Introduction
The tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL) is an attractive anticancer agent, since it
selectively induces apoptosis in a variety of cancer cells
(1–4). Furthermore, TRAIL exhibits potent
tumoricidal activity against tumor xenografts, without serious
side-effects in preclinical studies (2,3),
implicating the potential utility of TRAIL in the treatment of
human cancer.
TRAIL-induced apoptosis is initiated by the binding
of TRAIL to its functional death receptors, TRAIL-R1 (DR4) and
TRAIL-R2 (DR5). These TRAIL receptors have death domains (DD) in
their cytoplasmic tails, which recruit the adaptor protein
Fas-associated death domains (FADD) and trigger the formation of
the death-inducing signaling complex -forms of initiator caspases,
such as caspase-8, are activated and subsequently trigger the
activation of downstream effector caspases with or without
mitochondrial amplification via cleavage of the BH3-only Bcl-2
family member Bid. TRAIL can also bind the decoy receptors TRAIL-R3
(DcR1) and TRAIL-R4 (DcR2), which possess incomplete cytoplasmic
regions, and the soluble receptor osteoprotegerin. These receptors
may inhibit the apoptotic pathway by competing for TRAIL with
active death receptors (5). Higher
expression of decoy receptors in non-transformed cells is
considered one of the mechanisms that confer TRAIL selectivity to
cancer cells. In addition, multiple pathways, including cellular
FLICE-like inhibitory protein (c-FLIP), anti-apoptotic B-cell
lymphoma 2 proteins (Bcl-2), and X-linked inhibitor of apoptosis
protein (XIAP) pathways, may confer insensitivity to TRAIL in
non-transformed cells (6).
However, these mechanisms can also be utilized in resistant cancer
cells to counteract to the antitumor effect of TRAIL. In fact,
decoy R, c-FLIP and Bcl-2 are overexpressed in several
TRAIL-resistant cancer cells (7–9). In
addition, resistance to TRAIL could be gained at various other
steps in the signaling pathways of apoptosis.
Currently, TRAIL resistance in cancer cells has
emerged as the major obstacle to successful TRAIL therapy. To date,
the mechanisms underlying TRAIL resistance have been mostly studied
in TRAIL-sensitive cancer cells to understand how sensitive cells
acquire TRAIL resistance. However, it is also important to
understand how less sensitive cancer cells can gain much stronger
resistance to TRAIL after TRAIL exposure, since cancers frequently
display heterogeneous features, and even cancer cells with primary
resistance could develop stronger resistance via various mechanisms
after exposure to TRAIL. In this study, we observed an unexpected
role of DR4 in TRAIL-resistant HepG2 cells and examined the
mechanism underlying DR4-mediated resistance in HepG2-TR cells.
Materials and methods
Cell culture
The HepG2 HCC cell line, AGS and SNU601 gastric
cancer cell lines, and HCT116 colon cancer cell line were obtained
from the Korean Cell Line Bank (Seoul, Korea) and cultured in
RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with
10% (v/v) fetal bovine serum (FBS) and 1% antibiotics at 37°C in a
5% CO2 atmosphere. A TRAIL-resistant variant of HepG2
cells (HepG2-TR) was obtained by periodically exposing the HepG2
cell line to increasing concentrations (5–100 ng/ml) of recombinant
human TRAIL (rhTRAIL; a gift from T.H. Kim, Department of
Biochemistry and Molecular Biology, Chosun University, Gwangju,
Korea) and selecting for resistance to TRAIL-induced apoptosis.
Cell viability assays
For cell viability detection, cells were plated in a
96-well plate at a density of 1×104 cells/well,
incubated for 24 h, and then treated with rhTRAIL for 48 h. Next,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
solution (0.5 mg/ml) was added to the wells and plates were
incubated at 37°C in a CO2 incubator for 4 h. The plates
were centrifuged at 600 × g for 10 min and the culture medium was
removed. The cells were solubilized using dimethyl sulfoxide and
the solubilized formazan product was quantified using an
enzyme-linked immunosorbent assay plate reader at 595 nm. The
absorbance of the untreated cells was set to 100% and cell survival
was expressed as a percentage of this value.
Apoptosis analysis
For Hoechst 33342 staining, cells were stained with
1 μg/ml Hoechst 33342 for 15 min at room temperature in the dark,
and both the floating and attached cells were collected and
centrifuged. The pooled cell pellets were washed with ice-cold
phosphate-buffered saline (PBS), fixed in 3.7% formaldehyde on ice,
washed and resuspended in PBS, and then a fraction of the
suspension was centrifuged using a cyto-spinner (Shandon; Thermo
Fisher Scientific, Waltham, MA, USA). Slides were prepared, air
dried, mounted in anti-fade solution, and observed under a
fluorescence microscope (DM5000; Leica Microsystems, Washington DC,
USA), as described elsewhere. Any condensed/fragmented nuclei were
assessed as apoptotic cells. In total, 500 cells distributed across
random microscope viewing fields were counted, and the number of
apoptotic cells is expressed as a percentage of the total number of
cells counted. For the flow cytometric analysis, cells were washed
with 1% Triton X-100/PBS and fixed with cold methanol for 30 min.
After washing, fixed cells were incubated with 20 μg/ml DNAse-free
RNase A for 30 min at 37°C, then with 50 μg/ml propidium iodide for
an additional 40 min on ice. The cells were washed and subjected to
fluorescence-activated cell sorting analysis (NAVIOS flow
cytometer; Beckman Coulter, Inc., Brea, CA, USA).
Autophagy analysis
For autophagy detection, the CYTO-ID Autophagy
detection kit was used according to the manufacturer's instruction
(Enzo Life Sciences, Farmingdale, NY, USA). In brief, cells
cultured on a 96-well microplate were treated with TRAIL and a
positive control (a combination of rapamycin and chloroquine) for
48 h. After treatment, the medium was removed and cells were rinsed
with assay buffer. Then, cells were incubated with dual-color
detection solution (CYTO-ID Green/Hoechst 33342) diluted in assay
buffer at 37°C for 30 min, rinsed twice, and analyzed with a
fluorescence microplate reader. CYTO-ID Green was detected using a
FITC filter (Ex=488/Em=530) and Hoechst 33342 was read with a DAPI
filter set (Ex=340/Em=480). The relative green fluorescence
intensity was normalized against nuclear blue fluorescence
intensity. To observe autophagic vacuoles by fluorescence
microscopy, treated cells were incubated with 10 μM
monodansylcadaverine for 30 min, washed with PBS, and observed
under an inverted fluorescence microscope at 340/525 nm.
Immunoblotting
Equal amounts of protein extracts were
electrophoretically separated using 10–12% SDS-PAGE and transferred
to a nitrocellulose membrane using standard techniques. Antibodies
were used to probe for DR4, DR5 (ProSci, Inc., Poway, CA, USA),
caspase-3, caspase-8, phospho-JNK1/2, total JNK1/2 (Cell Signaling
Technology, Danvers, MA, USA), cytochrome c, caveolin-1
(Santa Cruz Biotechnology, Santa Cruz, CA, USA), and LC3 (Novus
Biologicals, Littleton, CO, USA). Anti-α-tubulin (BioGenex,
Fremont, CA, USA) was used as a loading control. Signals were
acquired using an Image Station 4000MM Image Analyzer (Kodak,
Rochester, NY, USA).
RNA interference (RNAi)
For the RNAi experiment, siRNA targeting DR4
(#1), 5′-CUGGAAAGUUCAUCUACUU (dtdt)-3′ (sense) and
5′-AAGUAGAUGAACUUUCCAG (dtdt)-3′ (antisense); DR5,
5′-CAGACUUGGUGCCCUUUG (dtdt)-3′ (sense) and 5′-UCAAAGGGCACCAAGUCUG
(dtdt)-3′ (antisense), and 5′-control siRNA, 5′-CCUACGCCAC
CAAUUUCGU(dtdt)-3′ (sense) and 5′-ACGAAAUUGGUG GCGUAGG (dtdt)-3′
(antisense) were purchased from Bioneer (Daejeon, Korea). siRNA
targeting DR4 (#2) (sc-35218) was purchased from Santa Cruz
Biotechnology. Cells were individually transfected with siRNA
oligonucleotides using the jetPEI transfection reagent
(Polyplus-transfection SA, Illkirch, France) and grown for 36 h
prior to the drug treatment.
Statistical analysis
All numerical data are reported as means ± SE. All
data represent the results of at least three independent
experiments. Student's t-tests were used to evaluate the
differences in means between control and treatment groups, and
one-way ANOVA was applied to analyze differences caused by gene
silencing or inhibitor treatment.
Results
Exposure to rhTRAIL increases the TRAIL
resistance of HepG2 cells
Consistent with previous studies reporting the
resistance of HCC cells to TRAIL (10–12),
human HCC HepG2 cells were relatively resistant to TRAIL-induced
cytotoxicity compared with other human cancer cell lines including
HCT116 colorectal cancer cells, and SNU601 and AGS gastric cancer
cells. HCT116 and SNU601 cells were sensitive to as low as 2 ng/ml
rhTRAIL, but HepG2 and AGS cells were less sensitive to TRAIL even
at 10 ng/ml rhTRAIL (Fig. 1A). In
this study, we found that HepG2 cells which show primary resistance
to TRAIL become more resistant after exposure to rhTRAIL. To
understand the mechanisms by which cancer cells gain stronger
resistance to TRAIL, we developed stably growing cells at high
concentrations of TRAIL (HepG2-TR) by exposing HepG2 cells to
stepwise increases in rhTRAIL concentrations (5–100 ng/ml). The
TRAIL-resistant cells were verified by detecting cell viability and
apoptosis after rhTRAIL application. As shown in Fig. 1B and C, <20% loss of viability
was detected in the HepG2-TR cells after exposure to 300 ng/ml
rhTRAIL for 24 h, in contrast to 80% loss of viability in parental
HepG2 cells. Consistently, apoptotic bodies in response to rhTRAIL
were significantly decreased in the HepG2-TR cells.
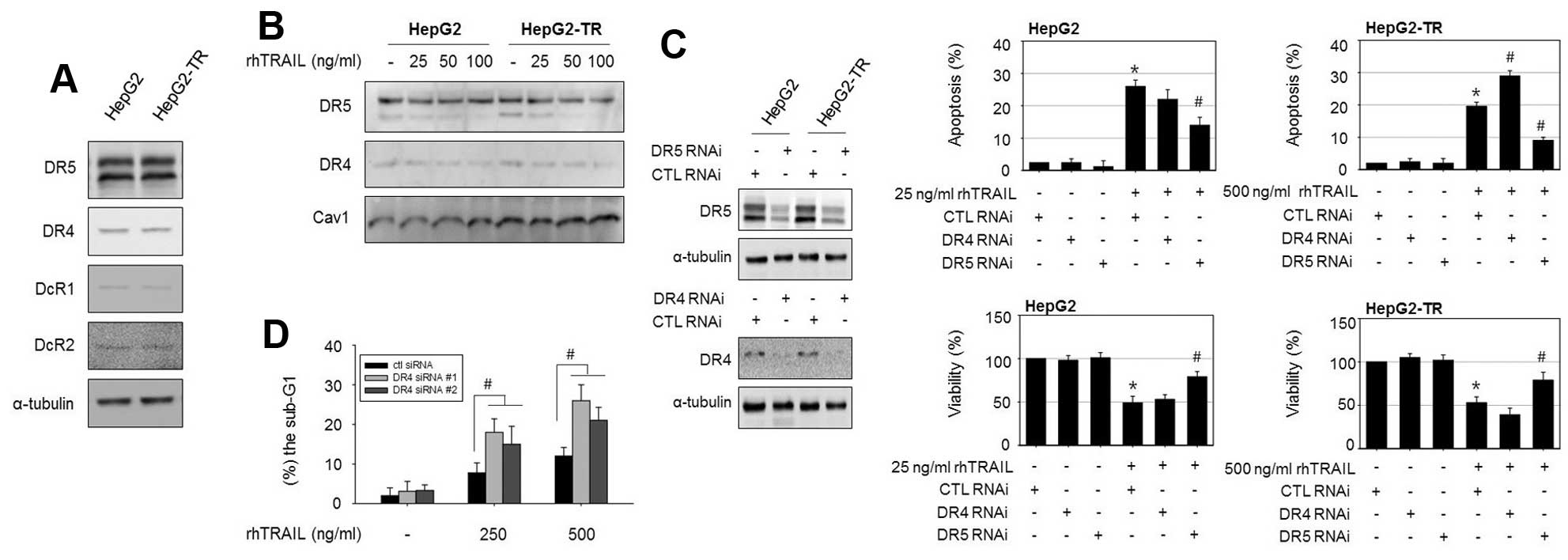
DR4 is involved in the resistance to
rhTRAIL-induced apoptosis in HepG2-TR cells
Alterations in the expression levels or functional
activity of TRAIL receptors could be linked to the sensitivity to
TRAIL in various cancer cell types. However, an immunoblotting
analysis revealed that the protein levels of the death receptors
DR4 and DR5 and decoy receptors DcR1 and DcR2 did not differ
between HepG2-TR cells and parental HepG2 cells (Fig. 2A). Next, we analyzed the membrane
expression of DR4 and DR5 in HepG2 cells and HepG2-TR cells using
membranous cell fractions because the membrane localization of DRs
is thought to be essential for TRAIL-induced apoptosis. However,
the membrane protein levels of DR4 and DR5 were also similar
between cell types (Fig. 2B). We
explored whether alterations in the functional activity of death
receptors are involved in the characteristic resistance of HepG2-TR
cells to rhTRAIL using small interfering RNAs. DR4 and
DR5 were silenced in both cells and TRAIL-induced apoptosis
and viability were examined. As shown in Fig. 2C, the transfection efficiency of
DR4 and DR5 siRNA was confirmed by immunoblotting. In parental
HepG2 cells, 25 ng/ml rhTRAIL triggered apoptosis in ~26% of cells,
and silencing of both DR4 and DR5 reduced
rhTRAIL-induced apoptosis. However, in the HepG2-TR cells, the
knockdown of DR4 increased apoptosis, while the knockdown of
DR5 attenuated TRAIL-induced apoptosis. Interestingly, we
observed that DR4 silencing sensitizes HepG2-TR cells, but
not parental HepG2 cells, to rhTRAIL. To further confirm the role
of DR4 in rhTRAIL-induced apoptosis in HepG2-TR cells, we used
additional DR4 siRNAs targeting different regions of the
DR4 gene and confirmed the apoptotic rate in response to
rhTRAIL by a flow cytometric analysis. Similarly, transfection with
DR4 siRNA (#1 and #2) elevated rhTRAIL-induced apoptosis
compared to control siRNA-transfected cells, as demonstrated by the
increased proportion of cells in the sub-G1 phase
(Fig. 2D). These results again
suggested that DR4 interference made the HepG2-TR cells more
sensitive to rhTRAIL-induced cytotoxicity.
rhTRAIL induces protective autophagy in
HepG2-TR cells
A recent study suggested that TRAIL resistance is
correlated with the activation of autophagy in various cancer
cells. To explore whether autophagy is linked with TRAIL resistance
in HepG2-TR cells, the cells were stained with
monodansylcadaverine, a fluorescent compound that labels autophagic
vacuoles. Upon treatment with rhTRAIL, the number of autophagic
vacuoles increased in the HepG2-TR cells, but not in the parental
HepG2 cells (Fig. 3A). In
addition, treatment with rhTRAIL elevated the protein levels of
LC3-II, a molecular indicator of autophagy, in HepG2-TR cells, but
did not have a significant effect on the levels of LC3II in HepG2
cells (Fig. 3B). These results
indicate that rhTRAIL triggers higher levels of autophagy in
HepG2-TR cells compared to the parental cells. In response to
cytotoxic stimuli, the activation of autophagy may function as a
protective mechanism for survival or as a mechanism of cell death,
depending on the cell type and genetic context. To determine the
role of autophagy in rhTRAIL-induced cell death in HepG2-TR cells,
we employed the autophagy inhibitor 3-methyladenine. While
3-methyladenine had little cytotoxicity, it increased TRAIL-induced
apoptosis considerably in HepG2-TR cells, as evidenced by an
increase in apoptotic nuclei, and enhanced rhTRAIL-induced
cytotoxicity, as evidenced by a reduction in cell viability
(Fig. 3C). To further confirm
these observations, siRNA targeting ATG5 was used to prevent
autophagy. The knockdown of ATG5, which was confirmed by western
blotting, significantly enhanced rhTRAIL-induced apoptosis and
reduced cell viability (Fig. 3D).
Combined, these results suggest that autophagy triggered by rhTRAIL
plays a protective role in HepG2-TR cells.
DR4 plays a role in rhTRAIL-induced
protective autophagy in HepG2-TR cells
In the present study, rhTRAIL triggered protective
autophagy in HepG2-TR cells, and DR4 was associated with the
resistance of HepG2-TR cells to rhTRAIL. Thus, we investigated
whether DR4 is related to the induction of autophagy in HepG2-TR
cells. To this end, we transfected the HepG2-TR cells with
DR4 siRNA, and assessed the occurrence of rhTRAIL-induced
autophagy based on fluorescence staining of autophagic vesicles. As
shown in Fig. 4A, the knockdown of
DR4 siRNA significantly reduced the number of
rhTRAIL-induced autophagic vesicles compared to that of the control
RNA transfected HepG2-TR cells. Furthermore, the silencing of
DR4 suppressed the rhTRAIL-mediated increase in the LC3II
protein level and elevated the cleavage of caspase-3 and release of
cytochrome c in HepG2-TR cells (Fig. 4B). However, the knockdown of
DR5 had no significant effect on the induction of autophagic
vesicles or the elevation of LC3II levels in response to rhTRAIL.
Thus, DR4 appears to play an anti-apoptotic role by contributing to
the induction of autophagy in HepG2-TR cells. Furthermore,
DR4 knockdown increased the levels of the active form of
caspase-8 and caspase-8 activity upon exposure to rhTRAIL in
HepG2-TR cells (Fig. 4C and D).
Therefore, rhTRAIL-mediated DR4 signaling may induce autophagy and
interfere with DISC activation in the resistant cells.
 | Figure 4DR4 mediates rhTRAIL-induced autophagy
in HepG2-TR cells. (A) HepG2-TR cells were transfected with DR4
siRNA, DR5 siRNA, or control siRNA, and exposed to 200 ng/ml
rhTRAIL for 24 h. Autophagy was detected using an autophagy assay
kit and results are presented as relative fluorescence units. As a
positive control, a combination of 1 μM rapamycin and 10 μM
chloroquine was used. *P<0.05 vs. control;
#P<0.05 vs. CTL RNAi transfected cells treated with
rhTRAIL. (B–D) HepG2-TR cells transfected with DR4 siRNA, DR5
siRNA, or control siRNA were exposed to 200 ng/ml rhTRAIL for 24 h,
and then subjected to immunoblotting to detect LC3II, cleaved
caspase-3, cytosolic cytochrome c, DR4, DR5 (B), and cleaved
caspase-8 (C), or to a caspase-8 activity assay (D).
#P<0.05 vs. CTL RNAi transfected cells treated with
rhTRAIL. |
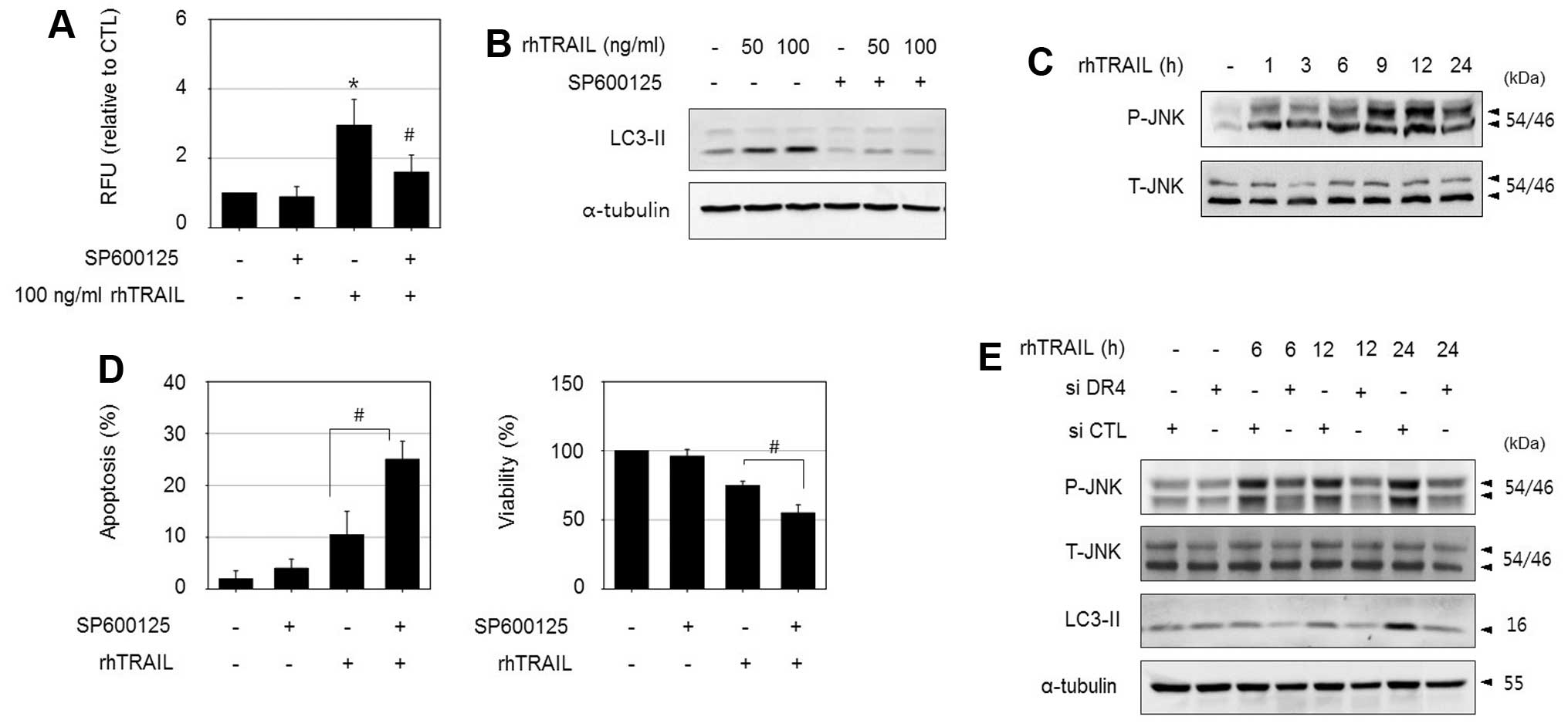
Activation of c-Jun N-terminal kinase
(JNK) is involved in DR4-mediated autophagy in HepG2-TR cells
In addition to the activation of the apoptotic
pathway via DISC formation, TRAIL activates pro-survival pathways
such as NF-κB, PI3K/Akt, ERK and JNK signaling, which confer
resistance to TRAIL (13–15). Furthermore, a number of studies
have shown that the prevention of MAPK pathways can sensitize some
TRAIL-resistant tumor cells to apoptosis, suggesting a possible
anti-apoptotic role of MAPK (16,17).
To determine the signaling molecules involved in
DR4-mediated autophagy in HepG2-TR cells, we investigated the roles
of several essential kinases. Among them, the JNK inhibitor
SP600125 significantly reduced rhTRAIL-induced autophagy in
HepG2-TR cells, as determined by an autophagy assay, and decreased
the levels of LC3II (Fig. 5A and
B). In addition, treatment with rhTRAIL increased
phosphorylated JNK levels based on an immunoblot assay (Fig. 5C). Consistent with the
cytoprotective role of autophagy, the suppression of autophagy by
combined treatment with SP600125 enhanced rhTRAIL-induced apoptosis
based on apoptotic body formation, and increased the sensitivity to
rhTRAIL as assessed by an MTT assay (Fig. 5D). Next, we examined whether JNK
activation is associated with the DR4 signaling pathway. Although
inhibition of JNK using SP600125 did not affect DR4 expression
level (data not shown), the silencing of DR4 reduced
rhTRAIL-induced JNK phosphorylation and suppressed LC3II
accumulation in HepG2-TR cells as demonstrated in Fig. 5E. These results indicate that JNK
is a downstream regulator of DR4 activation. Collectively, these
results suggest that the mechanism underlying TRAIL resistance is
at least partially mediated by DR4/JNK-induced autophagy in
HepG2-TR cells.
Discussion
Tumor cell resistance to antitumor drugs is a major
obstacle in cancer therapy, including TRAIL-based approaches. Some
cancer cells are intrinsically resistant to TRAIL, and even
initially sensitive cancer cells often acquire stronger resistance
during constant and repetitive exposure to TRAIL. The development
of TRAIL resistance is attributed to the genetic heterogeneity and
adaptive responses of cancer cells. These characteristics enable
cancer cells to survive toxic environments and lead to the
selection and expansion of survived cells after TRAIL treatment. In
addition, a variety of defects that block the apoptotic cascade may
be important factors in cancer cell resistance to TRAIL, and can be
caused at both the receptor signaling level, e.g., diminished
membrane surface expression of the TRAIL receptors DR4 or DR5
(18) and induction of c-FLIP, and
at the intrinsic mitochondrial level, e.g., overexpression of the
anti-apoptotic Bcl-2 family protein MCL-1 (19). Both DR4 and DR5 are transmembrane
receptors for TRAIL with cytoplasmic death domains, which can
transduce apoptotic signaling via DISC formation. They share a
sequence homology of 58% and induce apoptosis via similar
mechanisms. However, differences in the roles of DR4 and DR5 in the
induction of apoptosis have been demonstrated in various tumor cell
types (20–22), and it is uncertain whether it is
more advantageous to target one or both DRs for effective
treatment, particularly given that the effects appear to be tumor
cell-specific.
One of the essential factors that determine the
differential contribution of death receptors to TRAIL-induced
apoptosis may be the expression of DR4 and DR5. In agreement with
this hypothesis, several antitumor agents, including
ursodeoxycholic acid, andrographolide and curcumin, which increase
the expression of DR4 or DR5, promote the sensitivity of various
cancer cells to TRAIL (10,23,24).
In particular, the cell membrane expression of DRs appears to be
important, since their membrane distribution is necessary for
ligand binding to initiate apoptotic signaling. A decrease in the
cell surface expression of DR4 or DR5 is correlated with a decrease
in TRAIL sensitivity in myeloma cells and breast cancer cells
(18,25). Post-translational modifications,
such as glycosylation and palmitoylation, of DRs are also predicted
to be important mechanisms in the regulation of the sensitivity of
TRAIL-induced apoptosis (26).
Post-translational modification of DRs appears to be linked to the
activation of death receptors, including recruitment to membrane
rafts (27).
However, the TRAIL receptors DR4 and DR5 not only
triggered apoptosis, but also induced survival pathways in
resistant tumor cells, depending on cellular context. The known
non-canonical signaling events triggered by death receptors include
the activation of various kinases, such as RIP1, NF-κB, JNK, p38
kinase, ERK1/2, TAK1 and PI3K/Akt (28). In this study, we observed that DR4
is involved in TRAIL resistance in HepG2-TR cells. In parental
HepG2 cells, although DR4 was much less effective than DR5 in
TRAIL-induced apoptosis, DR4 did not confer resistance.
Nevertheless, rhTRAIL induced autophagy in HepG2-TR cells, as
detected by autophagic vacuoles and LC3II levels, and this was at
least partially regulated by DR4. The knockdown of DR4 not only
reduced the autophagic features, but also restored apoptosis and
cytotoxicity. These results indicate that DR4 function switched to
play an antagonistic role in TRAIL-induced apoptosis by triggering
protective autophagy in HepG2-TR cells. Similar to our results, an
anti-apoptotic role of DR4 has been reported in human lung cancer
cells, in which the geranylgeranyltransferase inhibitor GGTI298
effectively augmented TRAIL-induced apoptosis in resistant cells,
and in these conditions, the knockdown of DR5 attenuated apoptosis,
but the knockdown of DR4 sensitized cancer cells to
GGTI298/TRAIL-induced apoptosis (29).
Autophagy is an alternative mechanism for cell
death, referred to as type II cell death; however, it also acts as
an adaptive cell response mechanism, protecting cells from
bioenergetic stress. Various cytotoxic agents that induce apoptosis
activate protective autophagy in certain cancer cells (30,31).
TRAIL was also shown to be able to induce both apoptosis and
autophagy in cancer cells. Furthermore, autophagy attenuates
TRAIL-induced apoptosis and is related to the resistance of cancer
cells to TRAIL treatment. The prevention of a TRAIL-mediated
autophagic pathway by silencing autophagic genes facilitates
TRAIL-induced apoptosis (32).
These results are in accordance with our findings that the
occurrence of autophagy in response to TRAIL confers resistance to
HepG2-TR cells.
Although the mechanism by which DR4 signaling
triggers autophagy is not fully understood, it appears that a
downstream effector of DR4 switched to an autophagic regulator from
an apoptotic DISC factor during the adaptive response to rhTRAIL,
as evidenced by the suppression of caspase-8 activation in HepG2-TR
cells. In an analysis of the mechanisms involved in DR4-mediated
autophagic signaling in HepG2-TR cells, we found that JNK
activation is linked to the induction of autophagy. Previous
studies demonstrated that TRAIL can activate the JNK pathway in
several cancer cell lines via dual effects, cell death or survival,
depending on the cellular context (33). In the present study, JNK appeared
to mediate DR4-signalled autophagy, as evidenced by suppression of
JNK phosphorylation after DR4 silencing and decrease in autophagy
by JNK inhibition. Thus, the DR4/JNK axis may play an essential
role in the TRAIL-mediated autophagic pathway in HepG2-TR
cells.
However, the induction of DR4-mediated autophagy may
not be the sole explanation for the resistance of HepG2-TR cells
because blocking DR4 did not completely reduce cell viability.
Previously, the inhibition of proteasomes by MG132 or PS-341
rescues TRAIL sensitivity in TRAIL-resistant HepG2 cells (34), suggesting that proteasomes is
another resistance mechanism. It was also hypothesized that
TRAIL-resistant HepG2 cells can be sensitized by decrease in cFLIP
expression and caspase-8 activation (11), which is consistent with our results
regarding the suppression of caspase-8 activity in resistant cells,
although the resistance mechanism is different. This disparity
could be explained by the different conditions with respect to
TRAIL exposure; differences in the concentration and treatment
duration may induce different tolerance mechanisms. Thus, even
within a cell line, there appears to be various resistance
mechanisms depending on micro-environmental conditions. The results
of this study extend our understanding of TRAIL resistance
mechanisms and may contribute to development of strategies to
overcome TRAIL resistance.
Acknowledgements
The present study was supported by a research fund
from the Chosun University, 2014 (K207007001). We thank Professor
Tae-Hyoung Kim for the kind gift of rhTRAIL and Ms. Jeong-Eun Choi
for her excellent technical assistance.
References
|
1
|
Pitti RM, Marsters SA, Ruppert S, Donahue
CJ, Moore A and Ashkenazi A: Induction of apoptosis by Apo-2
ligand, a new member of the tumor necrosis factor cytokine family.
J Biol Chem. 271:12687–12690. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ashkenazi A, Pai RC, Fong S, Leung S,
Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert
A, et al: Safety and antitumor activity of recombinant soluble Apo2
ligand. J Clin Invest. 104:155–162. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Walczak H, Miller RE, Ariail K, Gliniak B,
Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, et al:
Tumoricidal activity of tumor necrosis factor-related
apoptosis-inducing ligand in vivo. Nat Med. 5:157–163. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wiley SR, Schooley K, Smolak PJ, Din WS,
Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA,
et al: Identification and characterization of a new member of the
TNF family that induces apoptosis. Immunity. 3:673–682. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Falschlehner C, Emmerich CH, Gerlach B and
Walczak H: TRAIL signalling: Decisions between life and death. Int
J Biochem Cell Biol. 39:1462–1475. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
van Dijk M, Halpin-McCormick A, Sessler T,
Samali A and Szegezdi E: Resistance to TRAIL in non-transformed
cells is due to multiple redundant pathways. Cell Death Dis.
4:e7022013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rippo MR, Moretti S, Vescovi S, Tomasetti
M, Orecchia S, Amici G, Catalano A and Procopio A: FLIP
overexpression inhibits death receptor-induced apoptosis in
malignant mesothelial cells. Oncogene. 23:7753–7760. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu X, Yue P, Khuri FR and Sun SY: Decoy
receptor 2 (DcR2) is a p53 target gene and regulates
chemosensitivity. Cancer Res. 65:9169–9175. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hetschko H, Voss V, Horn S, Seifert V,
Prehn JH and Kögel D: Pharmacological inhibition of Bcl-2 family
members reactivates TRAIL-induced apoptosis in malignant glioma. J
Neurooncol. 86:265–272. 2008. View Article : Google Scholar
|
|
10
|
Zhou J, Lu GD, Ong CS, Ong CN and Shen HM:
Andrographolide sensitizes cancer cells to TRAIL-induced apoptosis
via p53-mediated death receptor 4 up-regulation. Mol Cancer Ther.
7:2170–2180. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ganten TM, Haas TL, Sykora J, Stahl H,
Sprick MR, Fas SC, Krueger A, Weigand MA, Grosse-Wilde A, Stremmel
W, et al: Enhanced caspase-8 recruitment to and activation at the
DISC is critical for sensitisation of human hepatocellular
carcinoma cells to TRAIL-induced apoptosis by chemotherapeutic
drugs. Cell Death Differ. 11(Suppl 1): S86–S96. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pathil A, Armeanu S, Venturelli S,
Mascagni P, Weiss TS, Gregor M, Lauer UM and Bitzer M: HDAC
inhibitor treatment of hepatoma cells induces both
TRAIL-independent apoptosis and restoration of sensitivity to
TRAIL. Hepatology. 43:425–434. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song JH, Tse MC, Bellail A, Phuphanich S,
Khuri F, Kneteman NM and Hao C: Lipid rafts and nonrafts mediate
tumor necrosis factor related apoptosis-inducing ligand induced
apoptotic and nonapoptotic signals in non small cell lung carcinoma
cells. Cancer Res. 67:6946–6955. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu J, Zhou JY, Wei WZ and Wu GS:
Activation of the Akt survival pathway contributes to TRAIL
resistance in cancer cells. PLoS One. 5:e102262010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang L, Dittmer MR, Blackwell K, Workman
LM, Hostager B and Habelhah H: TRAIL activates JNK and NF-κB
through RIP1-dependent and -independent pathways. Cell Signal.
27:306–314. 2015. View Article : Google Scholar
|
|
16
|
Secchiero P, Gonelli A, Carnevale E,
Milani D, Pandolfi A, Zella D and Zauli G: TRAIL promotes the
survival and proliferation of primary human vascular endothelial
cells by activating the Akt and ERK pathways. Circulation.
107:2250–2256. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang XD, Borrow JM, Zhang XY, Nguyen T
and Hersey P: Activation of ERK1/2 protects melanoma cells from
TRAIL-induced apoptosis by inhibiting Smac/DIABLO release from
mitochondria. Oncogene. 22:2869–2881. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Y and Zhang B: TRAIL resistance of
breast cancer cells is associated with constitutive endocytosis of
death receptors 4 and 5. Mol Cancer Res. 6:1861–1871. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Han J, Goldstein LA, Gastman BR and
Rabinowich H: Interrelated roles for Mcl-1 and BIM in regulation of
TRAIL-mediated mitochondrial apoptosis. J Biol Chem.
281:10153–10163. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
MacFarlane M, Inoue S, Kohlhaas SL, Majid
A, Harper N, Kennedy DB, Dyer MJ and Cohen GM: Chronic lymphocytic
leukemic cells exhibit apoptotic signaling via TRAIL-R1. Cell Death
Differ. 12:773–782. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lemke J, Noack A, Adam D, Tchikov V,
Bertsch U, Röder C, Schütze S, Wajant H, Kalthoff H and Trauzold A:
TRAIL signaling is mediated by DR4 in pancreatic tumor cells
despite the expression of functional DR5. J Mol Med (Berl).
88:729–740. 2010. View Article : Google Scholar
|
|
22
|
Kelley RF, Totpal K, Lindstrom SH, Mathieu
M, Billeci K, Deforge L, Pai R, Hymowitz SG and Ashkenazi A:
Receptor-selective mutants of apoptosis-inducing ligand 2/tumor
necrosis factor-related apoptosis-inducing ligand reveal a greater
contribution of death receptor (DR) 5 than DR4 to apoptosis
signaling. J Biol Chem. 280:2205–2212. 2005. View Article : Google Scholar
|
|
23
|
Lim SC, Duong HQ, Choi JE, Lee TB, Kang
JH, Oh SH and Han SI: Lipid raft-dependent death receptor 5 (DR5)
expression and activation are critical for ursodeoxycholic
acid-induced apoptosis in gastric cancer cells. Carcinogenesis.
32:723–731. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jung EM, Park JW, Choi KS, Park JW, Lee
HI, Lee KS and Kwon TK: Curcumin sensitizes tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis
through CHOP-independent DR5 upregulation. Carcinogenesis.
27:2008–2017. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gómez-Benito M, Martinez-Lorenzo MJ, Anel
A, Marzo I and Naval J: Membrane expression of DR4, DR5 and
caspase-8 levels, but not Mcl-1, determine sensitivity of human
myeloma cells to Apo2L/TRAIL. Exp Cell Res. 313:2378–2388. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gonzalvez F and Ashkenazi A: New insights
into apoptosis signaling by Apo2L/TRAIL. Oncogene. 29:4752–4765.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Linderoth E, Pilia G, Mahajan NP and Ferby
I: Activated Cdc42-associated kinase 1 (Ack1) is required for tumor
necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor
recruitment to lipid rafts and induction of cell death. J Biol
Chem. 288:32922–32931. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Azijli K, Weyhenmeyer B, Peters GJ, de
Jong S and Kruyt FA: Non-canonical kinase signaling by the death
ligand TRAIL in cancer cells: Discord in the death receptor family.
Cell Death Differ. 20:858–868. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen S, Fu L, Raja SM, Yue P, Khuri FR and
Sun SY: Dissecting the roles of DR4, DR5 and c-FLIP in the
regulation of geranylgeranyltransferase I inhibition-mediated
augmentation of TRAIL-induced apoptosis. Mol Cancer. 9:232010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Han J, Hou W, Goldstein LA, Lu C, Stolz
DB, Yin XM and Rabinowich H: Involvement of protective autophagy in
TRAIL resistance of apoptosis-defective tumor cells. J Biol Chem.
283:19665–19677. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mahalingam D, Keane M, Pirianov G, Mehmet
H, Samali A and Szegezdi E: Differential activation of JNK1
isoforms by TRAIL receptors modulate apoptosis of colon cancer cell
lines. Br J Cancer. 100:1415–1424. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ganten TM, Koschny R, Haas TL, Sykora J,
Li-Weber M, Herzer K and Walczak H: Proteasome inhibition
sensitizes hepatocellular carcinoma cells, but not human
hepatocytes, to TRAIL. Hepatology. 42:588–597. 2005. View Article : Google Scholar : PubMed/NCBI
|