Introduction
Cervical cancer (CC) is the fourth most common tumor
type and leading cause of cancer death among women worldwide. Its
incidence rate has increased in recent years (1,2).
Thus, to develop better prognostic and therapeutic strategies for
CC, insight into the molecular and biologic mechanisms of
tumorigenesis is critical.
Recent research suggests that
Ezrin-radixin-moesin-binding phosphoprotein 50 (EBP50, also named
NHERF1, NHERF), a scaffold protein containing two tandem
PSD-95/Discs Large/ZO-1 (PDZ) domains, plays an important role in
tumor development and progression. EBP50 exerts tumor suppressing
function in breast cancer, pancreatic cancer, biliary cancer,
hepatocellular cancer, prostate cancer and glioblastoma (2–7). In
addition, EBP50 was also reported to play the tumor-promoting
roles. For example, EBP50 overexpression enhances cell invasion in
breast cancer cells, and higher EBP50 level is associated with
increasing tumor cytohistological grade, aggressive clinical
behavior, and unfavorable prognosis in breast cancer (8–10).
EBP50 could function as a positive regulator of Wnt signaling and
might cause a malignant phenotype in hepatocellular carcinoma cells
(11). EBP50 overexpression is
involved in GBM invasiveness (12). However, the functional expression
of EBP50 in CC cell proliferation and progression have not been
reported.
Epidermal growth factor receptor (EGFR) was reported
to correlate with cervical cancer (13). Overactivation of EGFR signaling is
a hallmark of cancer and therapy strategy toward EGFR inhibition in
cervical cancer has been ongoing (1). However, signaling proteins that
connect the EGFR oncogenic cascade are poorly characterized
(14). Scaffold proteins formed
multiprotein complexes that were central to accurate coordination
of signaling pathways (15).
Abnormal expression of scaffold proteins has been linked to
different types of cancer in human (16). EBP50 was reported to regulate EGFR
signaling (4,17,18).
EBP50 overexpression induced a sustained activation of EGFR by
increasing the level of EGFR at the HeLa cell surface (17). However, EBP50 is also reported to
suppress EGFR activity by depleting the amount of EGFR at the cell
surface (4). In addition, EBP50
blocked EGFR phosphorylation to inhibit EGF-induced breast cancer
cell proliferation (18). Thus,
the effect of EBP50 in regulating EGFR signaling was controversial.
Especially, the regulatory effect of EBP50 on EGFR signaling in CC
patients remains unclear.
In this study, we first found low expression of
EBP50 in CC samples and EBP50 expression level negatively
correlated with CC cell proliferation, cell cycle and EGFR
signaling activation. EBP50 knockdown abolished the inhibition on
EGF-induced ERK signaling activation. In order to verify EBP50
regulated EGFR signaling via interaction, we constructed EBP50
mutant DD (S279D/S301D) which disrupted the interaction with EGFR.
The overexpression of DD mutant attenuated its inhibition on
EGFR-mediated signaling, revealing EBP50 regulated EGFR signaling
via interaction with EGFR. Further evidence showed that EBP50 could
predict the prognosis of CC patients after ruling out the patients
with mutation/copy number alteration of egfr/ErbB gene and
(chemo)radiation, which led to continuous activation of egfr
gene affecting patient prognosis, respectively. EBP50 could be the
precise therapeutic target and prognostic marker for CC
patients.
Materials and methods
Tissue microarray data
The IHC-based protein expression data including
high-resolution images were viewed and downloaded from the Human
Protein Atlas web portal (www.proteinatlas.org).
The Cancer Genome Atlas (TCGA) data
The TCGA data about mRNA (RNA Seq v2) and protein
(RPPA) expression levels in cervical cancer patients were obtained
from https://www.synapse.org/. The EBP50 mRNA
level and c-Raf_pS338 protein level were used in this study.
Clinical data was downloaded from cBioPortal database (www.cbioportal.org).
Gene set enrichment analysis
The association between phenotypes, biological
processes/pathway and EBP50 expression level was analyzed using
gene set enrichment analysis (GSEA v2.2, http://www.broad.mit.edu/gsea/). GSEA calculates a
gene set enrichment score (ES) that estimates whether genes from
pre-defined gene set [obtained from the Molecular Signatures
Database, MSigDB, http://software.broadinstitute.org;genesets:REGULATION_OF_CELL_PROLIFERATION,
POSITIVE_REGULATION_OF_CELL_CYCLE (annotated by the GO term GO:
0042127 and GO: 0045787, respectively), EGFR_UP.V1_DN,
REACTOME_PERK_REGULATED_GENE_EXPRESSION, EGFR_UP.V1_UP] are
enriched among the highest- (or lowest-) ranked genes or
distributed randomly. Default settings were used. Thresholds for
significance were determined by permutation analysis (1,000
permutations). False discovery rate (FDR) was calculated. A gene
set is considered significantly enriched when the FDR score is
<0.05.
Plasmids
shEBP50 constructs (pSuper.puro shEBP50) were kind
gifts of Dr M.J. Wheelock (University of Nebraska Medical Center,
Omaha, NE, USA).
GST-tagged EBP50_WT and GST-tagged EBP50 mutant
(S279D/S301D, DD) plasmids, pBK-CMV-Hemagglutinin (HA)-EBP50_WT and
pBK-CMV-HA-EBP50 mutant (DD) expression plasmids were kindly
provided by Dr Randy Hall from Emory University.
Cell culture, transfection and cell
treatments
The human cervical carcinoma cell line HeLa was
obtained from the American Type Culture Collection (ATCC, Manassas,
VA, USA). HeLa was grown in Dulbecco’s modified Eagle’s medium
(DMEM, Gibco) (19) at 37°C and 5%
CO2. The media were supplemented with 10% fetal bovine
serum (FBS, Hyclone, Logan, UT, USA) and 1% antibiotic-antimycotic
agent (Life Technologies, Inc., Grand Island, NY, USA). Cells were
grown to 80% confluency for use. Transfections were performed by
Lipofectamine 2000 (Invitrogen, CA, USA) with plasmid DNA following
the protocol as reported before (3).
HeLa cells were serum starved overnight, then
treated with 100 ng/ml EGF (Sigma-Aldrich Chemical Corp., St.
Louis, MO, USA) for 5 min at 37°C to detect the effect of EBP50
knockdown, EBP50_WT or EBP50_DD overexpression on EGFR-mediated
signal transduction pathways.
Stable transfection
For stable knockdown, shEBP50 constructs were
transfected into HeLa cells following the protocol. Two days
following transfection, cells were transferred to 90-mm plates and
cultured in selection medium with 0.5 μg/ml puromycin
(Sigma-Aldrich) to obtain EBP50 knockdown cells. Resistant colonies
formed were harvested and cultured for at least a month, then the
fractions were used for analysis of EBP50 expression by western
blotting, with GAPDH expression as a protein loading control.
Stably-transfected cells were maintained and passaged in culture
medium with puromycin (0.25 μg/ml) (18). HeLa cells stably knocked down with
the shEBP50 plasmids were called HeLa_shEBP50.
Western blotting
Samples were run on 10% sodium dodecyl sulfate
(SDS)-polyacrylamide gels (PAGE) and transferred to PVDF membranes.
The blots were blocked in blocking buffer (5% non-fat dry milk in
TBST buffer) for 1 h at room temperature and then incubated with
primary antibodies (1:1,000) in blocking buffer overnight at 4°C.
The blots were washed three times with TBST buffer and incubated
for 1 h at room temperature with a horseradish peroxidase
(HRP)-conjugated anti-mouse IgG or anti-rabbit IgG secondary
antibody (1:3000; Amersham Biosciences, Piscataway, NJ, USA) in
blocking buffer. Finally, the blots were washed three times with
TBST buffer and visualized via enzyme-linked chemiluminescence
using the electrochemiluminescence (ECL) kit (Applygen Technologies
Inc., Beijing, China) (3). The
levels of immunoreactivity were semi-quantitatively analyzed by NIH
ImageJ 1.62 software.
The primary antibody specific for EBP50 was
purchased from BD Biosciences (San Jose, CA, USA), HA was from MBL
(Nagoya, Japan). Other primary antibodies specific for GAPDH,
phospho-EGFR (Tyr1173), phospho-ERK1/2 (Thr202/Tyr204), total EGFR
and ERK1/2 were all bought from Cell Signaling Technology (Beverly,
MA, USA).
GST pull-down assay
Glutathione S-transferase (GST) fusion proteins were
purified from bacteria using glutathione-Sepharose 4B beads
(Sigma-Aldrich) according to the manufacturer’s protocol. The GST
pull-down assay was performed as described previously (20). Briefly, equal amounts of GST or
GST-EBP50_WT, GST-EBP50_DD fusion protein beads, were incubated
with equal amounts of cell lysates. After incubation at 4°C for 2
h, the beads were washed with ice-cold wash buffer (100 mM NaCl, 10
mM HEPES, pH 7.4, 5 mM EDTA, 1 mM benzamidine, 3% BSA and 0.1%
Tween-20). Proteins were then eluted with SDS sample buffer (50 mM
Tris/HCl, 100 mM DTT, 2% SDS, 0.1% bromophenol blue and 10%
glycerol), and detected with western blotting.
Co-immunoprecipitation assay
Co-immunoprecipitation was performed as described
before (21). Briefly, COS-7 cells
transiently transfected with HA-EBP50_WT or HA-EBP50_DD were
harvested and then lysed in 1 ml of ice-cold lysis buffer (10 mM
HEPES, 50 mM NaCl, 5 mM EDTA, 1 mM benzamidine, 0.5% Triton X-100,
pH 7.4). Lysates were solubilized, clarified and then incubated
with anti-HA or anti-EGFR antibody, prebounded with protein A&G
beads (Calbiochem, CA, USA). After washing with an ice-cold lysis
buffer three times, the immunoprecipitated proteins were eluted
from the beads with SDS sample loading buffer. The eluted samples
were then analyzed by western blotting.
Statistical analyses
Statistical analyses were performed using the SPSS
18.0 (SPSS Inc, Chicago, IL, USA) and Graphpad Prism 5 (Graphpad
software Inc, San Diego, CA, USA). Group distributions were
compared using the Student’s t-test. A value of P<0.05 was
considered statistically significant. The association between the
EBP50 expression level and patient’s overall survival (OS) was
assessed by Kaplan-Meier method.
Results
EBP50 expression negatively correlates
with cell proliferation and cell cycle in cervical cancer (CC)
patients
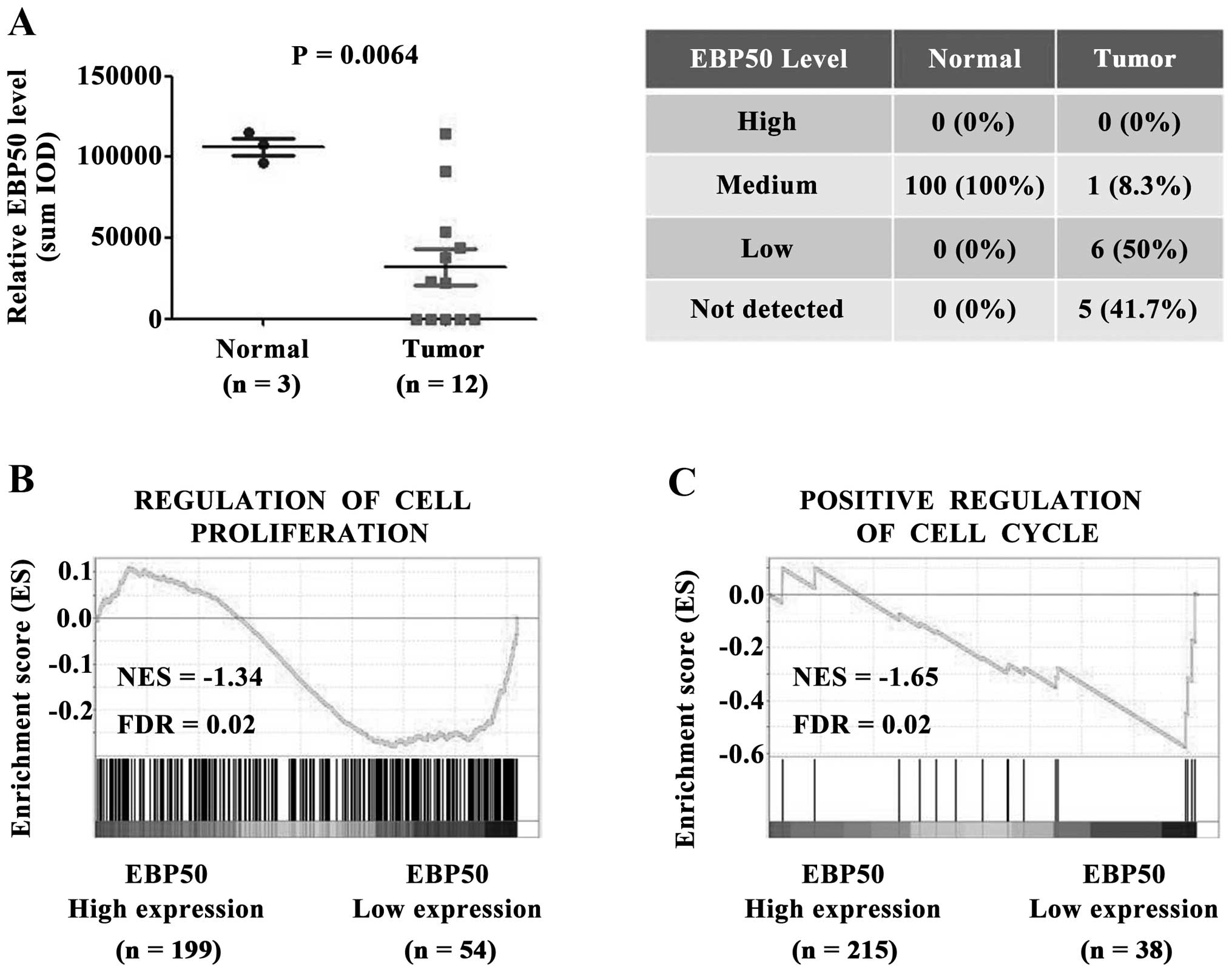
To investigate the role of EBP50 in CC, the
expression level of EBP50 in CC tissues was first analyzed.
Immunohistochemistry data from tissue microarrays of the Human
Protein Atlas dataset were obtained to analyze the expression level
of EBP50 between cervical cancer and normal cervix tissues. EBP50
protein expression level was significantly downregulated in CC
tissues compared with the normal controls (P<0.01, Fig. 1A). In 41.7% of tumor tissues EBP50
was not detected. 50% of tumor tissues weakly expressed EBP50, and
8.3% of tumor tissues expressed EBP50 in medium level (Fig. 1A). In contrast with tumor tissues,
normal cervical tissues expressed EBP50 in medium level at 100%.
Taken together, EBP50 was expressed in medium level for all normal
cervix tissues, whereas weak or no expression was detected in large
part of CC tissues despite medium level of EBP50 in CC tissue of
one case. EBP50 expression level was significantly downregulated in
CC tissues.
Low expression level of EBP50 in CC tissues
indicated that EBP50 played a role in CC tumorigenesis, so we
further studied the biological effect of EBP50 in CC samples. The
CC patients in the TCGA dataset was divided into high EBP50
expression and low EBP50 expression groups, and the correlations
between cell proliferation/cell cycle and EBP50 expression were
further analyzed using the GSEA method. As shown in Fig. 1B, the gene set of cell
proliferation and cell cycle, was highly enriched in EBP50 low
expression group (Fig. 1C, FDR
<0.05). These data suggested a negative correlation between
EBP50 expression level and cell proliferation or cell cycle in
clinical CC samples. Low expression level of EBP50 in CC tissues
contributed to CC cell proliferation or cell cycle.
EBP50 expression is negatively associated
with EGFR signaling activation in cervical cancer
To investigate the mechanism by which EBP50
suppressed CC cell proliferation and cell cycle, we studied the
regulatory effect of EBP50 on EGFR signaling since EGFR is an
important growth promoting factor in CC (22,23)
and EBP50 was reported to regulate EGFR signaling via its
interaction with EGFR (4,17). We performed GSEA to identify the
correlation between EBP50 expression level and EGFR signaling. EGFR
gene set (EGFR_UP.V1_DN, the downregulated gene set after EFGR
pathway was activated, indicating the activation of EGFR pathway)
was highly enriched in the EBP50 high expression group (Fig. 2A, FDR <0.05), revealing that
EBP50 negatively correlated with EGFR signaling activation. To
further investigate the effect of EBP50 expression on EGFR-mediated
signal activation, the regulatory effect of EBP50 on c-Raf and ERK,
which are the downstream molecules for EGFR-mediated signaling,
were further analyzed. TCGA dataset was used to analyze the
correlation between EBP50 and c-Raf_pS338 expression levels. EBP50
expression level is negatively correlated with c-Raf_pS338
expression levels (Fig. 2B). ERK
activation gene set was enriched in the low EBP50 expression group
(Fig. 2C, FDR <0.05), further
suggesting a negative correlation between EBP50 expression level
and EGFR/ERK signaling in clinical CC samples.
To verify the regulatory effect of EBP50 on
EGFR-mediated ERK signaling, we further knocked down the expression
of EBP50 in HeLa cells to detect EGF-induced ERK phosphorylation
level. As shown in Fig. 2D, EBP50
knockdown relieved its inhibition on EGF-induced ERK
phosphorylation level in HeLa cells. After 5 min of EGF
stimulation, ERK phosphorylation level in HeLa cells increased
2.1-fold over basal level, whereas ERK phosphorylation level in
EBP50 knockdown HeLa cells increased 7.5-fold over basal level.
These results confirmed that EBP50 inhibited EGF-induced ERK
phosphorylation in CC cells.
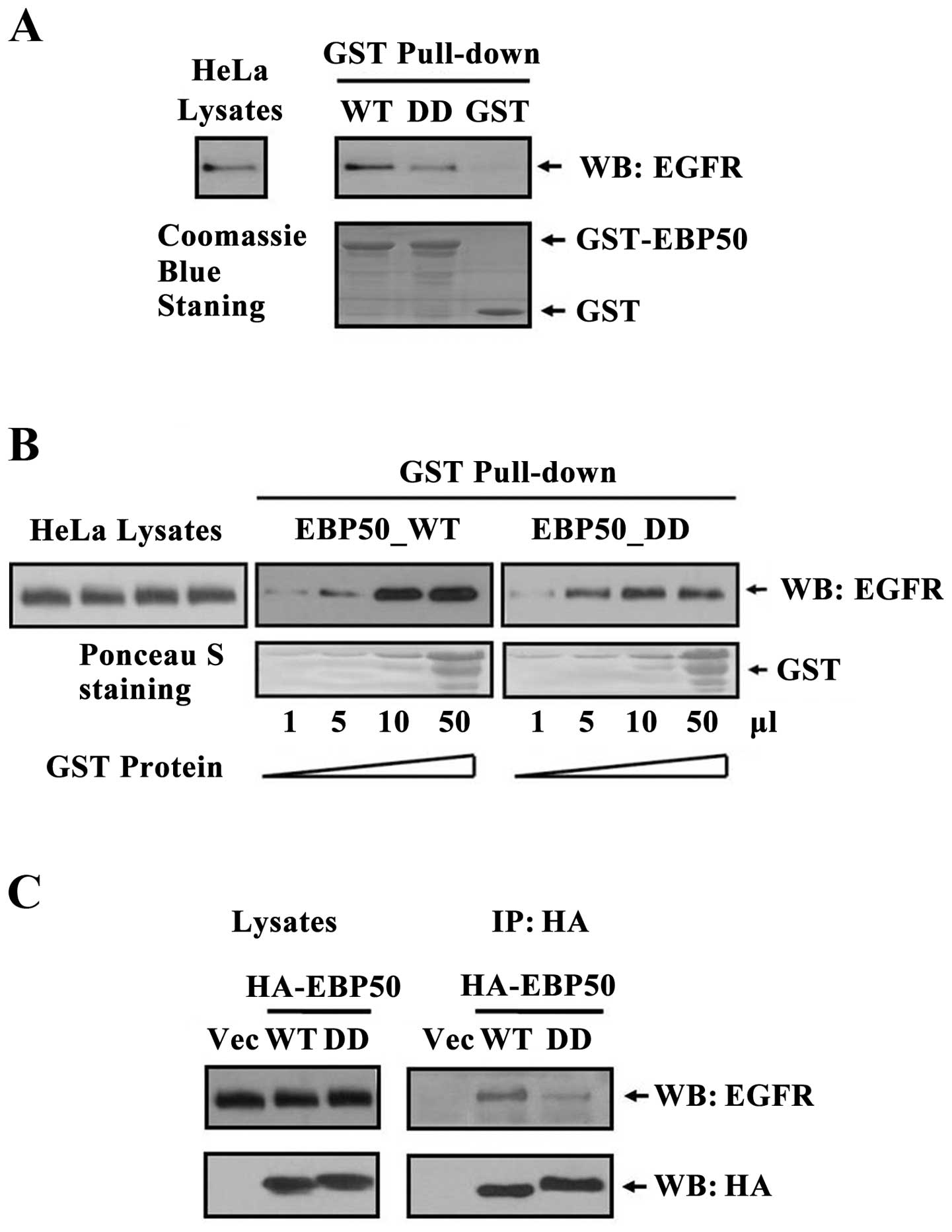
EBP50 mutant DD disrupts its interaction
with EGFR
EBP50 regulated multiple signaling pathways, such as
EGFR, PDGFR, PTEN and Wnt signaling pathways (11,17,24–28).
Thus, that EBP50 knockdown regulated EGF-induced ERK signaling can
not exclude other signaling pathway-mediated ERK signaling. To
verify EBP50 regulated EGFR signaling via its interaction with EGFR
in CC tissues, we constructed EBP50 mutant DD (29) and detected their interaction by GST
pull-down assay. As shown in Fig.
3A, the amount of endogenous EGFR in HeLa cells pulled down by
GST-EBP50_DD was much less than that by GST-EBP50_WT, suggesting
that EBP50_DD mutation retarded the association of EGFR and
EBP50.
To further test whether EBP50_DD mutation retarded
the association of EGFR and EBP50, we then used different dose of
GST-EBP50_WT and GST-EBP50_DD (1–50 μl) fusion protein to pull down
endogenous EGFR from the same amount of HeLa cell lysates,
respectively. With the increase of both GST-EBP50_WT and
GST-EBP50_DD fusion protein amount, the amount of EGFR pulled down
increased correspondingly. However, the amount of EGFR pulled down
by GST-EBP50_DD was less than that by GST-EBP50_WT in each dose
(Fig. 3B), further revealing
EBP50_DD mutation retarded the association of EGFR and EBP50.
To verify the results, we used
co-immunoprecipitation assay to further investigate the disruption
effect of EBP50_DD mutation on its interaction with EGFR in
cellular context. Results showed that less EGFR
co-immunoprecipitated with EBP50_DD than EBP50_WT (Fig. 3C), which was consistent with GST
pull-down results, and again demonstrated that EBP50_DD mutation
disrupted the interaction of EGFR with EBP50.
EBP50 regulates EGFR/ERK signaling via
its interaction with EGFR
To confirm the inhibitory effect of EBP50 on
EGFR/ERK signaling by interacting with EGFR in CC cells, we
transiently transfected HA-EBP50_WT and HA-EBP50_ DD plasmids into
HeLa cells, respectively. As shown in Fig. 4, EBP50_DD attenuated the inhibition
of EBP50 on EGFR-mediated ERK phosphorylation in HeLa cells. After
5 min of EGF stimulation, EGFR phosphorylation level was increased
1.3-fold over basal level and ERK phosphorylation level was
increased 3.3-fold over basal level in HA-EBP50_DD transfected HeLa
cells, whereas EGFR phosphorylation level was increased 1-fold over
basal level and ERK phosphorylation level was increased 2.2-fold
over basal level in HA-EBP50_WT transfected HeLa cells,
respectively. The activation levels of EGFR and ERK in HA-EBP50_DD
transfected HeLa cells were higher than those in HA-EBP50_WT
transfected HeLa cells. The result indicated that disrupted
interaction of EBP50 with EGFR led to decreased inhibition on EGFR
signaling. This result further verified EBP50 inhibited
EGFR-mediated ERK phosphorylation via interaction with EGFR in CC
cells.
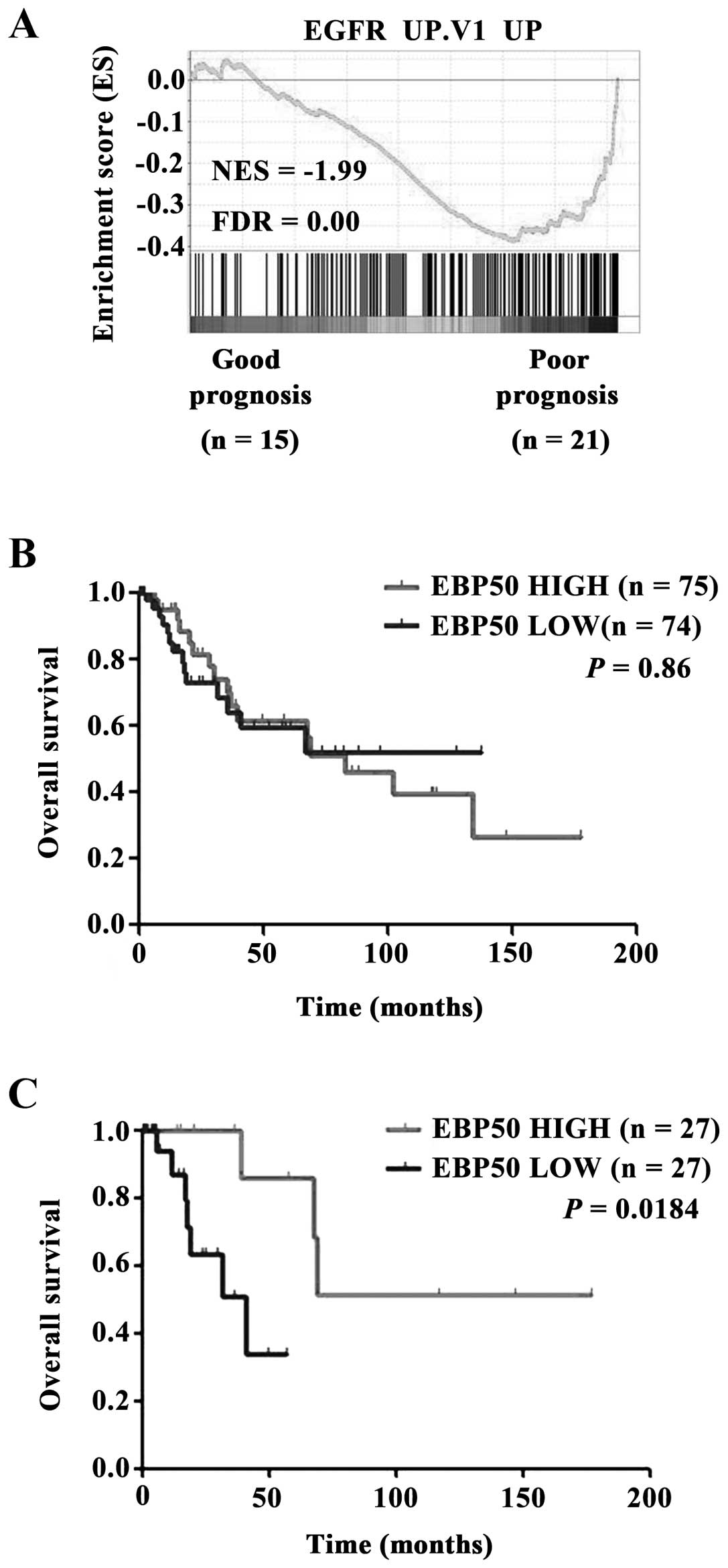
Low EBP50 expression level is correlated
with poor prognosis in cervical cancer
Activation of EGFR was associated with poor
prognosis in CC (30). The
analysis of TCGA dataset revealed EGFR pathway was over-activated
in CC patients with poor prognosis (3 years, dead) compared with
those with good prognosis (5 years, living) (Fig. 5A). The clinical prognosis relevance
of EBP50 expression level with CC patients was also evaluated.
Unexpectedly, EBP50 expression level in TCGA dataset had no
significant prognosis predictive ability for all CC patients
(Fig. 5B). Considering that
EGFR/ErbB gene mutation or copy number alteration (CNA) will
lead to continuous activation of EGFR signaling and (chemo)
radiation will influence the prognosis of CC patients. When CC
patients with EGFR/ErbB gene mutation or copy number
alteration (CNA) and (chemo)radiation were omitted, Kaplan-Meier
survival analysis showed that high EBP50 expression group had
better outcomes than low EBP50 expression group in terms of
survival duration (Fig. 5C,
P<0.05). These results further indicated that EBP50 affected
prognosis of CC patients via regulating EGFR signaling pathway.
Discussion
EBP50 is an adaptor protein consisting of two PDZ
domains and one ezrin-binding region. Through these functional
domains, EBP50 interacts with many proteins and regulates their
functions (11,17,24,25,28,31).
For example, EBP50 can enhance the stability of phosphatase and
tensin homologue deleted on chromosome 10 (PTEN) protein and
recruit PTEN to cell membrane in breast cancer cells (24,31),
stabilize β-catenin at cell membrane of mouse embryonic fibroblast
(MEF) cells (11,28), promote platelet-derived growth
factor receptor (PDGFR) phosphorylation in normal cells (25,28).
In this study, we found that EBP50 expression negatively correlated
with EGFR-mediated ERK activation. EBP50 knockdown abolished its
inhibition on EGF-induced ERK activation. PTEN and PDGFR, can also
regulate ERK signaling (32–34).
EBP50 could regulate EGFR-mediated ERK signaling via direct
interaction with EGFR (17) or
indirectly recruiting PTEN to EGFR (35). To elucidate whether EBP50 modulate
ERK signaling via interaction with EGFR, we constructed EBP50
mutant DD which destroyed the interaction with EGFR. The
overexpression of EBP50 mutant DD attenuated the inhibition of
EBP50 on EGF-induced EGFR and ERK activation in CC cells, further
verifying EBP50 regulated EGFR signaling via direct interaction
with EGFR in CC.
We speculated the following mechanisms by which
scaffold protein EBP50 regulated EGFR signaling via direct
interaction. Scaffold proteins were shown to promote conformational
changes of their binding partners (36,37).
It was possible that when EBP50 bound with EGFR, the conformation
of EGFR changed and EGFR was not easily phosphorylated. EBP50
altered the subcellular localization or membrane localization of
EGFR in biliary carcinoma cells (4). In CC cells, EBP50 might also alter
the subcellular localization or membrane localization of EGFR to
regulate EGFR-mediated signaling. These aspects need to be further
investigated.
Previously we reported that EBP50 overexpression in
HeLa cells could suppress HeLa cell proliferation and
anchorage-independent growth (3).
In this study, we found that EBP50 was significantly downregulated
in CC tissues. EBP50 expression negatively correlated with CC cell
proliferation and cell cycle by interacting with EGFR and
suppressing EGFR signaling. We further found EBP50 could predict
prognosis of CC patients without continuous activation
mutation/copy number alteration of egfr/ErbB gene and
(chemo)radiation which affected patient prognosis. These data
supported a novel tumor suppressor role of EBP50 in cervical cancer
and new mechanism by which EBP50 interacted with EGFR and inhibited
EGFR-mediated signaling.
In cervical cancer samples, EGFR signaling was
overactivated in about 33% of CC patients (30,38).
Activated EGFR predicted poor response to (chemo)radiation and
survival in CC, EGFR pathway was a promising therapeutic target for
CC patients (30). The mechanisms
for EGFR signaling activation were diverse, including egfr
activating mutations (39,40), egfr gene amplification
(39,41–45)
and disorder of EGFR signaling regulation (4,18).
In 32.63% of CC patients egfr gene activating mutations were
found (46). In addition, TCGA
dataset showed that ErbB and egfr alterations
(including mutation and amplification) were detected in ~17.7 and
4% of CC cases, respectively. EBP50 failed to predict the prognosis
of all CC patients. However, after ruling out patients with
continuous activation mutation/copy number alteration of
egfr/ErbB gene and (chemo)radiation which affected patient
prognosis, EBP50 showed the prognosis predictive effect for CC
patients. This further revealed predictive role of low EBP50
expression level for CC patients with poor prognosis was dependent
on its interaction with EGFR and its regulatory role on EGFR
signaling pathway.
In conclusion, this study demonstrated that EBP50
expression negatively correlated with CC cell proliferation, cell
cycle and EGFR signaling activation. EBP50 knockdown abolished the
inhibition on EGF-induced ERK signaling activation. The
overexpression of EBP50 mutant DD disrupted the interaction with
EGFR attenuated its inhibition on EGFR-mediated signaling,
revealing EBP50 regulated EGFR signaling via interaction with EGFR.
Further evidence showed that EBP50 could predict the prognosis of
CC patients after ruling out the patients with continuous
activation mutation/copy number alteration of egfr/ErbB gene
and (chemo)radiation affecting patient prognosis. Our findings
provided further insights into the molecular pathogenesis of
cervical cancer and EBP50 could be a novel, precise therapeutic
target and prognostic marker for CC patients.
Acknowledgements
This study was supported by the National Natural
Science Foundation of the People’s Republic of China (no. 30900247,
81372739).
Abbreviations:
|
EBP50
|
ezrin-radixin-moesin-binding
phospho-protein-50
|
|
EGFR
|
epidermal growth factor receptor
|
|
CC
|
cervical cancer
|
|
PTEN
|
phosphatase and tensin homolog deleted
on chromosome ten
|
|
PDGFR
|
platelet-derived growth factor
receptor
|
References
|
1
|
Zhang W, Jiang Y, Yu Q, Qiang S, Liang P,
Gao Y, Zhao X, Liu W and Zhang J: EGFR promoter methylation, EGFR
mutation, and HPV infection in Chinese cervical squamous cell
carcinoma. Appl Immunohistochem Mol Morphol. 23:661–666. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ji M, Fan D, Yuan L, Zhang Y, Dong W and
Peng X: EBP50 inhibits pancreatic cancer cell growth and invasion
by targeting the β-catenin/E-cadherin pathway. Exp Ther Med.
10:1311–1316. 2015.PubMed/NCBI
|
|
3
|
Zheng JF, Sun LC, Liu H, Huang Y, Li Y and
He J: EBP50 exerts tumor suppressor activity by promoting cell
apoptosis and retarding extracellular signal-regulated kinase
activity. Amino Acids. 38:1261–1268. 2010. View Article : Google Scholar
|
|
4
|
Clapéron A, Guedj N, Mergey M, Vignjevic
D, Desbois-Mouthon C, Boissan M, Saubaméa B, Paradis V, Housset C
and Fouassier L: Loss of EBP50 stimulates EGFR activity to induce
EMT phenotypic features in biliary cancer cells. Oncogene.
31:1376–1388. 2012. View Article : Google Scholar
|
|
5
|
Molina JR, Agarwal NK, Morales FC, Hayashi
Y, Aldape KD, Cote G and Georgescu MM: PTEN, NHERF1 and PHLPP form
a tumor suppressor network that is disabled in glioblastoma.
Oncogene. 31:1264–1274. 2012. View Article : Google Scholar
|
|
6
|
Peng XL, Ji MY, Yang ZR, Song J and Dong
WG: Tumor suppressor function of ezrin-radixin-moesin-binding
phospho-protein-50 through β-catenin/E-cadherin pathway in human
hepatocellular cancer. World J Gastroenterol. 19:1306–1313. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ma Q, Jiao Y, Hao Y, Yan S, Lyu N, Gao H,
Li D, Liu Q, Zheng J and Song N: Targeting of NHERF1 through RNA
interference inhibits the proliferation and migration of metastatic
prostate cancer cells. Oncol Lett. 11:1149–1154. 2016.PubMed/NCBI
|
|
8
|
Bellizzi A, Malfettone A, Cardone RA and
Mangia A: NHERF1/EBP50 in breast cancer: Clinical perspectives.
Breast Care (Basel). 5:86–90. 2010. View Article : Google Scholar
|
|
9
|
Song J, Bai J, Yang W, Gabrielson EW, Chan
DW and Zhang Z: Expression and clinicopathological significance of
oestrogen-responsive ezrin-radixin-moesin-binding phosphoprotein 50
in breast cancer. Histopathology. 51:40–53. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cardone RA, Bellizzi A, Busco G, Weinman
EJ, Dell’Aquila ME, Casavola V, Azzariti A, Mangia A, Paradiso A
and Reshkin SJ: The NHERF1 PDZ2 domain regulates
PKA-RhoA-p38-mediated NHE1 activation and invasion in breast tumor
cells. Mol Biol Cell. 18:1768–1780. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shibata T, Chuma M, Kokubu A, Sakamoto M
and Hirohashi S: EBP50, a beta-catenin-associating protein,
enhances Wnt signaling and is over-expressed in hepatocellular
carcinoma. Hepatology. 38:178–186. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kislin KL, McDonough WS, Eschbacher JM,
Armstrong BA and Berens ME: NHERF-1: Modulator of glioblastoma cell
migration and invasion. Neoplasia. 11:377–387. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yan J, Zhang Y, Ren C, Shi W and Chen L:
Involvement of nuclear protein C23 in activation of EGFR signaling
in cervical cancer. Tumour Biol. 37:905–910. 2016. View Article : Google Scholar
|
|
14
|
Shostak K, Zhang X, Hubert P, Göktuna SI,
Jiang Z, Klevernic I, Hildebrand J, Roncarati P, Hennuy B, Ladang
A, et al: NF-κB-induced KIAA1199 promotes survival through EGFR
signalling. Nat Commun. 5:52322014. View Article : Google Scholar
|
|
15
|
Bhattacharyya RP, Reményi A, Yeh BJ and
Lim WA: Domains, motifs, and scaffolds: The role of modular
interactions in the evolution and wiring of cell signaling
circuits. Annu Rev Biochem. 75:655–680. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Clapéron A and Therrien M: KSR and CNK:
Two scaffolds regulating RAS-mediated RAF activation. Oncogene.
26:3143–3158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lazar CS, Cresson CM, Lauffenburger DA and
Gill GN: The Na+/H+ exchanger regulatory
factor stabilizes epidermal growth factor receptors at the cell
surface. Mol Biol Cell. 15:5470–5480. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yao W, Feng D, Bian W, Yang L, Li Y, Yang
Z, Xiong Y, Zheng J, Zhai R and He J: EBP50 inhibits EGF-induced
breast cancer cell proliferation by blocking EGFR phosphorylation.
Amino Acids. 43:2027–2035. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Forni C, Braglia R, Lentini A, Nuccetelli
M, Provenzano B, Tabolacci C and Beninati S: Role of
transglutaminase 2 in quercetin-induced differentiation of B16-F10
murine melanoma cells. Amino Acids. 36:731–738. 2009. View Article : Google Scholar
|
|
20
|
Sun L, Zheng J, Wang Q, Song R, Liu H,
Meng R, Tao T, Si Y, Jiang W and He J: NHERF1 regulates actin
cytoskeleton organization through modulation of α-actinin-4
stability. FASEB J. 30:578–589. 2016. View Article : Google Scholar
|
|
21
|
Yang L, Zheng J, Xiong Y, Meng R, Ma Q,
Liu H, Shen H, Zheng S, Wang S and He J: Regulation of
β2-adrenergic receptor cell surface expression by interaction with
cystic fibrosis trans-membrane conductance regulator-associated
ligand (CAL). Amino Acids. 47:1455–1464. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu SF, Qian WY, Zhang JW, Yang YB, Liu Y,
Dong Y, Zhang ZB, Zhu YP and Feng YJ: Network motifs in the
transcriptional regulation network of cervical carcinoma cells
respond to EGF. Arch Gynecol Obstet. 287:771–777. 2013. View Article : Google Scholar
|
|
23
|
Herbst RS: Review of epidermal growth
factor receptor biology. Int J Radiat Oncol Biol Phys. 59(Suppl):
21–26. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang L, Wang Y, Chen P, Hu J, Xiong Y,
Feng D, Liu H, Zhang H, Yang H and He J: Na(+)/H(+) exchanger
regulatory factor 1 (NHERF1) is required for the
estradiol-dependent increase of phosphatase and tensin homolog
(PTEN) protein expression. Endocrinology. 152:4537–4549. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maudsley S, Zamah AM, Rahman N, Blitzer
JT, Luttrell LM, Lefkowitz RJ and Hall RA: Platelet-derived growth
factor receptor association with Na(+)/H(+) exchanger regulatory
factor potentiates receptor activity. Mol Cell Biol. 20:8352–8363.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pan Y, Weinman EJ and Dai JL:
Na+/H+ exchanger regulatory factor 1 inhibits
platelet-derived growth factor signaling in breast cancer cells.
Breast Cancer Res. 10:R52008. View
Article : Google Scholar
|
|
27
|
Wheeler DS, Barrick SR, Grubisha MJ,
Brufsky AM, Friedman PA and Romero G: Direct interaction between
NHERF1 and Frizzled regulates β-catenin signaling. Oncogene.
30:32–42. 2011. View Article : Google Scholar
|
|
28
|
Kreimann EL, Morales FC, de Orbeta-Cruz J,
Takahashi Y, Adams H, Liu TJ, McCrea PD and Georgescu MM: Cortical
stabilization of beta-catenin contributes to NHERF1/EBP50 tumor
suppressor function. Oncogene. 26:5290–5299. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
He J, Lau AG, Yaffe MB and Hall RA:
Phosphorylation and cell cycle-dependent regulation of
Na+/H+ exchanger regulatory factor-1 by Cdc2
kinase. J Biol Chem. 276:41559–41565. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Noordhuis MG, Eijsink JJ, Ten Hoor KA,
Roossink F, Hollema H, Arts HJ, Pras E, Maduro JH, Reyners AK, de
Bock GH, et al: Expression of epidermal growth factor receptor
(EGFR) and activated EGFR predict poor response to (chemo)radiation
and survival in cervical cancer. Clin Cancer Res. 15:7389–7397.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takahashi Y, Morales FC, Kreimann EL and
Georgescu MM: PTEN tumor suppressor associates with NHERF proteins
to attenuate PDGF receptor signaling. EMBO J. 25:910–920. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim HA, Kim KJ, Seo KH, Lee HK and Im SY:
PTEN/MAPK pathways play a key role in platelet-activating
factor-induced experimental pulmonary tumor metastasis. FEBS Lett.
586:4296–4302. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shen H, Zhu F, Liu J, Xu T, Pei D, Wang R,
Qian Y, Li Q, Wang L, Shi Z, et al: Alteration in Mir-21/PTEN
expression modulates gefitinib resistance in non-small cell lung
cancer. PLoS One. 9:e1033052014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li QL, Gu FM, Wang Z, Jiang JH, Yao LQ,
Tan CJ, Huang XY, Ke AW, Dai Z, Fan J, et al: Activation of
PI3K/AKT and MAPK pathway through a PDGFRβ-dependent feedback loop
is involved in rapamycin resistance in hepatocellular carcinoma.
PLoS One. 7:e333792012. View Article : Google Scholar
|
|
35
|
Zheng J, Dai Y, Yang Z, Yang L, Peng Z,
Meng R, Xiong Y and He J: Ezrin-radixin-moesin-binding
phosphoprotein-50 regulates EGF-induced AKT activation through
interaction with EGFR and PTEN. Oncol Rep. 35:530–537. 2016.
|
|
36
|
Dard N and Peter M: Scaffold proteins in
MAP kinase signaling: More than simple passive activating
platforms. BioEssays. 28:146–156. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cortese MS, Uversky VN and Dunker AK:
Intrinsic disorder in scaffold proteins: Getting more from less.
Prog Biophys Mol Biol. 98:85–106. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bumrungthai S, Munjal K, Nandekar S,
Cooper K, Ekalaksananan T, Pientong C and Evans MF: Epidermal
growth factor receptor pathway mutation and expression profiles in
cervical squamous cell carcinoma: Therapeutic implications. J
Transl Med. 13:2442015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Giaccone G: Epidermal growth factor
receptor inhibitors in the treatment of non-small-cell lung cancer.
J Clin Oncol. 23:3235–3242. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hynes NE and Lane HA: ERBB receptors and
cancer: The complexity of targeted inhibitors. Nat Rev Cancer.
5:341–354. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Marquez A, Wu R, Zhao J, Tao J and Shi Z:
Evaluation of epidermal growth factor receptor (EGFR) by
chromogenic in situ hybridization (CISH) and immunohistochemistry
(IHC) in archival gliomas using bright-field microscopy. Diagn Mol
Pathol. 13:1–8. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Baselga J and Arteaga CL: Critical update
and emerging trends in epidermal growth factor receptor targeting
in cancer. J Clin Oncol. 23:2445–2459. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Takehana T, Kunitomo K, Suzuki S, Kono K,
Fujii H, Matsumoto Y and Ooi A: Expression of epidermal growth
factor receptor in gastric carcinomas. Clin Gastroenterol Hepatol.
1:438–445. 2003. View Article : Google Scholar
|
|
44
|
Al-Kuraya K, Schraml P, Torhorst J, Tapia
C, Zaharieva B, Novotny H, Spichtin H, Maurer R, Mirlacher M,
Köchli O, et al: Prognostic relevance of gene amplifications and
coamplifications in breast cancer. Cancer Res. 64:8534–8540. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fallon KB, Palmer CA, Roth KA, Nabors LB,
Wang W, Carpenter M, Banerjee R, Forsyth P, Rich K and Perry A:
Prognostic value of 1p, 19q, 9p, 10q, and EGFR-FISH analyses in
recurrent oligodendrogliomas. J Neuropathol Exp Neurol. 63:314–322.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Qureshi R, Arora H, Biswas S, Perwez A,
Naseem A, Wajid S, Gandhi G and Rizvi MA: Mutation analysis of EGFR
and its correlation with the HPV in Indian cervical cancer
patients. Tumour Biol. Jan 14–2016.(Epub ahead of print).
View Article : Google Scholar : PubMed/NCBI
|