Introduction
Glioblastoma multiforme (GBM) is the most prevalent
and aggressive malignant tumour of the central nervous system.
Despite progress in therapeutic strategies combining surgery,
radio-chemotherapy and immunotherapy, patients with GBM still have
a dismal prognosis (1,2). Therefore, elucidation of the
molecular mechanisms underlying the development and progression of
GBM is needed to explore new specific therapeutic strategies.
Macrophage migration inhibitory factor (MIF) was
first identified due to its role in inhibition of macrophage
migration (3). After decades of
research, MIF has been shown to play a vital role in both immune
response and tumourigenesis (4).
In addition to inhibition of macrophage migration, MIF blocks NK
cell activity (5), decreases acute
inflammation and mediates chronic inflammation, which partly
promotes tumourigenesis (6–8). As
a promotor of tumourigenesis, MIF is strongly expressed in various
types of malignant cancers, including GBM (9,10).
MIF exhibits a broad spectrum of pro-neoplastic activities,
enhancing tumour cell proliferation (11), inducing angiogenesis (12), and reducing tumour cell death by
inhibition of p53 (13). In recent
studies, MIF has been shown to contribute to malignant progression
of GBM. These results indicate that MIF is a promising target for
anti-GBM therapy. However, the signalling pathways activated by MIF
have not been completely elucidated thus far.
Autophagy is a highly conserved cellular catabolic
pathway that monitors, degrades and recycles intracellular proteins
and organelles in lysosomes to support metabolism and promotes cell
survival (14,15). It is believed to play a contextual
role in cancer (16). In 40–75% of
specific cancers, such as human prostate, breast, and ovarian
cancers, the essential autophagy gene ATG6/BECN1 was lost (17–19).
However, in many cancers, autophagy was a tumour promoter that was
involved in tumour initiation and development (20). High levels of autophagy in glioma,
especially under stressful microenvironments, promote malignant
progression. To date, autophagy has been suggested to be a
potential target for glioma therapy.
Rho-associated coiled-coil containing kinases
(ROCKs) are serine/threonine kinases that are central regulators of
the actomyosin cytoskeleton (20).
There are two mammalian ROCK homologs, ROCK1 and 2. ROCKs were
initially recognized as activated Rho GTPase-binding proteins. The
Rho GTPase family is known for its regulation of actin cytoskeleton
organization and dynamics (21).
RhoA and B, which belong to the Rho GTPase proteins, are the best
characterized ROCK regulators (22,23).
Activation of ROCKs promotes the formation of stress fibres and
actomyosin contraction via phosphorylation of numerous downstream
target proteins, including the myosin regulatory light chain (MLC)
and the myosin-binding subunit (MYPT1) (24–26).
In addition, extensive studies have demonstrated that ROCK1 has a
diverse range of functions in tumourigenesis, including cell
contraction, migration, apoptosis, survival, and proliferation.
Dendritic cells (DCs) are professional
antigen-presenting cells of the immune system that are derived from
hematopoietic progenitor cells (HPCs) in the bone marrow. They
initiate and modulate the immune response (27). In the periphery, immature DCs
(iDCs) derived from proliferating progenitors capture and process
antigens. As a consequence of antigen deposition and inflammation,
DCs begin to mature, expressing lymphocyte co-stimulatory
molecules, migrating to lymphoid organs and secreting cytokines to
initiate immune responses. Many studies have shown that DCs can
identify and attack infiltrating tumour cells to control tumour
regrowth through immunological memory and immune surveillance
(28,29). These studies and others have
challenged the traditional notion of CNS ‘immune privilege’, which
is an imprecise characterization of the CNS immune environment
(30). Immunotherapy for GBM
appears to be a meaningful treatment approach to promote long-term
survival, and DCs play a vital role in this strategy (31).
In the present study, we identified MIF as a
promoter of autophagy in GBM cells. Furthermore, we verified that
ROCK1 was involved in MIF-induced progression of glioma. In
addition, we explored the potential immune functions of MIF and
found that MIF played an inhibitory role in the immune response of
DCs. Our research highlighted the different roles of MIF in glioma
progression and the immune system response.
Materials and methods
Ethics statement
This study was approved by the Institutional Review
Board of Shandong University and written informed consent was
obtained from all patients, and the Hospital Ethics Committee
approved the experiments.
Tissue samples and cell lines
Human glioma cell lines (U87 and U251) were
purchased from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China). Both U87 and U251 cells had been recently
authenticated based on cross-species checks, DNA authentication and
quarantine. The cell lines were grown in Dulbecco's modified
Eagle's medium (DMEM; HyClone, Logan, UT, USA) supplemented with
10% fetal bovine serum (Gibco BRL, Gaithersburg, MD, USA) in a
humidified incubator with 5% CO2 at 37°C. Six normal
brain tissues were collected from patients undergoing internal
decompression surgery following severe traumatic brain injury.
Twenty-five human glioma tissues, including 12 low-grade glioma
tissues (5 grade-I and 7 grade-II tumours) and 13 high-grade glioma
tissues (3 grade-III and 10 grade-IV tumours) were obtained from
the Department of Neurosurgery, Qilu Hospital of Shandong
University.
Chemical reagents, siRNA and
transfections
Recombinant human MIF (rhMIF; Peprotech, Inc., Rocky
Hill, NJ, USA), Y27632 (Selleck Chemicals, Houston, TX, USA), IL-4,
GM-CSF (both from Peprotech, Inc.), LPS (Sigma-Aldrich), CCL21
(Peprotech, Inc.) and DAPI (Beyotime Institute of Biotechnology,
Shanghai, China) were obtained from indicated companies. siRNA
negative control and siMIF were designed and purchased from
GenePharma (Shanghai, China). The siRNA sequences used were the
following: siRNA negative control forward,
5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′; siMIF forward,
5′-CCGAUGUUCAUCGUAAACATT-3′ and reverse,
5′-UGUUUACGAUGAACAUCGGTT-3′. Cell transfection experiments were
performed with nucleic acids using Lipofectamine 2000 (Invitrogen
Life Technologies, Carlsbad, CA, USA) according to the
manufacturer's instructions.
RhoA GTPase activity assay
The activity of RhoA and Rac1 GTPases in cell
lysates from control and MIF-treated glioblastoma cells was
assessed using a G-LISA® assay to measure the GTP-bound
form of RhoA and Rac1 (Cytoskeleton, Inc., Denver, CO, USA).
Briefly, glioblastoma cells were incubated with MIF (100 ng/ml, 30
min). Cell lysates were incubated on RhoA and Rac1 GTPase affinity
plates and color developed using HRP detection reagent mixture.
Samples were read on a microplate reader (Bio-Rad, Berkeley, CA,
USA).
RNA isolation, reverse transcription, and
quantitative real-time PCR
qPCR was conducted to measure the expression levels
of DCs. Total RNA was isolated using RNAiso Plus (Takara). Total
RNA (0.5–1 μg) was reverse-transcribed with a ReverTra Ace qPCR RT
kit (FSQ-101, Toyobo) according to the manufacturer's protocol to
synthesize cDNA. Real-time PCR was performed using a SYBR Premix Ex
TaqTM kit (Toyobo) with specific primers. The primers
used were the following: CD80 forward, 5′-AAACTCGCATCTACTGGCAAA-3′
and reverse, 5′-GGTTCTTGTACTCGGGCCATA-3′; CD83 forward,
5′-AAGGGGCAAAATGGTTCTTTCG-3′ and reverse, 5′-GCA
CCTGTATGTCCCCGAG-3′; CD86 forward, 5′-CTGCTCAT CTATACACGGTTACC-3′
and reverse, 5′-GGAAACGTCGT ACAGTTCTGTG-3′. The reactions were
performed by using a LightCycler 2.0 Instrument (Roche Applied
Science, Mannheim, Germany). mRNA levels were normalized to GAPDH.
All data for each sample were collected in triplicate. The fold
changes were calculated by relative quantification
(2−ΔΔCt).
GFP-LC3 stable cell lines and
quantitative GFP-LC3 analysis
To obtain U251 and U87 GFP-LC3 stable cell lines, we
cloned LC3 inserts to the plenty-N-GFP vectors and generated
lentiviruses by GenePharma. U87 and U251 cells were infected with
the lentiviruses and selected stable clones with G418 (Sangon
Biotech Co., Ltd., Shanghai, China). U87 and U251 GFP-LC3 stable
cell lines were transfected with siMIF or treated with rhMIF and
fixed in 4% paraformaldehyde. GFP-LC3 puncta formation assay was
determined by capturing images using Olympus microscope (DP72;
Olympus, Tokyo, Japan). Cells with ≥5 puncta were considered as
GFP-LC3 puncta-positive cells. The percentage of GFP-LC3
puncta-positive cells was quantified by counting 200 GFP-LC3 stable
cells.
Staining of F-actin fibre formation using
rhodamine-phalloidin
Glioblastoma cells and DCs plated on 24-well plate
were grown for 24 h and then cultured in serum-free medium for 16
h. After incubating with or without rhMIF (100 ng/ml, 24 h),
glioblastoma cells were washed once with phosphate-buffered saline
(PBS) and DCs were dropped on a glass slide. Then they were fixed
with 4% formaldehyde in PBS for 10 min. The fixed cells were washed
twice with PBS and permeabilized with 0.3% Triton X-100 in PBS.
Cells were then stained with rhodamine-phalloidin (Cytoskeleton,
Inc.) in PBS and cell nuclei were counterstained with DAPI. Images
were captured by Olympus microscope.
Western blot analysis
Total protein was extracted from tissues and cells
using RIPA buffer (Beyotime Institute of Biotechnology) with 1%
phenylmethyl sulfonyl fluoride, and protein concentration was
determined by the BCA method (Beyotime Institute of Biotechnology).
Proteins were separated using 10–15% SDS-PAGE and transferred onto
polyvinylidene difluoride membranes (Millipore, USA). The membranes
were blocked by 5% skim milk blocking buffer for 1 h and then
incubated in the primary antibodies at 4°C overnight. After washing
with TBST, the blots were incubated with horseradish
peroxidase-conjugated secondary antibodies at room temperature for
1 h. Finally, protein bands were visualized by enhanced
chemiluminescence (ECL) (Millipore) and detected using an ECL
detection system (Thermo Fisher Scientific, Inc., Beijing, China)
and quantified with Quantity One software. The following primary
antibodies were used: rabbit anti-LC3B, p62 and p-MYPT1 were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA);
rabbit anti-ROCK1 was purchased from Abcam (Cambridge, UK); rabbit
anti-MYPT1 was purchased from ProteinTech Group, Inc. (Wuhan,
China); rabbit anti-GAPDH was purchased from Goodhere Biotechnology
Co., Ltd. (Hangzhou, China). The relative integrated density values
were measured based on the GADPH protein as the control.
Immunohistochemistry
Paraffin-embedded samples were sliced and mounted on
microscopic slides. Rabbit anti-MIF (1:200 dilutions), anti-LC3B
(1:200 dilutions) and anti-CD1a (ZSGB-Bio, Beijing, China)
antibodies were used as the primary antibodies. Heat-induced
epitope retrieval was performed with a microwave in 10 mmol/l
citric acid buffer at pH 7.2. The samples were incubated with the
antibody overnight in a humidified chamber at 4°C followed by
incubation with a horseradish peroxidase-conjugated secondary
antibody (ZSGB-Bio). Finally, 3,3′-diaminobenzidine
tetrahydrochloride (DAB) was used to reveal the signal. The total
immunostaining score was estimated using both the percentage of
positively stained tumour cells and the staining intensity. The
percentage positivity was scored as ‘0’ (<5%, negative), ‘1’
(5–25%, sporadic), ‘2’ (25–50%, focal), or ‘3’ (>50%, diffuse).
The staining intensity was scored as ‘0’ (no staining), ‘1’ (weakly
stained), ‘2’ (moderately stained), or ‘3’ (strongly stained). Both
the percentage of positive cells and the staining intensity were
evaluated under double-blind conditions. The immunostaining score
was calculated as the percentage positive score multiplied by the
staining intensity score and ranged from 0 to 9.
In vitro migration assay
DC migration was assessed as previously described
(32). Briefly, the lower chamber
of 24 Transwell plates with polycarbonate membranes and 5-μm pore
size (Corning, Inc.) was filled with 200 μl of RPMI-1640 media
containing 10% FBS and CCL21 (100 ng/ml). Next, iDC and mature DC
(mDC) (1×105/100 μl medium) were seeded in the upper
chamber, and plates were incubated for 2 h at 37°C. Cells in the
lower chamber were counted. Glioblastoma cell migration was
evaluated using a Transwell chamber (8-μm pore size; Corning,
Inc.). Cells that did migrate to the lower surface were fixed and
stained with eosin solution and counted under a microscope
(Olympus). Five random views were used to count the cells, and the
independent experiments were repeated three times.
Colony formation assay
Glioblastoma cells (500) were seeded in 6-well
plates and cultured for 4 weeks at DMEM containing 10% FBS with
indicated treatments. Then the colonies were washed three times
with PBS, and fixed with 75% ethanol for 10 min, dried and stained
with 0.1% crystal violet solution (Beyotime Institute of
Biotechnology) for 10 min. Images were taken and the colonies were
counted under the light microscope.
Generation and culture of
monocyte-derived DCs
PBMCs were isolated from leukocyte-enriched buffy
coats of healthy volunteers by centrifugation with Ficoll-Paque
Plus (Sigma-Aldrich). Monocytes were enriched from PBMCs by
positive selection using anti-CD14-conjugated magnetic MicroBeads
(Miltenyi Biotec). Monocytes were cultured at 1×106/ml
in complete RPMI medium containing 1,000 U/ml GM-CSF and 500 U/ml
IL-4 under 37°C, 5% humidified CO2 for 5 days to
generate iDCs. In the middle of the 5 days, half of the medium was
replaced by fresh medium containing cytokines. To induce
maturation, 1 μg/ml LPS was added in iDCs for another 2 days. To
investigate the influence of MIF on DC maturation, 100 ng/ml rhMIF
were added during or after the maturation.
Statistical analyses
Data analyses were conducted with SPSS 16.0 (SPSS,
Inc., Chicago, IL, USA) and GraphPad Prism 5 (GraphPad Software,
Inc., La Jolla, CA, USA). Data were analyzed using one-way ANOVA,
Student's two-tailed t-test. Data are presented as the mean ±
standard deviation (SD) of three independent experiments, followed
by Dunnett's test for multiple comparisons of the means. All tests
were two-tailed, and p<0.05 was considered statistically
significant.
Results
MIF activates the RhoA-ROCK1 pathway in
glioblastoma cells
We first investigated whether RhoA-ROCK1 activity
was enhanced by MIF in glioblastoma cells. G-LISA assays were
performed to measure the Rho GTPase activity. The RhoA GTPase
activity increased following stimulation with rhMIF, but Rac1
GTPase was not affected in glioblastoma cells (Fig. 1A). Next, three GBM cell lines were
treated with rhMIF, and actin filaments were detected by
rhodamine-phalloidin staining. Actin polymerization was strongly
enhanced in GBM cells treated with rhMIF as there was increased
F-actin stress fibre formation compared to that of the control,
indicating that ROCK1 was activated by rhMIF (Fig. 1B). To further confirm the activity
of ROCK1, the phosphorylation of MYPT1 was assessed by western blot
analysis. MIF significantly increased the phosphorylation of the
MYPT1 in a dose-dependent manner (Fig.
1C). In contrast, the expression of ROCK1 was unchanged
following rhMIF stimulation, suggesting that ROCK1 was regulated
only at the enzyme level. These data demonstrated that MIF
activates the canonical RhoA-ROCK1 pathway.
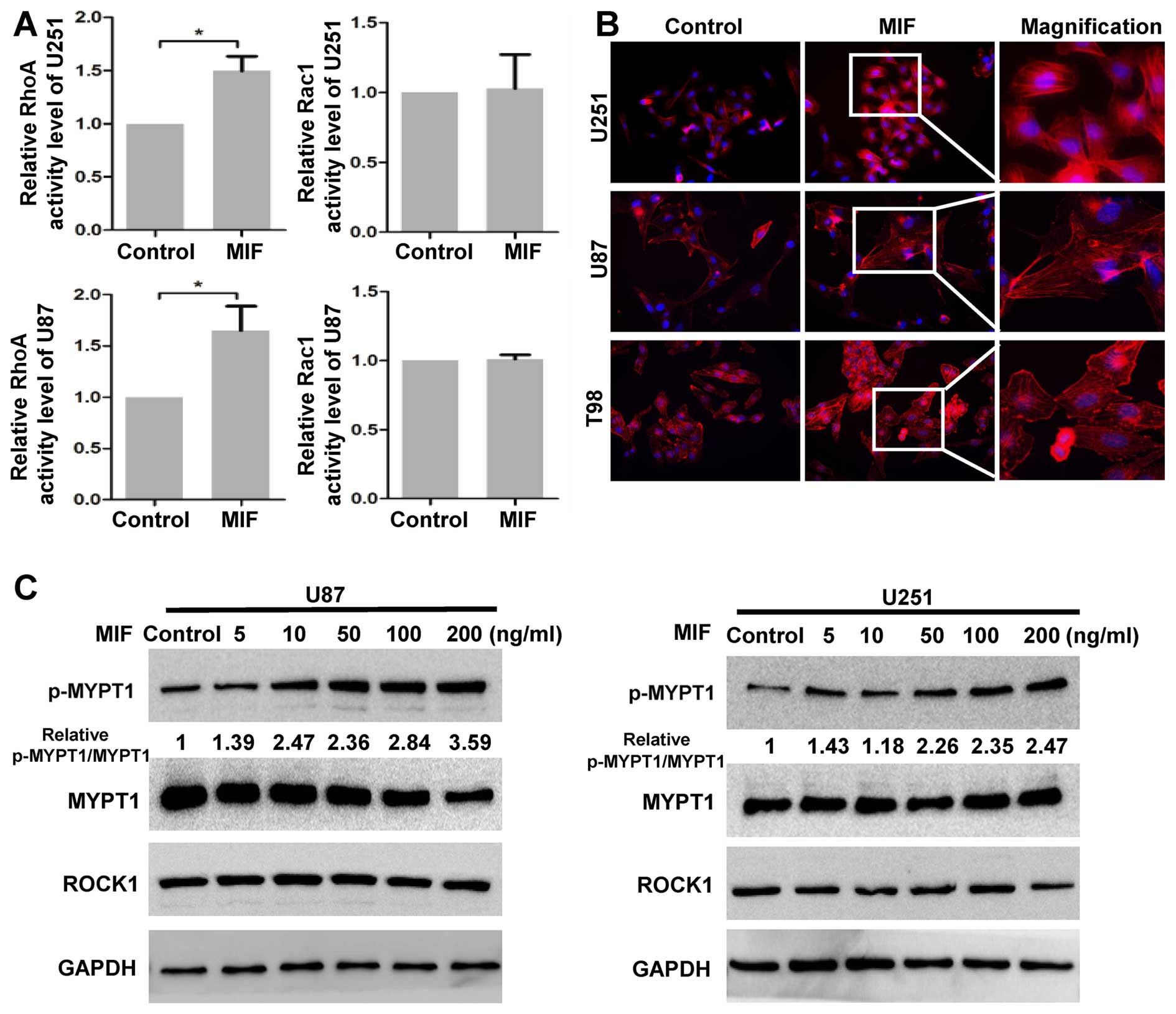 | Figure 1MIF promotes the activity of ROCK1.
(A) U87 and U251 cells were untreated or treated with the rhMIF
(100 ng/ml) for 30 min. The activation of the RhoA and Rac1 GTPases
was assessed by G-LISA assays. The data are shown as the mean ± SD
of independent experiments, n=3. (B) After no treatment or exposure
to rhMIF for 24 h, the actin filaments of U87 and U251 cells were
stained with rhodamine-phalloidin (red), and nuclei were stained
with DAPI (blue). Representative magnifications are shown. (C) U87
and U251 cells were subjected to different concentrations of MIF
(5, 10, 50, 100 and 200 ng/ml) for 12 h. The expression levels of
p-MYPT1, MYPT1, ROCK1 and GAPDH were determined by western blot
analysis. *P<0.05, Student's two-tailed t-test. MIF,
macrophage migration inhibitory factor; ROCK, Rho-associated
coiled-coil containing kinase; rhMIF, recombinant human MIF; SD,
standard deviation. |
MIF influences autophagy levels in
glioma
To explore the role of MIF in autophagic activity in
glioblastoma, we performed LC3 conversion and GFP-LC3 puncta
formation assays in both U87 and U251 cell lines. siMIF used to
neutralize the level of endogenous MIF was transfected into U87 and
U251 cells. The expression of LC3B-II decreased, suggesting that
MIF knockdown suppressed autophagy in glioblastoma cells (Fig. 2A). We also examined the
localization of GFP-LC3 by fluorescence microscopy in
siMIF-transfected U87 and U251 cells stably expressing the GFP-LC3
fusion protein. There was a significant decrease in GFP-LC3 puncta
in siMIF-transfected cells compared with that in the negative
control cells (Fig. 2B). Both cell
lines were stimulated by rhMIF, and autophagy was enhanced, as
indicated by the increased expression of LC3B-II and percentage of
GFP-LC3 puncta-positive cells (Fig. 2A
and B). However, the effect of rhMIF on autophagy of
glioblastoma cells seemed not to be quite in a dose-dependent
manner.
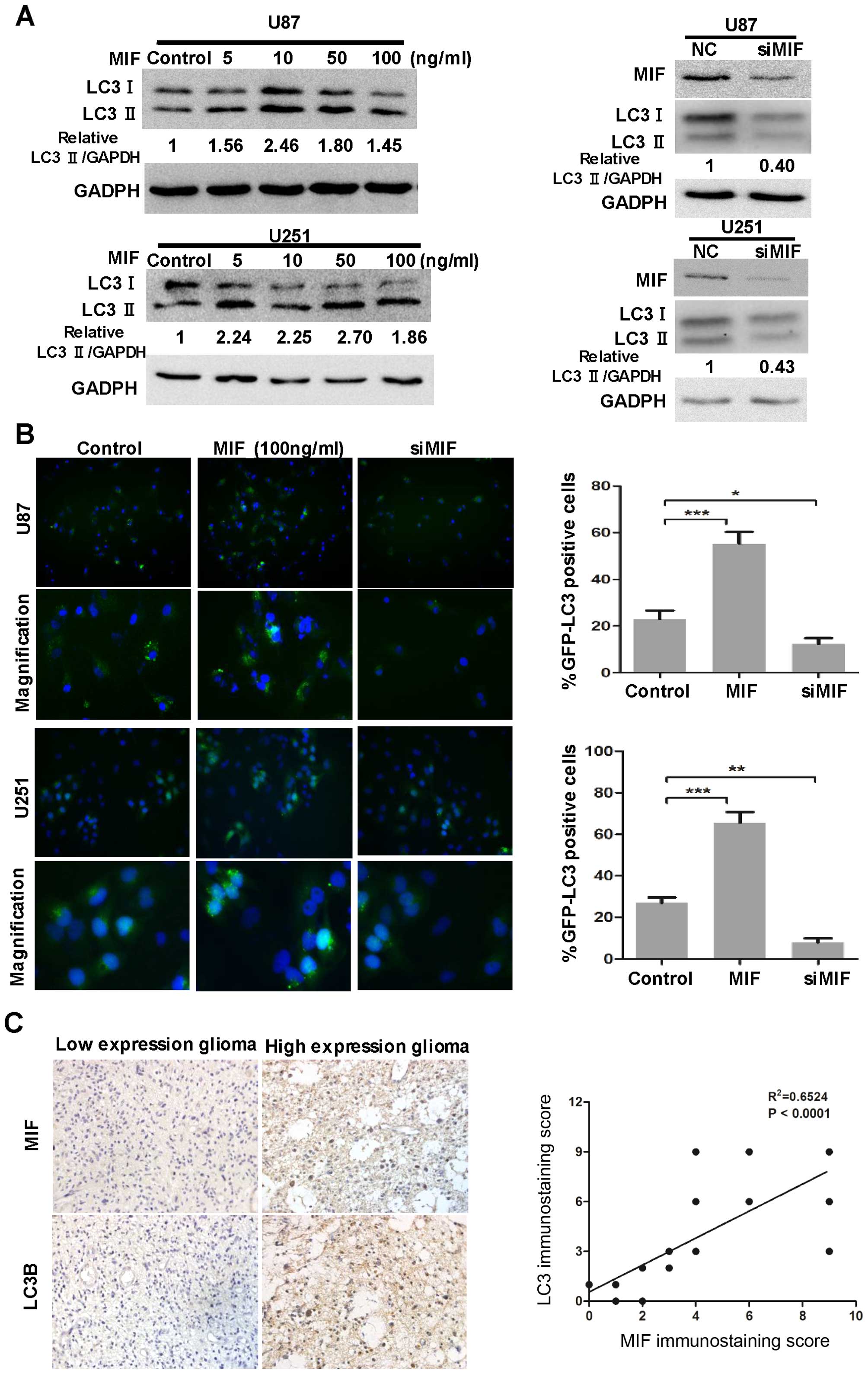 | Figure 2MIF influences the autophagic
activity in glioblastoma. (A) U87 and U251 cells were exposed to
different concentrations of rhMIF (5, 10, 50 and 100 ng/ml) for 24
h. U87 and U251 cells were transfected with siMIF (100 nM) for 24
h. Then, MIF, LC3B and GAPDH levels were determined by western blot
analysis. (B) U87 and U251 cells stably expressing GFP-LC3 were
stimulated with rhMIF or transfected with siMIF for 24 h and then
fixed. Representative images are shown. Quantitative analysis of
the rates of GFP-LC3 puncta-positive cells is shown. The data are
shown as the mean ± SD of independent experiments, n=3. (C)
Representative images of glioma tissues showed immunostaining of
MIF and LC3B (magnification, ×400). Correlation of MIF expression
with LC3B was analyzed in 25 glioma specimens. The linear
regression coefficient and statistical significance are shown.
*P<0.05, **p<0.01,
***p<0.001, one-way ANOVA. MIF, macrophage migration
inhibitory factor; rhMIF, recombinant human MIF; SD, standard
deviation. |
Finally, to assess the clinical relevance of the
above observations, we investigated the expression of MIF and LC3B
in glioma specimens. Glioma tissues with strong MIF
immunohistochemical signals had high expression of LC3B and vice
versa (Fig. 2C). The linear
regression analysis indicated the positive correlation of MIF with
LC3B. These results demonstrated a positive relationship between
MIF and autophagy in glioma.
MIF enhances the autophagy, migration and
colony formation of glioblastoma cells by activating ROCK1
Although several studies have identified ROCK1 as a
regulator of autophagy, whether MIF enhances autophagy via ROCK1 is
unknown. Therefore, we used Y27632, an inhibitor of ROCK1 activity,
and siROCK1 to confirm the role of ROCK1 in MIF-induced autophagy.
Western blot analyses were performed to detect autophagy. The
increased expression of LC3B-II induced by MIF was significantly
reversed by Y27632 and ROCK1 knockdown (Fig. 3B). In addition, U87 cells stably
expressing GFP-LC3 were treated with MIF and then fixed to assess
the actin filaments by rhodamine-phalloidin staining. The F-actin
stress fibre formation was localized in GFP-LC3 puncta-positive
cells, suggesting that glioblastoma cells with high ROCK1 activity
had increased autophagy (Fig.
3A).
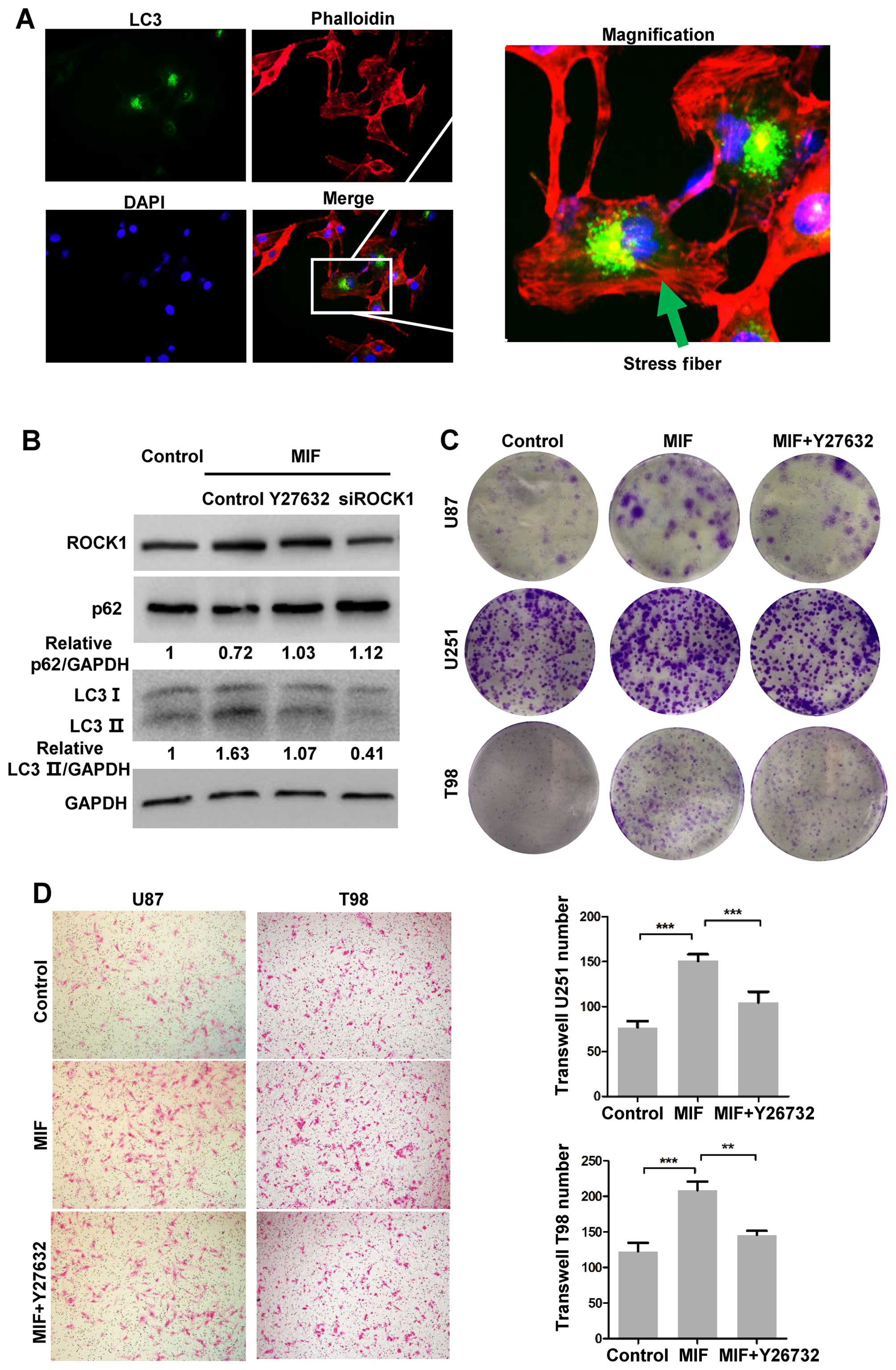 | Figure 3MIF increases autophagy by activating
ROCK1. (A) U87 cells stably expressing GFP-LC3 were cultured with
MIF for 24 h and fixed. The actin filaments were stained by
rhodamine-phalloidin. The nuclei were stained by DAPI.
Representative images are shown. (B) U87 cells were treated with
rhMIF, and the activity of ROCK1 was inhibited by Y260072 or
siROCK1. LC3B, p62 and GAPDH expressions were detected by western
blot analysis. (C) Colony formation assays were performed using
U87, U251 and T98 cells stimulated by MIF (10 ng/ml) with or
without Y270026 for 4 weeks. Representative images are shown. (D)
U87 and T98 cells exposed to MIF (100 ng/ml) with or without
Y270062 were used to perform Transwell assays for 18 h.
Representative images are shown. The data are shown as the mean ±
SD of independent experiments, n=3. **P<0.01,
***p<0.001, one-way ANOVA. MIF, macrophage migration
inhibitory factor; ROCK, Rho-associated coiled-coil containing
kinase; rhMIF, recombinant human MIF; SD, standard deviation. |
Furthermore, we explored whether ROCK1 activity
contributes to increased migration and colony formation induced by
MIF. The migration of U87, U251 and T98 cells was measured by
Transwell assays. rhMIF promoted the migration of all three cell
lines, and as expected, Y27632 suppressed the MIF-induced migration
(Fig. 3D). Y27632 partly blocked
the MIF-induced colony formation (Fig.
3C). These data demonstrated that the activity of ROCK1 played
a crucial role in MIF-induced tumourigenesis.
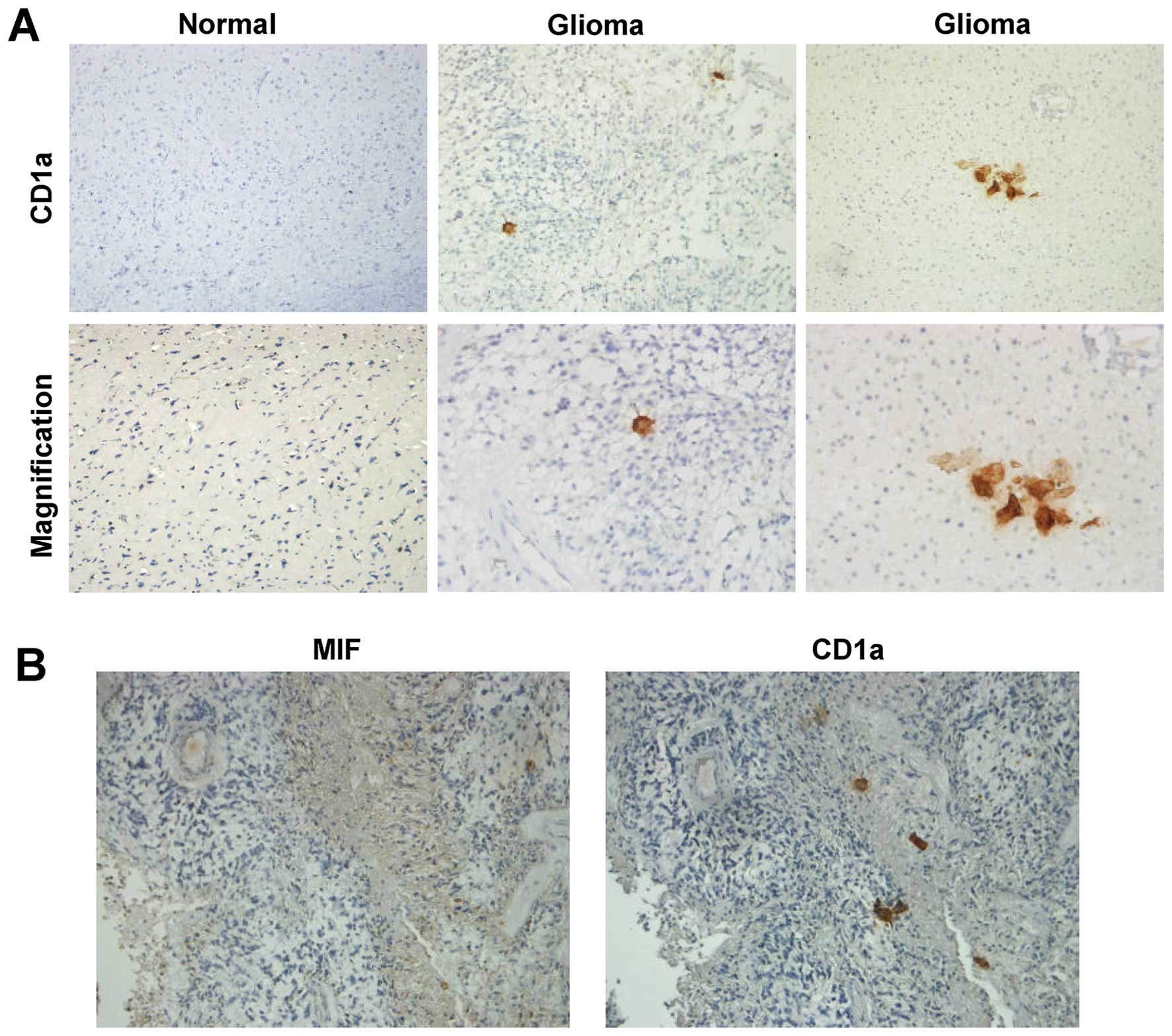
DCs infiltrate glioma tissues
A previous report identified DCs in brain tissues
(33). To determine whether DCs
infiltrated glioma tissues, we used CD1a, a marker of DCs, to
detect DCs in glioma specimens by immunohistochemistry (34). Few CD1a-positive cells were found
in glioma specimens, while in normal brain tissues, there were no
CD1a-positive cells (Fig. 4A).
This suggested that DCs could infiltrate gliomas through
blood-brain barrier (BBB) disruption or an unknown mechanism.
Moreover, in serial sections of glioma tissue, CD1a-positive cells
were localized in regions with high levels of MIF (Fig. 4B). These data indicated that MIF
may suppress the migration of DCs to impede initiation of immune
responses.
MIF inhibits the migration of both iDCs
and mDCs
To study the effects of MIF on DCs, we generated
iDCs and mDCs from PBMCs of healthy individuals (Fig. 5A). The cellular morphology of the
iDCs and mDCs was observed (Fig.
5B). The effect of MIF on migration of DCs was assessed using
Transwell assays. The migration of iDCs and mDCs towards CCL21 was
significantly decreased in MIF-treated DCs compared to that of the
controls (Fig. 5C). Furthermore,
to verify whether MIF regulates actin rearrangement in DCs,
rhodamine-phalloidin staining was performed. There were strong
phalloidin-stained puncta in MIF-treated DCs, suggesting a
deficiency of the intact F-actin ring (Fig. 5D).
 | Figure 5MIF suppresses the migration of DCs.
(A) A schematic diagram of the induction of iDCs and mDCs is shown.
(B) Images of iDCs and mDCs are exhibited (magnification, ×400).
(C) iDCs and mDCs were treated with rhMIF, and Transwell assays
were performed. The data are shown as the mean ± SD of independent
experiments, n=3. (D) iDCs and mDCs were stained by
rhodamine-phalloidin. Representative images are shown.
*P<0.05, **p<0.01, Student's two-tailed
t-test. MIF, macrophage migration inhibitory factor; DCs, dendritic
cells; iDCs, immature DCs; mDCs, mature DCs; rhMIF, recombinant
human MIF; SD, standard deviation. |
MIF counteracts iDC maturation and mDC
function
To further characterize the immunological impact of
MIF on DCs, we studied its effects on DC maturation and the
expression of co-stimulatory molecules. iDCs were cultured with MIF
or supernatants from siMIF-transfected or negative control U87
cells for 3 days in the presence of LPS, which promoted maturation
(Fig. 6A). Subsequently, the
expression of the maturation markers CD83, CD80 and CD86 was
quantified by qPCR. In the presence of MIF, the two co-stimulatory
molecule markers, but not CD83, were significantly decreased
compared to that of the no MIF treatment, indicating that MIF had
an inhibitory role in the maturation of iDCs (Fig. 6C). Similarly, cultures of iDCs with
supernatant from siMIF-transfected U87 cells showed slightly
increased expression of CD80 (Fig.
6C), while increased expression of CD83 and CD86 was not
detected (data not shown). Moreover, we cultured the mDCs in the
presence or absence of MIF for 24 h, and the expression of the
markers was also downregulated (Fig.
6B). These data demonstrated that MIF plays an inhibitory role
in DC maturation and function.
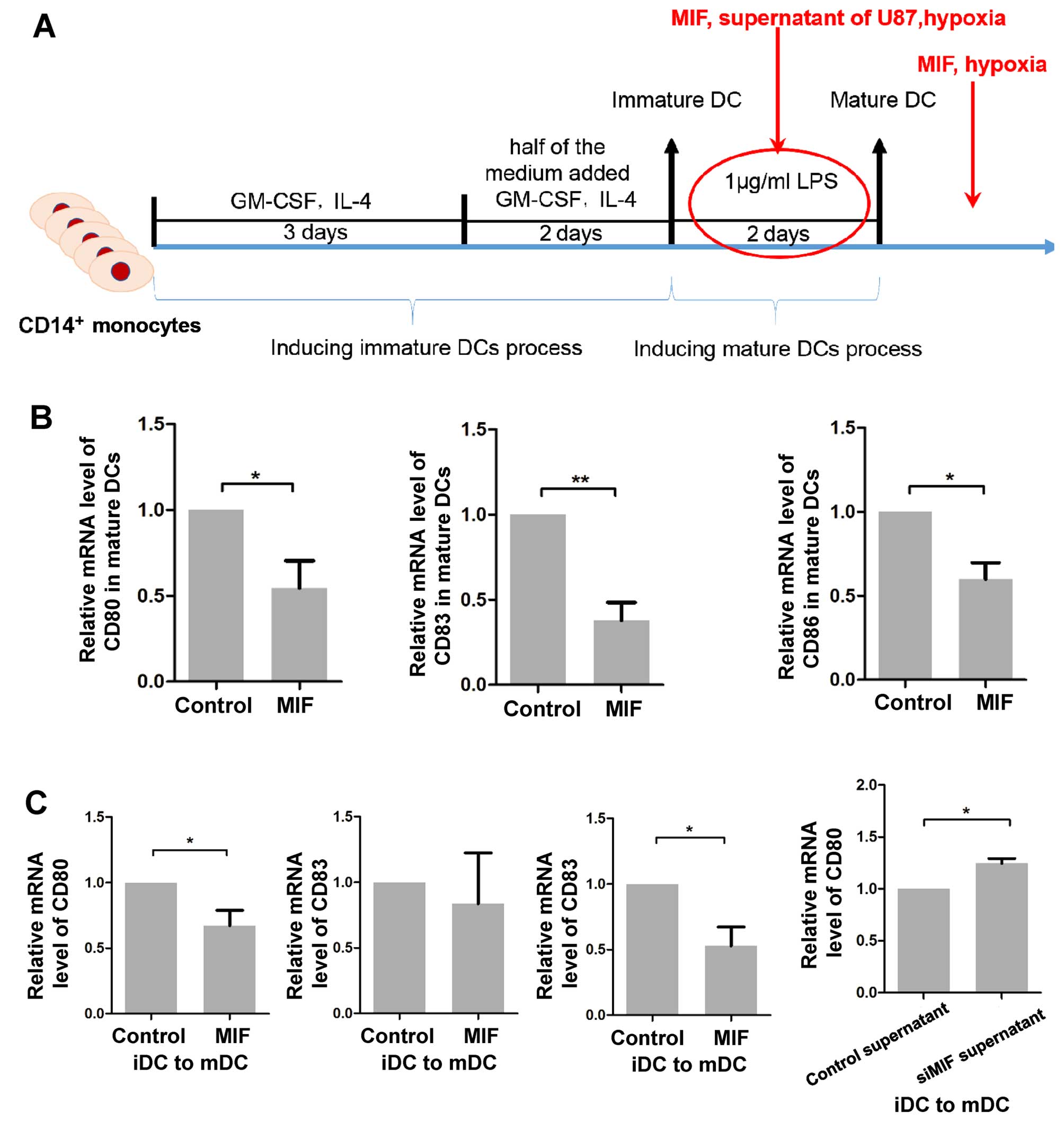 | Figure 6MIF suppresses the maturation and
function of DCs. (A) A schematic diagram of the intervention during
or after the induction of mDCs from iDCs is shown. (B) mDCs were
treated with rhMIF for 24 h. Cells were collected for qPCR analysis
to quantify the expression of CD80, CD83 and CD86. The data are
shown as the mean ± SD of independent experiments, n=3. (C) rhMIF
was added as indicated during the maturation of iDCs, and the DCs
were then collected to measure the expression of CD80, CD83 and
CD86. The data are shown as the mean ± SD of independent
experiments, n=3. *P<0.05, **p<0.01,
Student's two-tailed t-test. MIF, macrophage migration inhibitory
factor; DCs, dendritic cells; mDCs, mature DCs; iDCs, immature DCs;
rhMIF, recombinant human MIF; SD, standard deviation. |
Discussion
MIF is a pleiotropic cytokine that has important
pro-inflammatory and pro-tumourigenesis roles. Intriguingly,
increasing data have attributed the immune evasion of glioblastoma
to MIF. The involvement of MIF in both tumourigenesis and tumour
immune escape indicates that it may be a promising target for
anti-glioblastoma treatment. In this study, we found that MIF
augmented autophagy of glioblastoma cells by activating the
RhoA-ROCK1 pathway. In addition, we identified the inhibitory
immunological effect of MIF on DCs.
Overexpression of MIF is associated with malignancy,
recurrence and poor prognosis of patients with gliomas (35). Extensive studies have shown that
MIF contributes to malignant progression of glioma via various
processes, including angiogenesis, migration, invasion and contact
inhibition. Most recently, MIF has been shown to facilitate brain
tumourigenesis (36) and maintain
the tumourigenic capacity of brain tumour-initiating cells
(37), which supports its
pro-tumourigenic role in glioma. Autophagy as a process of cellular
self-digestion has been considered a pro-survival response to
glioma treatment (38,39). MIF was associated with autophagy.
However, to date, whether MIF is implicated in autophagy and its
underlying mechanism has not yet been determined in glioma.
There are conflicting reports on whether MIF
increases or decreases autophagy in other tumours. In breast
cancer, Wu et al demonstrated that MIF is a strong
suppressor of autophagy, leading to resistance to
chemotherapy-induced autophagic cell death (40). In contrast, several studies showed
that MIF induced autophagy via reactive oxygen species generation
and contributed to anti-concanavalin A-induced apoptosis by
upregulating autophagy (41,42).
Moreover, MIF knockout was associated with loss in cardiac
autophagy during ageing (43). In
our study, we found that exogenous MIF promoted autophagy in
glioblastoma cells. Since the identification of MIF, it has been
reported to be involved in pleiotropic cytokine signalling via
multiple receptors, such as CD74, CD44 and CXC chemokine receptors
(44–46). The opposing effects of autophagy in
different cells may be attributed to various receptor patterns.
Interestingly, Rho GTPases have been reported to be involved in
MIF-induced tumour invasion (47,48).
Most importantly, ROCK1, the downstream effector of the RhoA, was
identified as a critical regulator of autophagy during metabolic
stress (49). Our study confirmed
that MIF increased the RhoA-ROCK1 activation but not the expression
level of ROCK1, as determined by the G-LISA assay and the
phosphorylation of MYPT1. Moreover, ROCK1 has been shown to be
involved in the migration and proliferation of glioblastoma
(50,51). These results led us to hypothesize
that MIF enhances autophagy, and even malignant progression, by
activating ROCK1 in glioma. Knockdown of ROCK1 or inhibition of
ROCK1 reversed the autophagy, migration and colony formation
induced by MIF. Interestingly, although U251 showed higher ability
of colony formation in the control group than the other two cell
lines, U251 was also significantly influenced by MIF stimulation
and the ROCK1 activity. These results demonstrate the important
role of ROCK1 in regulating the MIF-induced malignant progression
of glioma.
Although therapeutic strategies have improved in the
past decades, the average overall survival for patients with
glioblastoma is still very poor (52). Immune therapy has attracted
increased attention and has emerged as a promising adjuvant
treatment for glioblastoma. There is clear evidence for the immune
escape role of MIF in gliomas (53,54).
In glioma, MIF suppressed the antitumoural effects of NK cells and
downregulated the activating immune receptor (NKG2D) on NK cells
and CD8+ T cells. In addition, a recent study showed
that MIF suppressed the immune response by supporting
immune-suppressive myeloid-derived suppressor cells in the GBM
tumour microenvironment. However, the association between DCs,
initiators of the immune response, and MIF remains unclear. First,
we found infiltration of DCs in gliomas.
Intriguingly, CD1a-positive cells were localized in
regions with high expression of MIF, suggesting an inhibitory
effect of MIF on migration of DCs. To address this hypothesis, we
sorted the CD14+ monocytes from PBMCs and used them to
generate iDCs and mDCs. Transwell assays confirmed that MIF had a
strong inhibitory effect on the migration of both iDCs and mDCs.
Additionally, the intact F-actin ring appeared to be dysfunctional
with MIF treatment. Because DCs migrate through pathological
tissues before reaching their final destination in the lymph nodes,
the inhibitory effect of MIF can delay or prevent DCs from
returning to the lymph nodes to stimulate the immune response.
Considering that DC maturation is required for the initiation of an
immune response, the effect of MIF on maturation and function of
DCs was measured by assessing the maturation marker, CD83, and
co-stimulatory molecules (CD80 and CD86) (55). Accordingly, incubation of mDCs with
MIF substantially decreased the expression of CD83, CD80 and CD86,
reflecting a significant immunosuppressive effect of MIF on mDCs.
In addition, MIF apparently impaired the LPS-induced maturation of
DCs, confirmed by the decreased expression of co-stimulatory
molecules, but not CD83. To assess the glioma microenvironment, we
used the supernatant of U87 cells, which has been reported to have
higher MIF RNA levels than U251 cells, for further experiments.
Consistent with previous observations, incubation of iDCs cultured
with LPS and supernatant from knockdown MIF U87 cells slightly
augmented the expression of CD80 compared to that of the control
supernatant. The supernatant contains much lower concentrations of
MIF than the exogenous MIF, indicating that CD80 was more sensitive
to the effect of MIF.
In summary, we showed that MIF is a critical
mediator of autophagy in glioblastoma. We identified ROCK1 as a
potent downstream effector of MIF that further regulates autophagy.
Moreover, ROCK1 may also be a core regulator that promotes
MIF-induced malignant progression of glioblastoma. In addition, we
showed that MIF plays an important role in the escape of DC
surveillance. It suppressed the maturation and function of DCs,
which was reflected by the downregulation of the maturation marker
CD83 and the co-stimulatory molecules CD80 and CD86. However, the
detailed mechanism underlying the inhibitory effect of MIF on DCs
was not elucidated in this study, and further investigations should
be performed to define this mechanism. Finally, we showed that MIF
directly or indirectly contributes to an immune microenvironment
that favours glioblastoma progression.
Acknowledgements
We thank Professor Xun Qu for helpful comments and
advice on this study. This study was supported by grants from the
National Natural Science Foundation of China (nos. 81101594,
81372719, 81172403, 81300510, 81402077, 81571284 and 91542115) and
Taishan Scholars of Shandong Province of China (no.
ts201511093).
References
|
1
|
Meyer MA: Malignant gliomas in adults. N
Engl J Med. 359:1850author reply 1850. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ohgaki H and Kleihues P: Population-based
studies on incidence, survival rates, and genetic alterations in
astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol.
64:479–489. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bloom BR and Bennett B: Mechanism of a
reaction in vitro associated with delayed-type hypersensitivity.
Science. 153:80–82. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Calandra T and Roger T: Macrophage
migration inhibitory factor: A regulator of innate immunity. Nat
Rev Immunol. 3:791–800. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Apte RS, Sinha D, Mayhew E, Wistow GJ and
Niederkorn JY: Cutting edge: Role of macrophage migration
inhibitory factor in inhibiting NK cell activity and preserving
immune privilege. J Immunol. 160:5693–5696. 1998.PubMed/NCBI
|
|
6
|
Daun JM and Cannon JG: Macrophage
migration inhibitory factor antagonizes hydrocortisone-induced
increases in cytosolic IkappaBalpha. Am J Physiol Regul Integr Comp
Physiol. 279:R1043–R1049. 2000.PubMed/NCBI
|
|
7
|
Bucala R and Donnelly SC: Macrophage
migration inhibitory factor: A probable link between inflammation
and cancer. Immunity. 26:281–285. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roger T, Chanson AL, Knaup-Reymond M and
Calandra T: Macrophage migration inhibitory factor promotes innate
immune responses by suppressing glucocorticoid-induced expression
of mitogen-activated protein kinase phosphatase-1. Eur J Immunol.
35:3405–3413. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meyer-Siegler KL, Iczkowski KA, Leng L,
Bucala R and Vera PL: Inhibition of macrophage migration inhibitory
factor or its receptor (CD74) attenuates growth and invasion of
DU-145 prostate cancer cells. J Immunol. 177:8730–8739. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bacher M, Eickmann M, Schrader J, Gemsa D
and Heiske A: Human cytomegalovirus-mediated induction of MIF in
fibroblasts. Virology. 299:32–37. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mitchell RA, Metz CN, Peng T and Bucala R:
Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic
phospholipase A2 activation by macrophage migration inhibitory
factor (MIF). Regulatory role in cell proliferation and
glucocorticoid action. J Biol Chem. 274:18100–18106. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bacher M, Schrader J, Thompson N, Kuschela
K, Gemsa D, Waeber G and Schlegel J: Up-regulation of macrophage
migration inhibitory factor gene and protein expression in glial
tumor cells during hypoxic and hypoglycemic stress indicates a
critical role for angiogenesis in glioblastoma multiforme. Am J
Pathol. 162:11–17. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lue H, Thiele M, Franz J, Dahl E,
Speckgens S, Leng L, Fingerle-Rowson G, Bucala R, Lüscher B and
Bernhagen J: Macrophage migration inhibitory factor (MIF) promotes
cell survival by activation of the Akt pathway and role for
CSN5/JAB1 in the control of autocrine MIF activity. Oncogene.
26:5046–5059. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rabinowitz JD and White E: Autophagy and
metabolism. Science. 330:1344–1348. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: A double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Aita VM, Liang XH, Murty VV, Pincus DL, Yu
W, Cayanis E, Kalachikov S, Gilliam TC and Levine B: Cloning and
genomic organization of beclin 1, a candidate tumor suppressor gene
on chromosome 17q21. Genomics. 59:59–65. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:1845–1846. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo JY, Xia B and White E:
Autophagy-mediated tumor promotion. Cell. 155:1216–1219. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hall A: The cytoskeleton and cancer.
Cancer Metastasis Rev. 28:5–14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Matsui T, Amano M, Yamamoto T, Chihara K,
Nakafuku M, Ito M, Nakano T, Okawa K, Iwamatsu A and Kaibuchi K:
Rho-associated kinase, a novel serine/threonine kinase, as a
putative target for small GTP binding protein Rho. EMBO J.
15:2208–2216. 1996.PubMed/NCBI
|
|
23
|
Leung T, Manser E, Tan L and Lim L: A
novel serine/threonine kinase binding the Ras-related RhoA GTPase
which translocates the kinase to peripheral membranes. J Biol Chem.
270:29051–29054. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Amano M, Chihara K, Kimura K, Fukata Y,
Nakamura N, Matsuura Y and Kaibuchi K: Formation of actin stress
fibers and focal adhesions enhanced by Rho-kinase. Science.
275:1308–1311. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Riento K and Ridley AJ: Rocks:
Multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol.
4:446–456. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Amano M, Nakayama M and Kaibuchi K:
Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell
polarity. Cytoskeleton Hoboken. 67:545–554. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Banchereau J and Steinman RM: Dendritic
cells and the control of immunity. Nature. 392:245–252. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lai A, Tran A, Nghiemphu PL, Pope WB,
Solis OE, Selch M, Filka E, Yong WH, Mischel PS, Liau LM, et al:
Phase II study of bevacizumab plus temozolomide during and after
radiation therapy for patients with newly diagnosed glioblastoma
multiforme. J Clin Oncol. 29:142–148. 2011. View Article : Google Scholar :
|
|
29
|
Ghulam Muhammad AK, Candolfi M, King GD,
Yagiz K, Foulad D, Mineharu Y, Kroeger KM, Treuer KA, Nichols WS,
Sanderson NS, et al: Antiglioma immunological memory in response to
conditional cytotoxic/immune-stimulatory gene therapy: Humoral and
cellular immunity lead to tumor regression. Clin Cancer Res.
15:6113–6127. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Perng P and Lim M: Immunosuppressive
mechanisms of malignant gliomas: Parallels at non-CNS sites. Front
Oncol. 5:1532015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Van Gool S, Maes W, Ardon H, Verschuere T,
Van Cauter S and De Vleeschouwer S: Dendritic cell therapy of
high-grade gliomas. Brain Pathol. 19:694–712. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shao Q, Ning H, Lv J, Liu Y, Zhao X, Ren
G, Feng A, Xie Q, Sun J, Song B, et al: Regulation of Th1/Th2
polarization by tissue inhibitor of metalloproteinase-3 via
modulating dendritic cells. Blood. 119:4636–4644. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Laperchia C, Allegra Mascaro AL, Sacconi
L, Andrioli A, Mattè A, De Franceschi L, Grassi-Zucconi G,
Bentivoglio M, Buffelli M and Pavone FS: Two-photon microscopy
imaging of thy1GFP-M transgenic mice: A novel animal model to
investigate brain dendritic cell subsets in vivo. PLoS One.
8:e561442013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Coventry BJ, Austyn JM, Chryssidis S,
Hankins D and Harris A: Identification and isolation of CD1a
positive putative tumour infiltrating dendritic cells in human
breast cancer. Adv Exp Med Biol. 417:571–577. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang XB, Tian XY, Li Y, Li B and Li Z:
Elevated expression of macrophage migration inhibitory factor
correlates with tumor recurrence and poor prognosis of patients
with gliomas. J Neurooncol. 106:43–51. 2012. View Article : Google Scholar
|
|
36
|
Ghoochani A, Schwarz MA, Yakubov E,
Engelhorn T, Doerfler A, Buchfelder M, Bucala R, Savaskan NE and
Eyüpoglu IY: MIF-CD74 signaling impedes microglial M1 polarization
and facilitates brain tumorigenesis. Oncogene. May 9–2016.(Epub
ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fukaya R, Ohta S, Yaguchi T, Matsuzaki Y,
Sugihara E, Okano H, Saya H, Kawakami Y, Kawase T, Yoshida K, et
al: MIF maintains the tumorigenic capacity of brain
tumor-initiating cells by directly inhibiting p53. Cancer Res.
76:2813–2823. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xue H, Yuan G, Guo X, Liu Q, Zhang J, Gao
X, Guo X, Xu S, Li T, Shao Q, et al: A novel tumor-promoting
mechanism of IL6 and the therapeutic efficacy of tocilizumab:
Hypoxia-induced IL6 is a potent autophagy initiator in glioblastoma
via the p-STAT3-MIR155-3p-CREBRF pathway. Autophagy. 12:1129–1152.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu R, Li J, Zhang T, Zou L, Chen Y, Wang
K, Lei Y, Yuan K, Li Y, Lan J, et al: Itraconazole suppresses the
growth of glioblastoma through induction of autophagy: Involvement
of abnormal cholesterol trafficking. Autophagy. 10:1241–1255. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu MY, Fu J, Xu J, O'Malley BW and Wu RC:
Steroid receptor coactivator 3 regulates autophagy in breast cancer
cells through macrophage migration inhibitory factor. Cell Res.
22:1003–1021. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lai YC, Chuang YC, Chang CP and Yeh TM:
Macrophage migration inhibitory factor has a permissive role in
concanavalin A-induced cell death of human hepatoma cells through
autophagy. Cell Death Dis. 6:e20082015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chuang YC, Su WH, Lei HY, Lin YS, Liu HS,
Chang CP and Yeh TM: Macrophage migration inhibitory factor induces
autophagy via reactive oxygen species generation. PLoS One.
7:e376132012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xu X, Hua Y, Nair S, Bucala R and Ren J:
Macrophage migration inhibitory factor deletion exacerbates
pressure overload-induced cardiac hypertrophy through mitigating
autophagy. Hypertension. 63:490–499. 2014. View Article : Google Scholar :
|
|
44
|
Leng L, Metz CN, Fang Y, Xu J, Donnelly S,
Baugh J, Delohery T, Chen Y, Mitchell RA and Bucala R: MIF signal
transduction initiated by binding to CD74. J Exp Med.
197:1467–1476. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shi X, Leng L, Wang T, Wang W, Du X, Li J,
McDonald C, Chen Z, Murphy JW, Lolis E, et al: CD44 is the
signaling component of the macrophage migration inhibitory
factor-CD74 receptor complex. Immunity. 25:595–606. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bernhagen J, Krohn R, Lue H, Gregory JL,
Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, et
al: MIF is a noncognate ligand of CXC chemokine receptors in
inflammatory and atherogenic cell recruitment. Nat Med. 13:587–596.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rendon BE, Roger T, Teneng I, Zhao M,
Al-Abed Y, Calandra T and Mitchell RA: Regulation of human lung
adenocarcinoma cell migration and invasion by macrophage migration
inhibitory factor. J Biol Chem. 282:29910–29918. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sun B, Nishihira J, Yoshiki T, Kondo M,
Sato Y, Sasaki F and Todo S: Macrophage migration inhibitory factor
promotes tumor invasion and metastasis via the Rho-dependent
pathway. Clin Cancer Res. 11:1050–1058. 2005.PubMed/NCBI
|
|
49
|
Gurkar AU, Chu K, Raj L, Bouley R, Lee SH,
Kim YB, Dunn SE, Mandinova A and Lee SW: Identification of ROCK1
kinase as a critical regulator of Beclin1-mediated autophagy during
metabolic stress. Nat Commun. 4:21892013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang P, Lu Y, Liu XY and Zhou YH:
Knockdown of Rho-associated protein kinase 1 suppresses
proliferation and invasion of glioma cells. Tumour Biol.
36:421–428. 2015. View Article : Google Scholar
|
|
51
|
Huang D, Qiu S, Ge R, He L, Li M, Li Y and
Peng Y: miR-340 suppresses glioblastoma multiforme. Oncotarget.
6:9257–9270. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Omuro A and DeAngelis LM: Glioblastoma and
other malignant gliomas: A clinical review. JAMA. 310:1842–1850.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mittelbronn M, Platten M, Zeiner P,
Dombrowski Y, Frank B, Zachskorn C, Harter PN, Weller M and
Wischhusen J: Macrophage migration inhibitory factor (MIF)
expression in human malignant gliomas contributes to immune escape
and tumour progression. Acta Neuropathol. 122:353–365. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Otvos B, Silver DJ, Mulkearns-Hubert EE,
Alvarado AG, Turaga SM, Sorensen MD, Rayman P, Flavahan WA, Hale
JS, Stoltz K, et al: Cancer stem cell-secreted macrophage migration
inhibitory factor stimulates myeloid derived suppressor cell
function and facilitates glioblastoma immune evasion. Stem Cells.
34:2026–2039. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Etminan N, Peters C, Lakbir D, Bünemann E,
Börger V, Sabel MC, Hänggi D, Steiger HJ, Stummer W and Sorg RV:
Heat-shock protein 70-dependent dendritic cell activation by
5-aminolevulinic acid-mediated photodynamic treatment of human
glioblastoma spheroids in vitro. Br J Cancer. 105:961–969. 2011.
View Article : Google Scholar : PubMed/NCBI
|