viability was measured at 0, 1, 2,
3, 4 and 5 days with Cellomics Arrayscan to observe the cell growth
with GFP signal. Consequently, the statistical analysis of the data
collected was performed to create a growth curve for the six days.
Each experiment was performed in triplicate.
MTT assay
The effects of AURKA silence on proliferation of
A549 cells were analyzed by MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)
(Sigma Chemical Co.). The A549 cells were plated at a final
concentration of 5×103 cells/well in 96-well culture
plates for different culture times. MTT (10 μl) (5 mg/ml in PBS)
was added to each well and incubated for an additional 3 h at 37°C.
The formazan crystals were dissolved in 100 μl of DMSO, and the
absorbance was read at 490 nm by an ELISA reader (ELx808, Bio-Tek
Instruments, Winooski, VT, USA).
Colony-formation assay
H1299 cells in all experimental groups were
trypsinized and resuspended in complete medium. Two groups
(scr-siRNA, AURKA-siRNA) of H1299 cells were plated in 96-well
plates at the rate of 500 cells per perforation. Three compound
perforations were set in each experimental group. The medium was
changed and cells were monitored every 3 days. After 2 weeks of
culture, the cells were washed with PBS. These perforations were
scanned and photographed with Cellomics ArrayScan, the number and
size of clones within the perforations were analyzed.
Flow cytometry analysis
The infected cells were synchronized by exposure to
serum-free medium for 24 h to induce starvation. Then adherent
cells were harvested by trypsinization, washed twice with ice-cold
PBS, fixed in 70% ethanol and incubated for 30 min at 4°C. After
the ethanol was discarded by centrifugation, the fixed cells
suspended in PI/RNase/PBS (100 μg/ml propidium iodide and 10 μg/ml
RNase A) for 45 min at room temperature in the dark. After the
suspension was filtered through a 50-μm nylon mesh, the DNA content
of the stained nuclei was analyzed by a flow cytometer to determine
the percentage of cells for each phase of the cell cycle. Each
experiment was performed in triplicate.
A total of 1.0×106 cells were collected
and washed with ice-cold PBS for twice. Cells were resuspended in
100 μl of Annexin V binding buffer, incubated with APC labeled
Annexin V at room temperature for 15 min. Apoptotic cells were
detected by a flow cytometer. Each experiment was performed in
triplicate.
Western blot analysis
The cell pellets were lysed in lysis buffer that was
supplemented with protease and phosphatase inhibitor cocktails.
Total protein (50 μg) was separated by SDS-PAGE and electroblotted
onto nitrocellulose membranes after protein quantitation using
Coomassie brilliant blue assay. Membranes were blocked by 5%
non-fat dry milk and incubated for 1 h with mouse monoclonal
antibodies against Aurora-A, WEE1, CDK4, EGFR, PAK4, RAF-1, CCND2,
CCND3. After incubation with the secondary antibodies
(peroxidase-conjugated anti-mouse IgG) for 1 h, protein bands were
visualized by enhanced chemiluminescence. GAPDH was used as an
internal positive control.
BrdU incorporation assay
The A549 cells infected with control lentivirus or
AURKA-siRNA lentivirus were cultured for 72 h at 37°C in a
humidified incubator with 5% CO2. Then cells were
trypsinized, resuspended, spread onto 96-well plates and VCR was
added after 12 h. The treated cells were cultured for 24 and 96 h,
respectively, and incubated with BrdU for 4 h. Subsequently, the
cells were fixed, washed and incubated with mouse anti-BrdU
antibody for 1 h and horseradish peroxidase-conjugated secondary
antibodies for 30 min following the manufacturer’s protocol
(Chemicon International Inc., Temecula, CA, USA). The immune
complexes were detected by the subsequent
3,3′,5,5′-Tetramethyl-benzidine (TMB) substrate reaction, and the
levels of BrdU incorporated into cells were quantified by measuring
the absorbance at 490 nm using a microplate reader (Bio-Rad 680,
Bio-Rad, Hercules, CA, USA).
Chemotherapy and apoptosis assay
To investigate whether the transfection with
lentivirus encoding a AURKA siRNA increases the chemosensitivity of
lung adenocarcinoma cells, A549 cells were treated with VCR at 50,
100 nM for 48 h after transfection with AURKA-siRNA and scr-siRNA,
respectively.
Then A549 cells in each group were harvested and
cells stained with the Annexin V apoptosis kit (Invitrogen)
according to the manufacturer’s instructions. Analysis of apoptosis
were performed using a FACScan flow cytometer
(Becton-Dickinson).
Statistical analysis
All quantitative data are represented as mean ± SD
in this study. Statistical analysis was performed by Student’s
t-test and one-way ANOVA using GraphPad Prism 5.0 software.
p<0.05 was considered to be statistically significant.
Results
The relationship between AURKA
expression, pathological characteristics and survival of lung
adenocarcinoma patients
Clinical characteristics of the 101 lung
adenocarcinoma patients including age, gender, tumor
differentiation, lymph node status, are summarized in Table I. There were 56 males and 45
females, aged from 30 to 80 years, with a median age of 62 years.
Of the patients, 46 demonstrated no lymph node metastasis (N0),
whereas 55 were identified with lymph node involvement (N+). The
grades of differentiation were 24 with grade I (well
differentiated) and 77 with grade II or III (moderately to poorly
differentiated). No significant association was found between AURKA
overexpression and clinicopathological features such as age,
gender, tumor differentiation, lymph node status in our study.
 | Table IDistribution of Aurora-A status in
lung adenocarcinoma according to clinicopathological
characteristics. |
Table I
Distribution of Aurora-A status in
lung adenocarcinoma according to clinicopathological
characteristics.
|
Characteristics | Number of
patients | High | Low | p-value |
|---|
| Gender |
| Male | 56 | 28 | 28 | 0.742 |
| Female | 45 | 24 | 21 | |
| Age (years) |
| <60 | 41 | 17 | 24 | 0.317 |
| ≥60 | 60 | 30 | 30 | |
|
Differentiation |
| Well | 24 | 9 | 13 | 0.122 |
| Moderate-poor | 77 | 43 | 34 | |
| Nodal status |
| N0 | 46 | 24 | 22 | 0.956 |
| N1 | 55 | 29 | 26 | |
Finally, the AURKA expression and prognosis in lung
adenocarcinoma was tested. AURKA was highly expressed in lung
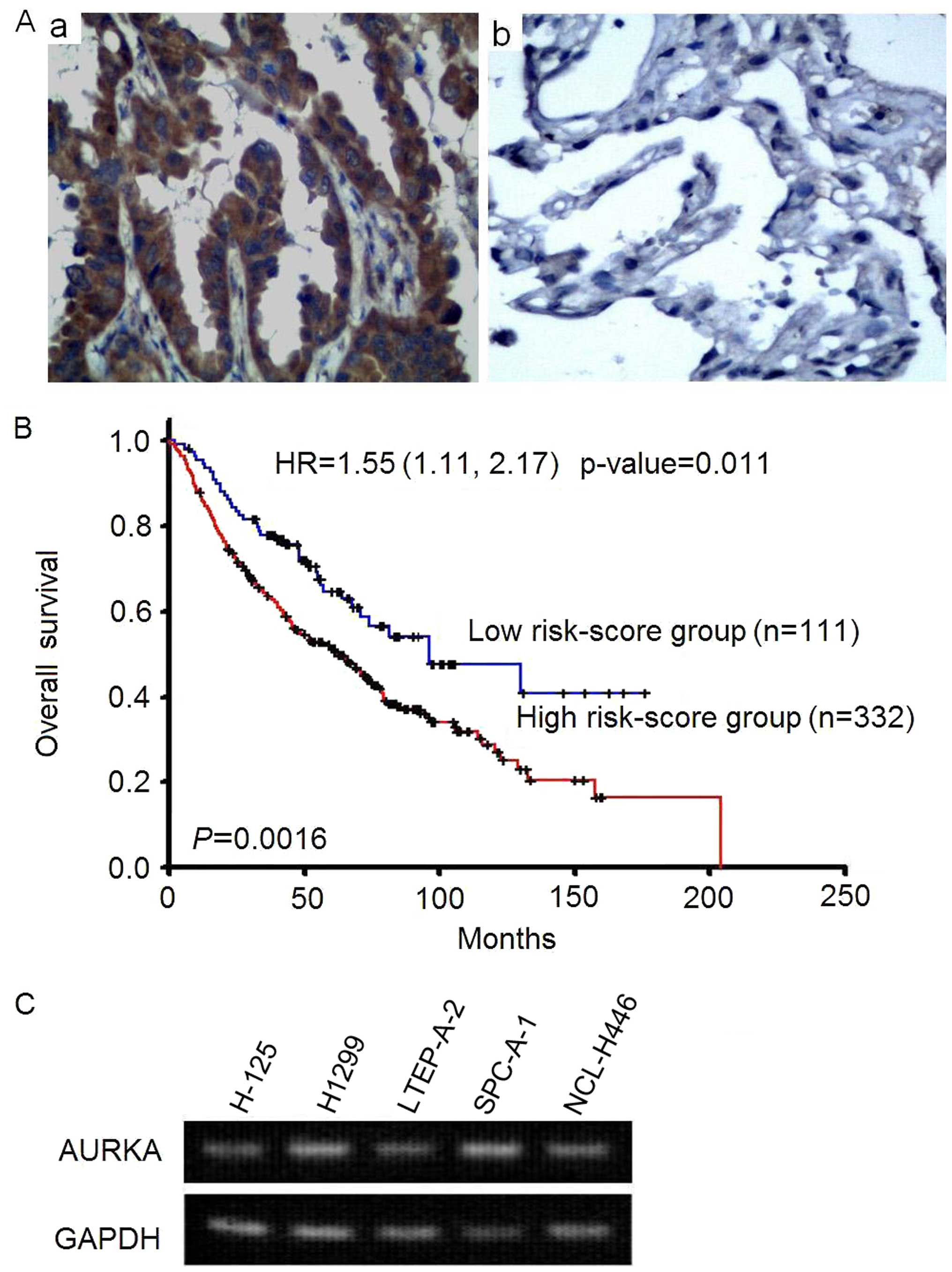
adenocarcinoma tissue (Fig. 1A).
Patients with high AURKA expression had shorter overall survival
than low expression group (p=0.0016, log-rank test). The adjusted
hazard ratio is 1.55 (95% CI=1.11–2.17, p=0.011) of AURKA
expression. It indicated that AURKA expression was an independent
prognostic factor after adjusted the effects of age, gender, and
stage (Fig. 1B).
To determine the expression of AURKA in lung
carcinoma cells, RT-PCR assay was performed. As showed in Fig. 1C, AURKA was highly expressed in
lung adenocarcinoma cell lines H-125, H1299, LTEP-A-2, SPC-A-1 and
human small cell lung cancer NCL-H446 cells. These results suggests
a correlation between the overexpression of AURKA and the
occurrence of NSCLC.
Lentivirus-mediated RNAi can efficiently
block expression of AURKA
To further illuminate the role of AURKA in lung
adenocarcinoma, we constructed the lentivirus-delivered
AURKA-specific siRNA vector (AURKA-siRNA) and scramble-siRNA vector
(scr-siRNA). Fluorescent microscope was used to investigate the
lentiviral infection efficiency. The results showed that >90% of
the cells exhibited the green fluorescence indicative of infection
after the transfection (Fig. 2A).
To determine the silencing efficiency, the expression levels of
AURKA mRNA and protein were detected by real-time PCR and western
blotting. The results indicated that the levels of RNA (Fig. 2B) and protein (Fig. 2C) expression of AURKA were
dramatically decreased in both H1299 and A549 cells compared to
scr-siRNA treatment groups. Thus, these results confirmed that the
AURKA-siRNA could downregulate the AURKA expression
effectively.
Inhibition of cell growth of lung
adenocarcinoma cells by depletion of AURKA
We tested the effect of AURKA-siRNA on the cell
viability of H1299 and A549 cells in vitro. Cellomics
analysis showed that AURKA knockdown significantly inhibited cell
growth of H1299 cells comparing with scr-siRNA treatment, and the
difference was more pronounced with time-dependent manner
(p<0.01) (Fig. 3A). The results
of the colony formation assay show that the number of colonies in
the AURKA-siRNA group (4.67±2.08) was significantly less than that
in the scr-siRNA group (19.33±2.52) in H1299 cells (p<0.01)
(Fig. 3B). These results
demonstrate that the reduction in AURKA expression decreases the
ability of H1299 cells to form colonies.
MTT assay was performed to study the effect of
AURKA-siRNA on A549 cell growth. As shown in Fig. 3C, A549 cells show a significant
(p<0.01) reduction in cell viability 5 days after infection. The
results suggests that silencing of AURKA gene inhibits the
proliferation of lung adenocarcinoma cells.
Knockdown of AURKA leads to alterations
in cell cycle of lung adenocarcinoma cells
In order to study the mechanisms underlying
RNAi-mediated proliferation inhibition, the changes in the cell
cycle was detected by flow cytometric analysis of the DNA content.
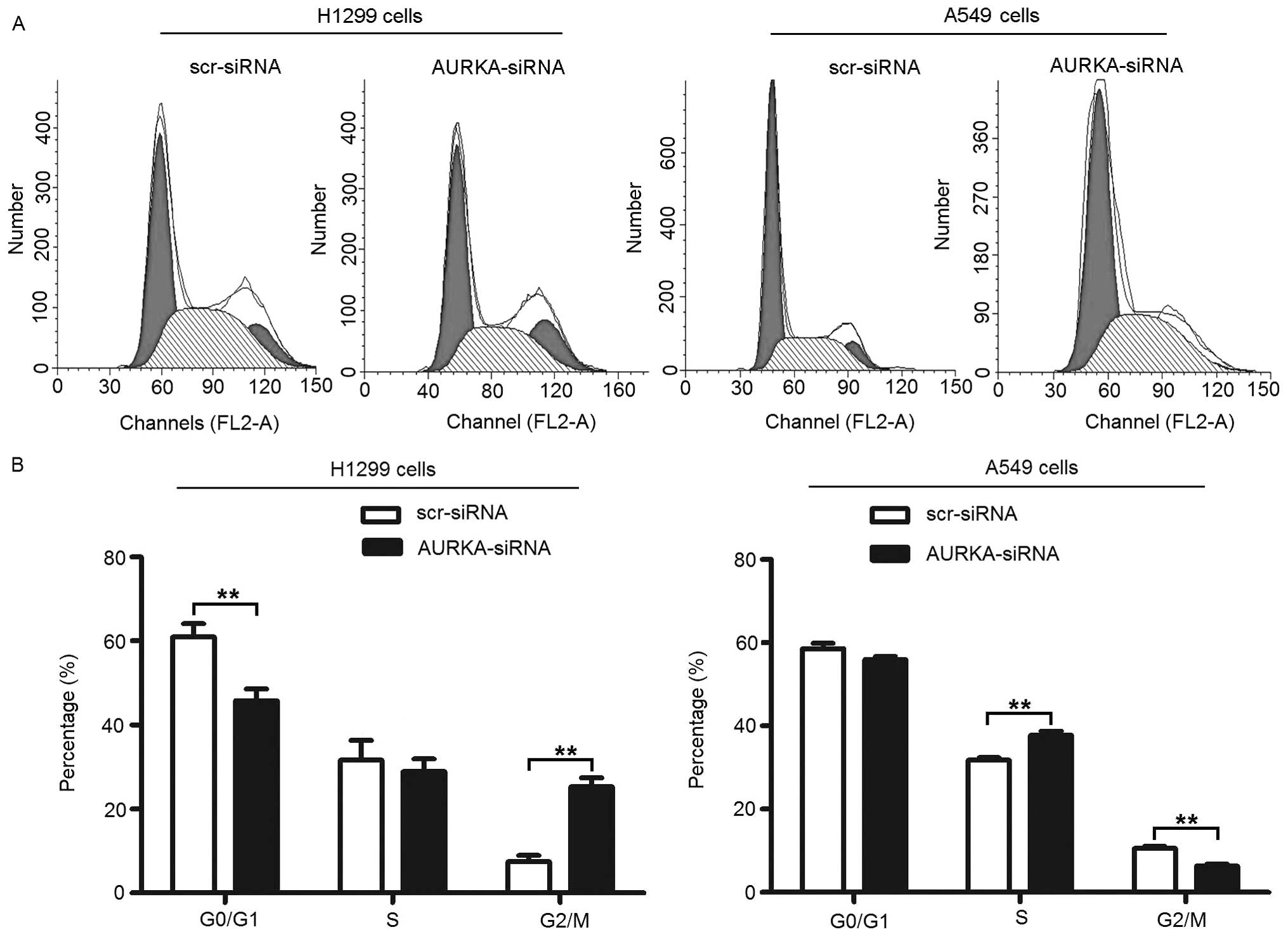
As shown in Fig. 4A and B,
treatment with AURKA-siRNA results in an increase in the percentage
of H1299 cells in the G2/M phase from 7.40±1.57% to 25.28±2.18%
(p<0.01). In accordance with this increase in the percentage of
cells in the G2/M phase, there was a significant decrease in the
percentage of cells in the G0/G1 phase from 60.94±3.14% to
45.82±2.75% (p<0.01), but no significant change in the
percentage of cells in the S phase from 31.66±4.69% to 28.90±3.04%
(p>0.05). Treatment with AURKA-siRNA also results in an decrease
in the percentage of A549 cells in the G2/M phase from 10.57±0.53%
to 6.27±0.57% (p<0.01). Again, there was also a significant
increase in the percentage of cells in the S phase from 31.77±0.61%
to 37.74±1.01% (p<0.01), but no significant change in the
percentage of cells in the G0/G1 phase from 58.63±1.30% to
55.99±0.67% (p>0.05).
These results suggest that depletion of AURKA
inhibits the cellular proliferation of lung adenocarcinoma cells
via G2/M and S phase arrest of the cell cycle in H1299 and A549
cells, respectively. The inconsistent results of cell cycling may
derive from the endogenous differences in cell cycle in different
cell types.
Induction of apoptosis in lung
adenocarcinoma cells by AURKA knockdown
To determine whether knockdown of AURKA could induce
cell apoptosis, flow cytometry was used to analyze the apoptosis of
lung adenocarcinoma cells after infection with AURKA-siRNA for 72
h. As shown in Fig. 5A and B, the
percentage of apoptotic H1299 cells was 4.14±0.46% in scr-siRNA
group and the percentage of apoptotic cells increased to
18.04±2.69% in AURKA-siRNA group (p<0.01) (Fig. 5A). The percentage of apoptotic A549
cells was 0.98±0.02% in scr-siRNA group cells and increased to
8.27±0.61% in AURKA-siRNA group (p<0.01) (Fig. 5B). These data suggest that the
depletion of AURKA specifically induced apoptosis of the lung
adenocarcinoma cells.
Suppression of AURKA alters the
expression levels of cell cycle-related genes
To test the possible mechanisms underlying the lung
adenocarcinoma cell proliferation inhibition and apoptosis after
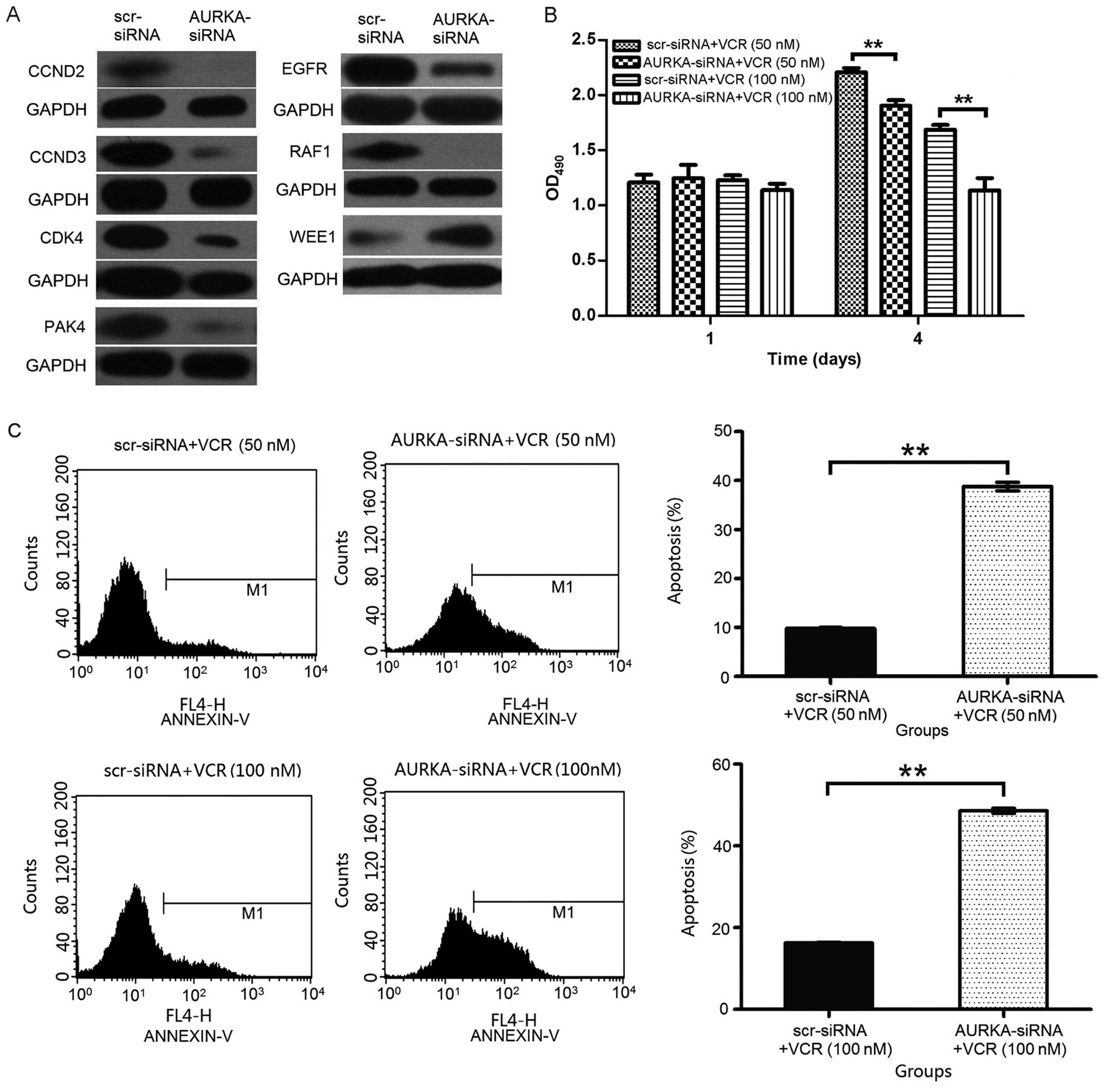
AURKA knockdown, we checked cell cycle-related gene expression. Our
results reveals that the knockdown of AURKA downregulated RAF-1,
CCND2, CCND3, CDK4, PAK4, EGFR and upregulated WEE1 expression in
H1299 cells. These results indicate that the heightened apoptosis
associated with AURKA downregulation may be partly mediated by cell
cycle-related proteins in H1299 cells (Fig. 6A).
Knockdown of AURKA enhances chemotherapy
sensitivity to VCR in human lung carcinoma A549 cells
The cooperative effects of AURKA knockdown and VCR
on repressing lung adenocarcinoma cell proliferation were
investigated in this study. AURKA knockdown cooperated with VCR to
inhibit A549 cell proliferation. As showed in Fig. 6B, at both low (50 nM) and high
concentration (100 nM) of VCR, AURKA depletion could cooperatively
inhibit A549 cell growth in vitro.
The number of apoptotic cells was determined by
Annexin V staining. A549 cells were transfected with scr-siRNA or
AURKA-siRNA, and then treated with VCR for 48 h. As shown in
Fig. 6C, the percentage of
apoptotic A549 cells was 9.79±0.18% in scr-siRNA + VCR (50 nM)
group and the percentage of apoptotic cells increased to
38.81±0.88% in AURKA-siRNA + VCR (50 nM) group (p<0.01). The
percentage of apoptotic A549 cells was 16.30±0.11% in scr-siRNA +
VCR (100 nM) group cells and increased to 48.58±0.63% in
AURKA-siRNA + VCR (100 nM) group (p<0.01). These data suggest
that the depletion of AURKA cooperatively induces apoptosis of the
lung adenocarcinoma cells.
Discussion
AURKA functions as an oncogene in several
malignancies and its overexpression is associated with a higher
grade of tumor and a poor prognosis. Aneuploidy is associated with
a poor outcome and a marker of metastasis in gastric carcinoma, a
correlation between aneuploidy and AURKA overexpression exists in
gastric cancer, clinical samples with gene amplification and
overexpression of AURKA showed aneuploidy and poor prognosis
(7,14). In this study, we also observed that
AURKA is highly expressed in lung adenocarcinoma tissue and its
overexpression is associated with shorter overall survival, which
indicates that AURKA plays important roles in the development of
lung adenocarcinoma.
AURKA promotes cell cycle progression by regulating
important mitotic events including spindle assembly, chromosome
maturation and mitotic entry (9,10).
Depletion of AURKA caused cell cycle arrest in G2/M and S phases in
H1229 and A549 cells, respectively. In addition, knockdown of AURKA
in such cells could induce apoptosis. Our results are similar as
other studies, which suggest that AURKA inhibition is potential
therapeutic for lung adenocarcinoma. This is first study that ties
AURKA with lung adenocarcinoma in tumor development and
treatment.
To elucidate the mechanisms of cell cycle arrest and
apoptosis after knockdown of AURKA, we checked the expression of
several cell cycle related genes. D-type cyclins (D1, D2, and D3)
are a family of key cell cycle regulators, as they can promote cell
cycle progression by binding to and activating cyclin-dependent
kinase 4 (cdk4)/cdk6. The activated cyclin D-cdk4/cdk6 complex can
then phosphorylate and deactivate the tumor suppressor protein pRB,
this phosphorylation in turn leads to the release and upregulation
of transcription factor E2F that promote progression from the G1 to
S phase of the cell cycle (15,16).
Aberrant expression of CCND2 can lead to unrestricted cell
proliferation. Its aberrant expression has been observed in various
cancers. Many studies have found that CCND2 is overexpressed or
amplified in many human cancers, such as CaP prostate cancer,
gastric cancer, ovarian and testicular tumors (17–19).
Cyclin D3 has been suggested to have a role in certain cancers.
Moreover, overexpression of cyclin D3 has been found in several
human cancers, such as renal cell carcinoma, pancreatic
adenocarcinoma and breast carcinoma (20–22).
CDK4 belongs to the cyclin dependent kinases family,
it has been found to promote cell proliferation by driving cell
cycle progression (23–25). Overexpression of CDK4 protein has
been described in many tumors, including oral squamous cell
carcinoma, pancreatic neuroendocrine tumor (NET), and lung cancer
(26–28). Patients with lung cancer with
higher CDK4 expression levels had a markedly shorter overall
survival time than those with low CDK4 expression (28).
PAK4 (P21-activated kinase 4), a subfamily of
serine/threonine protein kinases involved in cytoskeletal dynamics
and cell motility, plays a crucial role in oncogenic signaling
pathways. PAK4 is thought to regulate cancer cell progression
involving the c-Src/EGFR/cyclin D1 pathway (29,30).
PAK4 upregulation has been identified in many kinds of human cancer
cell lines and amplification of the chromosome region containing
PAK4 has been frequently observed in colorectal, pancreatic, and
ovarian cancer (31–34).
Epidermal growth factor receptor (EGFR), a receptor
tyrosine kinase (TK), is the expression product of oncogene c-erbB1
and plays essential roles in cell differentiation, proliferation,
development and maintenance in both cancerous and normal
physiological conditions. Expression of EGFR strongly affects the
outcomes of cancer patients in many cancer types. It has been found
to act as a powerful indicator with tumor progression and poor
survival (35,36). EGFR is frequently aberrantly
activated in NSCLC (37–39).
V-raf-1 murine leukemia viral oncogene homolog 1
(Raf-1) is a multifunctional protein with serine and threonine
kinase activity. It is a critical target of many growth factors in
various cell types. Raf-1 is at the apex of the mitogen activated
protein kinase (MEK)-ERK pathway, which controls a variety of
fundamental cellular including cell proliferation, survival and
migration including cell proliferation, migration, survival, and
transformation (40–42). In the present study, we found that
the knockdown of AURKA downregulated RAF-1, CCND2, CCND3, CDK4,
PAK4, EGFR, which indicates the potential mechanisms of AURKA
depletion-induced cell cycle arrest and apoptosis in NSCLC cell
lines.
WEE1, a tyrosine kinase regulator of the cell cycle,
has been associated with survival in several cancer types,
including malignant melanoma, breast cancer and glioblastoma. WEE1
was reported to be a safeguard against mitotic catastrophe in
instances of sensitive cell division. Its overexpression causes G2
arrest by promoting the inhibitory phosphorylation of
cyclin-dependent kinase (43–45).
Our results show that upregulated expression of WEE1 in H1299 cells
after AURKA knockdown, suggesting other mechanisms of AURKA
inhibition in treatment of lung adenocarcinoma.
VCR has been extensively used in clinic and its
anticancer mechanisms are through acting on tubulin, inhibiting the
cell mitosis and arresting cell cycling and proliferation, which
are similar with those of AURKA depletion. Therefore, we speculats
that silencing of AURKA gene may enhance VCR sensitivity and reduce
drug resistance. To further confirm that AURKA inhibition has
cooperative effects on repressing lung adenocarcinoma cell
proliferation, we treated A549 cells with different doses of VCR
combined with AURKA knockdown. The results are very interesting;
the AURKA knockdown enhanced the repressing effects of VCR on A549
cell proliferation. This is direct evidence that AURKA depletion
could combine with traditional chemotherapy drug for treating lung
adenocarcinoma.
In summary, this study firstly demonstrats that
AURKA is a therapeutic target for treatment of lung adenocarcinoma.
AURKA depletion could induce cell cycle arrest and apoptosis in
lung adenocarcinoma cells. Cooperative effects with VCR provided
direct evidence that AURKA is a target for lung adenocarcinoma
therapy. The detailed mechanisms should be elucidated and clinical
trials performed in the future.
Acknowledgements
This study was supported by a grant from the Natural
Science Foundation for the Youth (no. 81402220), Suzhou Planning
Project of Science and Technology (SYS201301) and the Science and
Technology Foundation of Kunshan City (no. ks1234). The authors
thank Dr Wenxiang Wei (Soochow University, Suzhou 215123, China)
for his sincere help and technical support.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chou HC and Chan HL: Effect of glutathione
reductase knockdown in response to UVB-induced oxidative stress in
human lung adenocarcinoma. Proteome Sci. 12:22014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pilotto S, Bria E, Peretti U, Massari F,
Garassino M, Pelosi G and Tortora G: Lung adenocarcinoma patient
refractory to gefitinib and responsive to crizotinib, with
concurrent rare mutation of the epidermal growth factor receptor
(L861Q) and increased ALK/MET/ROS1 gene copy number. J Thorac
Oncol. 8:e105–e106. 2013. View Article : Google Scholar
|
|
4
|
Onn A, Tsuboi M and Thatcher N: Treatment
of non-small-cell lung cancer: A perspective on the recent advances
and the experience with gefitinib. Br J Cancer. 91(Suppl 2):
S11–S17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fang W, Zhang J, Liang W, Huang Y, Yan Y,
Wu X, Hu Z, Ma Y, Zhao H, Zhao Y, et al: Efficacy of epidermal
growth factor receptor-tyrosine kinase inhibitors for Chinese
patients with squamous cell carcinoma of lung harboring EGFR
mutation. J Thorac Dis. 5:585–592. 2013.PubMed/NCBI
|
|
6
|
Shimada Y, Saji H, Nomura M, Matsubayashi
J, Yoshida K, Kakihana M, Kajiwara N, Ohira T and Ikeda N: Cancer
stem cell-related marker expression in lung adenocarcinoma and
relevance of histologic subtypes based on IASLC/ATS/ERS
classification. Onco Targets Ther. 6:1597–1604. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dar AA, Goff LW, Majid S, Berlin J and
El-Rifai W: Aurora kinase inhibitors - rising stars in cancer
therapeutics? Mol Cancer Ther. 9:268–278. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kallioniemi A, Kallioniemi OP, Piper J,
Tanner M, Stokke T, Chen L, Smith HS, Pinkel D, Gray JW and Waldman
FM: Detection and mapping of amplified DNA sequences in breast
cancer by comparative genomic hybridization. Proc Natl Acad Sci
USA. 91:2156–2160. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Romain C, Paul P, Kim KW, Lee S, Qiao J
and Chung DH: Targeting Aurora kinase-A downregulates cell
proliferation and angiogenesis in neuroblastoma. J Pediatr Surg.
49:159–165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thrane S, Pedersen AM, Thomsen MB,
Kirkegaard T, Rasmussen BB, Duun-Henriksen AK, Lænkholm AV, Bak M,
Lykkesfeldt AE and Yde CW: A kinase inhibitor screen identifies
Mcl-1 and Aurora kinase A as novel treatment targets in
anti-estrogen-resistant breast cancer cells. Oncogene.
34:4199–4210. 2015. View Article : Google Scholar
|
|
11
|
Elbashir SM, Harborth J, Lendeckel W,
Yalcin A, Weber K and Tuschl T: Duplexes of 21-nucleotide RNAs
mediate RNA interference in cultured mammalian cells. Nature.
411:494–498. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shedden K, Taylor JM, Enkemann SA, Tsao
MS, Yeatman TJ, Gerald WL, Eschrich S, Jurisica I, Giordano TJ,
Misek DE, et al; Director’s Challenge Consortium for the Molecular
Classification of Lung Adenocarcinoma. Gene expression-based
survival prediction in lung adenocarcinoma: A multi-site, blinded
validation study. Nat Med. 14:822–827. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zielske SP and Stevenson M: Importin 7 may
be dispensable for human immunodeficiency virus type 1 and simian
immunodeficiency virus infection of primary macrophages. J Virol.
79:11541–11546. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jeng YM, Peng SY, Lin CY and Hsu HC:
Overexpression and amplification of Aurora-A in hepatocellular
carcinoma. Clin Cancer Res. 10:2065–2071. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sherr CJ: G1 phase progression: Cycling on
cue. Cell. 79:551–555. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dong Q, Meng P, Wang T, Qin W, Qin W, Wang
F, Yuan J, Chen Z, Yang A and Wang H: MicroRNA let-7a inhibits
proliferation of human prostate cancer cells in vitro and in vivo
by targeting E2F2 and CCND2. PLoS One. 5:e101472010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mermelshtein A, Gerson A, Walfisch S,
Delgado B, Shechter-Maor G, Delgado J, Fich A and Gheber L:
Expression of D-type cyclins in colon cancer and in cell lines from
colon carcinomas. Br J Cancer. 93:338–345. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Susaki E, Nakayama K and Nakayama KI:
Cyclin D2 translocates p27 out of the nucleus and promotes its
degradation at the G0–G1 transition. Mol Cell Biol. 27:4626–4640.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hedberg Y, Roos G, Ljungberg B and
Landberg G: Cyclin D3 protein content in human renal cell carcinoma
in relation to cyclin D1 and clinico-pathological parameters. Acta
Oncol. 41:175–181. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ito Y, Takeda T, Wakasa K, Tsujimoto M and
Matsuura N: Expression and possible role of cyclin D3 in human
pancreatic adenocarcinoma. Anticancer Res. 21:1043–1048.
2001.PubMed/NCBI
|
|
22
|
Wong SC, Chan JK, Lee KC and Hsiao WL:
Differential expression of p16/p21/p27 and cyclin D1/D3, and their
relationships to cell proliferation, apoptosis, and tumour
progression in invasive ductal carcinoma of the breast. J Pathol.
194:35–42. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Retzer-Lidl M, Schmid RM and Schneider G:
Inhibition of CDK4 impairs proliferation of pancreatic cancer cells
and sensitizes towards TRAIL-induced apoptosis via downregulation
of survivin. Int J Cancer. 121:66–75. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Karim BO, Rhee KJ, Liu G, Zheng D and Huso
DL: Chemoprevention utility of silibinin and Cdk4 pathway
inhibition in Apc(−/+) mice. BMC Cancer. 13:1572013. View Article : Google Scholar
|
|
25
|
Chan KC, Ting CM, Chan PS, Lo MC, Lo KW,
Curry JE, Smyth T, Lee AW, Ng WT, Tsao GS, et al: A novel Hsp90
inhibitor AT13387 induces senescence in EBV-positive nasopharyngeal
carcinoma cells and suppresses tumor formation. Mol Cancer.
12:1282013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Poomsawat S, Buajeeb W, Khovidhunkit SO
and Punyasingh J: Alteration in the expression of cdk4 and cdk6
proteins in oral cancer and premalignant lesions. J Oral Pathol
Med. 39:793–799. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tang LH, Contractor T, Clausen R, Klimstra
DS, Du YC, Allen PJ, Brennan MF, Levine AJ and Harris CR:
Attenuation of the retinoblastoma pathway in pancreatic
neuroendocrine tumors due to increased cdk4/cdk6. Clin Cancer Res.
18:4612–4620. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu A, Wu B, Guo J, Luo W, Wu D, Yang H,
Zhen Y, Yu X, Wang H, Zhou Y, et al: Elevated expression of CDK4 in
lung cancer. J Transl Med. 9:382011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sørensen CS and Syljuåsen RG: Safeguarding
genome integrity: The checkpoint kinases ATR, CHK1 and WEE1
restrain CDK activity during normal DNA replication. Nucleic Acids
Res. 40:477–486. 2012. View Article : Google Scholar :
|
|
30
|
Callow MG, Clairvoyant F, Zhu S, Schryver
B, Whyte DB, Bischoff JR, Jallal B and Smeal T: Requirement for
PAK4 in the anchorage-independent growth of human cancer cell
lines. J Biol Chem. 277:550–558. 2002. View Article : Google Scholar
|
|
31
|
Siu MK, Chan HY, Kong DS, Wong ES, Wong
OG, Ngan HY, Tam KF, Zhang H, Li Z, Chan QK, et al: p21-activated
kinase 4 regulates ovarian cancer cell proliferation, migration,
and invasion and contributes to poor prognosis in patients. Proc
Natl Acad Sci USA. 107:18622–18627. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang HJ, Siu MK, Yeung MC, Jiang LL, Mak
VC, Ngan HY, Wong OG, Zhang HQ and Cheung AN: Overexpressed PAK4
promotes proliferation, migration and invasion of choriocarcinoma.
Carcinogenesis. 32:765–771. 2011.PubMed/NCBI
|
|
33
|
Liu Y, Xiao H, Tian Y, Nekrasova T, Hao X,
Lee HJ, Suh N, Yang CS and Minden A: The pak4 protein kinase plays
a key role in cell survival and tumorigenesis in athymic mice. Mol
Cancer Res. 6:1215–1224. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kimmelman AC, Hezel AF, Aguirre AJ, Zheng
H, Paik JH, Ying H, Chu GC, Zhang JX, Sahin E, Yeo G, et al:
Genomic alterations link Rho family of GTPases to the highly
invasive phenotype of pancreas cancer. Proc Natl Acad Sci USA.
105:19372–19377. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mitsudomi T and Yatabe Y: Epidermal growth
factor receptor in relation to tumor development: EGFR gene and
cancer. FEBS J. 277:301–308. 2010. View Article : Google Scholar
|
|
36
|
Cheng L, Zhang S, Alexander R, Yao Y,
MacLennan GT, Pan CX, Huang J, Wang M, Montironi R and
Lopez-Beltran A: The landscape of EGFR pathways and personalized
management of non-small-cell lung cancer. Future Oncol. 7:519–541.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pao W and Chmielecki J: Rational,
biologically based treatment of EGFR-mutant non-small-cell lung
cancer. Nat Rev Cancer. 10:760–774. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yamamoto H, Toyooka S and Mitsudomi T:
Impact of EGFR mutation analysis in non-small cell lung cancer.
Lung Cancer. 63:315–321. 2009. View Article : Google Scholar
|
|
39
|
Inamura K, Ninomiya H, Ishikawa Y and
Matsubara O: Is the epidermal growth factor receptor status in lung
cancers reflected in clinicopathologic features? Arch Pathol Lab
Med. 134:66–72. 2010.PubMed/NCBI
|
|
40
|
Alejandro EU, Kalynyak TB, Taghizadeh F,
Gwiazda KS, Rawstron EK, Jacob KJ and Johnson JD: Acute insulin
signaling in pancreatic beta-cells is mediated by multiple Raf-1
dependent pathways. Endocrinology. 151:502–512. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang H, Gambosova K, Cooper ZA, Holloway
MP, Kassai A, Izquierdo D, Cleveland K, Boney CM and Altura RA: EGF
regulates survivin stability through the Raf-1/ERK pathway in
insulin-secreting pancreatic β-cells. BMC Mol Biol. 11:662010.
View Article : Google Scholar
|
|
42
|
Yoon S and Seger R: The extracellular
signal-regulated kinase: Multiple substrates regulate diverse
cellular functions. Growth Factors. 24:21–44. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Magnussen GI, Holm R, Emilsen E, Rosnes
AK, Slipicevic A and Flørenes VA: High expression of Wee1 is
associated with poor disease-free survival in malignant melanoma:
Potential for targeted therapy. PLoS One. 7:e382542012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Murrow LM, Garimella SV, Jones TL, Caplen
NJ and Lipkowitz S: Identification of WEE1 as a potential molecular
target in cancer cells by RNAi screening of the human tyrosine
kinome. Breast Cancer Res Treat. 122:347–357. 2010. View Article : Google Scholar
|
|
45
|
Mir SE, De Witt Hamer PC, Krawczyk PM,
Balaj L, Claes A, Niers JM, Van Tilborg AA, Zwinderman AH, Geerts
D, Kaspers GJ, et al: In silico analysis of kinase expression
identifies WEE1 as a gatekeeper against mitotic catastrophe in
glioblastoma. Cancer Cell. 18:244–257. 2010. View Article : Google Scholar : PubMed/NCBI
|