Introduction
Colorectal cancer is the third most common cancer
worldwide (1), with nearly 1.36
million new cases diagnosed in 2012 (2). A major impediment in the success of
available therapies is the recurrent adaptation of cancer cells,
which evade from the tumor, and eventually reach and settle at
distant sites, leading to metastases, which are often considered as
the point of no return, and are associated with the worst outcome.
Therefore, understanding the mechanisms that drive resistance of
cancer cells bears special importance. Several such mechanisms have
been studied in depth in numerous experimental setups. These
include, but are not limited to: i) increased clearance of the
drugs, either through increased efflux, decreased influx or
increased metabolism that limit the in-cell life span of the
compounds; ii) decreased metabolic conversion/activation of
pro-drugs, which restricts the cytotoxic effect of the active
product; iii) increased repair capacity towards cytotoxic or
genotoxic damage; iv) decreased engagement of the apoptotic
machinery in response to drugs (3). A rational way to understand
resistance is to isolate, from cancer cells grown in vitro,
cells that are resistant to therapy, and to analyze their
phenotypic properties. In the context of colon cancer, as for other
epithelial cancers, it has been proposed that cancer cells may
originate from a small fraction of tumor initiating cells, or
cancer stem cells (CSC) that are located near the bottom of the
crypts (4,5). There is a set of therapeutic
strategies based on usage of anticancer agents such as oxaliplatin
or irinotecan, among many others, in colon cancer. Oxaliplatin, a
third generation platinum derivative, is frequently used for
treating advanced CRC, in association with 5-fluorouracil. This
platinum analogue inhibits tumor cell growth by covalent DNA
binding (6). Several studies
reported that platinum agents enter the cells by passive diffusion
and may be detoxified predominantly by the glutathione system
(3). Docetaxel (taxotere), an
analogue of taxol, inhibits cell replication upon promoting the
in vitro assembly of stable microtubules and inducing
microtubule-bundle formation (7).
It is used for the treatment of a number of solid tumors such as
breast, prostate, non-small cell lung cancer, and gastric
adenocarcinoma, but it had limited activity in colon cancer
patients (8). Passive diffusion is
believed to be the mechanism of uptake for docetaxel due to its
lipophilic characteristics, although the organic anion transporting
polypeptides (OATPs) could also play a role in its uptake (9). Docetaxel is mainly metabolized in the
liver by cytochrome P450 3A4 (CYP3A4), and to a minor extent by
CYP3A5 (10), and then eliminated
by the multidrug resistance protein 1 (MDR1, ABCB1, P-glycoprotein)
among others (11). However,
intrinsic or acquired resistance to these drugs is one of the major
obstacles in the use of these agents in therapy (11,12).
Platinum resistance is a process that could mainly include
deregulation in drug transport and detoxification, DNA repair and
impairment of apoptosis (3).
Likely, taxane resistance is associated with modifications in drug
efflux mechanisms, involving the adenosine triphosphate
(ATP)-binding cassette (ABC) transporter family (13). The link between CSC and drug
resistance has been reported in several studies (13,14).
The existence of CSC within solid tumors, such as breast (15), pancreas (16), brain (17) and colon (18,19)
or even in hematopoietic cancers (20) is rather accepted. CSC are
particularly resistant to drugs, in part as a result of high
expression levels of ATP-binding cassette (ABC) transporters, a
high DNA repair capacity and increased levels of anti-apoptotic
factors (21–23). In addition, signaling pathways
involved in differentiation and migration, such as the Wnt, Notch,
Hedgehog or BMP/TGFβ pathways, may be altered as a result of cancer
promoting mutations (5,24,25).
These pathways have been found to play a role in the regulation of
the self-renewal of CSC but also in the regulation of the
chemoresistant cells (26). In
this study, we generated cells resistant to oxaliplatin or
docetaxel, two cytotoxic drugs with distinct modes of action. We
characterized a number of phenotypic features, both directly
associated to chemo-resistance and related to CSC characteristics.
Our results confirm and extend previous studies, and further show
that acquired drug resistance depends more on the type of cancer
cell than on the drug type.
Materials and methods
Cell lines and culture conditions
HT29 cells were cultured in Dulbecco’s modified
Eagle’s medium (DMEM; 4.5 g/l glucose) (Lonza, Belgium)
supplemented with 10% fetal bovine serum (FBS) (Gibco Invitrogen,
USA), and HCT116 cells were maintained in Dulbecco’s modified
Eagle’s medium: nutrient mixture F-12 (DMEM/F-12) (Lonza, Belgium),
supplemented with 5% FBS. All cultures were incubated at 37°C in a
humidified atmosphere containing 5% CO2. The medium was
changed every two days, and cells were passaged using 0.05%
trypsin/EDTA (Gibco Invitrogen).
Cell viability and growth
Cell viability of HT29 and HCT116 cells was measured
by the colorimetric MTT
(3-(4,5-dimeth-ylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)
test (EMD Millipore, USA). HT29 or HCT116 cells (7,500) were seeded
in 100 μl medium into each well of 96-well plates and incubated for
24 h at 37°C. The medium was then changed with fresh medium and
exposed for 24, 48 or 72 h to the drugs at the following
concentrations: 5 nM docetaxel (Accord Healthcare, France), 10 μM
oxaliplatin (Teva Santé, France), 50 μM 5-fluorouracil (Pfizer,
USA) or 1 μM camptothecin (Sigma, USA). After the incubation
periods, 10 μl of MTT reagent (5 mg/ml in PBS) was added into each
well and cells were incubated at 37°C for 3 h to allow the MTT
cleavage to occur. The reaction was then stopped with 100 μl
isopropanol with 0.04 N hydrochloric acid (HCl). The absorbance was
measured within an hour, on a multiplate reader (Thermo Labsystems
Multiskan spectrum, UV/Visible Microplate Reader, USA) with a test
wavelength of 570 nm and a reference wavelength of 630 nm. For cell
growth analysis, 7,500 cells were seeded in 100 μl medium into each
well of 96-well plates and the MTT test was performed daily as
above.
Isolation of chemoresistant cells
HT29 and HCT116 cells were seeded at a density of
106 cells/57 cm2 plates and were then
continuously exposed to oxaliplatin (10 μM) or docetaxel (5
nM).
RNA extraction and reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA from parental and chemoresistant cells
were extracted using NucleoSpin RNA II columns according to the
manufacturer’s instructions (Macherey-Nagel, France). Two
micrograms of total RNA were used for cDNA synthesis with random
hexamers and reverse transcription was performed with the M-MULV
reverse transcriptase (NEB Biolabs, France) in a final volume of 25
μl. For PCR, 2 μl of cDNA was used with primers designed using the
‘Primer 3 Plus’ software (the primer sequences will be provided
upon request). The thermal cycling conditions were 94°C for 5 min,
followed by 23–35 cycles at 94°C for 30 sec, 59–61°C, for 50 sec
and 72°C for 1 min, and a final extension at 72°C for 5 min. The
housekeeping GAPDH or P0 genes were used as controls. The PCR
products were visualized on 2% agarose gels. Reactions were run in
three independent experiments.
Quantitative real-time PCR
The differential expression of all genes was
analyzed by real-time PCR (ABI 7000, Applied Biosystems, France).
PCR was performed with the Power SYBR-Green PCR Master Mix,
according to the manufacturer’s instructions (Applied Biosystems).
All conditions were normalized relative to the GAPDH control
transcript. The results were analyzed using the 2−ΔΔCt
method (27).
Total protein extraction and western
blotting
Cells were harvested, washed in PBS and homogenized
in lysis buffer (1% SDS, 1 mM Na3VO4, 10 mM
Tris pH 7.4) containing protease inhibitor cocktail (Roche, Meylan,
France) and kept for 20 min on ice. Genomic DNA was sheared by
repeated passing of the extracts through 25-gauge needles and then
centrifuged at 4200 g, for 1 min. Protein quantification was
carried out using the Bio-Rad DC Protein Assay system and the
absorbance was measured within one hour on a multiplate reader
(Thermo Labsystems Multiskan spectrum, UV/Visible Microplate
Reader, USA) at 750 nm. Fifty micrograms of protein extracts were
boiled in Laemmli buffer for 5 min, separated by SDS-PAGE using 8%
(MDR1 and E-cadherin) or 12% (ALDH1A1) polyacrylamide gels and
transferred onto a polyvinylidene fluoride membrane (GE Healthcare,
France) by electroblotting. Protein extracts used for detecting
MDR1 were not boiled. Membranes were saturated using Sea Block
Blocking Buffer (Thermo Scientific, France) for 1 h at room
temperature. Primary antibodies MDR1, ALDH1A1 (Cell Signaling
Technology, USA), E-cadherin and Hsc70 (Santa Cruz Biotechnology,
USA) were diluted in blocking buffer (Odyssey LI-COR Biosciences,
USA), containing 0.1% Tween-20®. Membranes were
incubated with the suitable primary antibody, at 4°C overnight,
then washed four times with PBS/0.1% Tween-20 and incubated with
the infrared absorbing secondary antibody (Odyssey®) for
1 h at room temperature. Membranes were washed again and protein
bands revealed using the Odyssey-LI-COR imaging system.
Cell cycle and flow cytometry
analysis
Cell cycle distribution of parental and resistant
cells was determined using the BD Accuri C6 flow cytometer (BD
Biosciences, USA). Cells were plated at a density of 106
cells/57 cm2 plates and cultured for 48 h before
analysis. Under these conditions, the cells were in the log phase
of the growth cycle. Cells were trypsinized and centrifuged. The
pellet was suspended in 1 ml of 70% cold ethanol (−20°C) and
incubated for ≥2 h at −20°C. After this incubation, cells were
washed twice with PBS and each pellet was suspended in 500 μl of a
PBS buffer containing 0.2% NP40 and 0.5 mg/ml of RNase A. At this
point, cells were incubated at room temperature for 15 min and then
on ice for 10 min. Finally, 50 μg/ml propidium iodide
(Sigma-Aldrich, USA) was added. Non-labeled corresponding cells
served as gating controls.
Anchorage-independent growth assay
Soft agar assays were performed to determine the
ability of parental and chemoresistant cells to grow under
anchorage-restricting conditions. Each well of a 6-well plate was
coated with 1 ml of the corresponding medium with 1 ml of 1% SeaKem
agarose (Lonza, USA). After 20 min of incubation at 37°C,
suspensions of 500 cells were added in 750 μl of medium with 250 μl
of 1% SeaKem agarose. Cells were incubated for 18 days under
standard conditions (37°C, 5% CO2) in 300 μl medium.
After the incubation period, cells were examined by
stereomicroscopy and the number of colonies was counted in each
well.
Cell proliferation
The proliferation rate of the parental and resistant
cells was analyzed over a 6-day culture period. For this, 24,000
cells per well were seeded in 6-well plates. Every day, 3 wells per
condition were counted using the BD Accuri C6 flow cytometer’s cell
counting function. Briefly, the supernatant was harvested and cells
washed with PBS. Then, cells were trypsinized with 300 μl of 0.05%
trypsin/EDTA per well. The single cell-suspensions were washed
twice with PBS, and suspended in a final volume of 150 μl PBS. The
total volume was passed through the cytometer and the cell number
was counted directly.
Wound-healing assay
Parental and resistant cells were seeded in 6-well
plates and allowed to adhere and spread to give a confluent
monolayer. At this point, a straight line was made with a p200
pipette tip to create a scratch (wound). Reference points were made
with an ultrafine tip marker. Cells were then washed gently with
PBS and cultured in their corresponding medium for the period of
the assay. Cells were observed using an inverted Zeiss Axio Vert.A1
microscope (x5) (Carl Zeiss, Germany). Photos were taken every day
on exactly the same field, with the microscopy camera Axiocam ERc
5s Rev 2.0 (Zeiss), starting from the day the scratch was made (day
0). The reference mark was left outside the captured field, and
scratch sizes were similar in the parental and resistant cells at
day 0. The images acquired for each condition were analyzed using
Zen Lite 2011 software. The distance (μm) between the two sides of
the scratch was measured every day, according to ref. 28.
Statistical analysis
Statistical significance was determined by Student’s
t-test. p-values <0.05 were considered as statistically
significant.
Results
Short-term cell viability analysis
In order to determine their drug sensitivity, HT29
and HCT116 cells were exposed to 5 nM docetaxel, 10 μM oxaliplatin,
50 μM 5-fluorouracil or 1 μM camptothecin for 24, 48 or 72 h. Cell
viability was assessed using the MTT test (Fig. 1). HT29 cells displayed nearly 80%
viability after docetaxel exposure for 24 h and essentially no
sensitivity to the three other molecules. After 48- and 72-h
treatment, cell viability decreased significantly in response to
docetaxel (68 and 45%, respectively), 5-fluorouracil (68 and 56%,
respectively) or camptothecin (42 and 23%, respectively).
Oxaliplatin reduced cell viability by 20% after 72 h. By contrast,
HCT116 cells were sensitive to the four molecules at 24 h, with 77,
89, 85 and 49% viability for docetaxel, oxaliplatin, 5-fluorouracil
and camptothecin, respectively. For the 48- and 72-h conditions,
HCT116 cell viability decreased even further with 29, 31, 30 and 3%
viability at 72 h for docetaxel, oxaliplatin, 5-fluorouracil and
camptothecin, respectively.
Isolation of drug-resistant cells
To determine the effects of drug resistance on the
cellular phenotype, we selected HCT116 and HT29-resistant
derivatives. Although we could not isolate camptothecin or
5-fluorouracil-resistant cells, we successfully recovered docetaxel
(5 nM) or oxaliplatin (10 μM)-resistant cell populations following
2 months of continuous exposure to either drug, after which no more
signs of cell death were observed. Stably resistant cell
populations were isolated after 7–9 weeks.
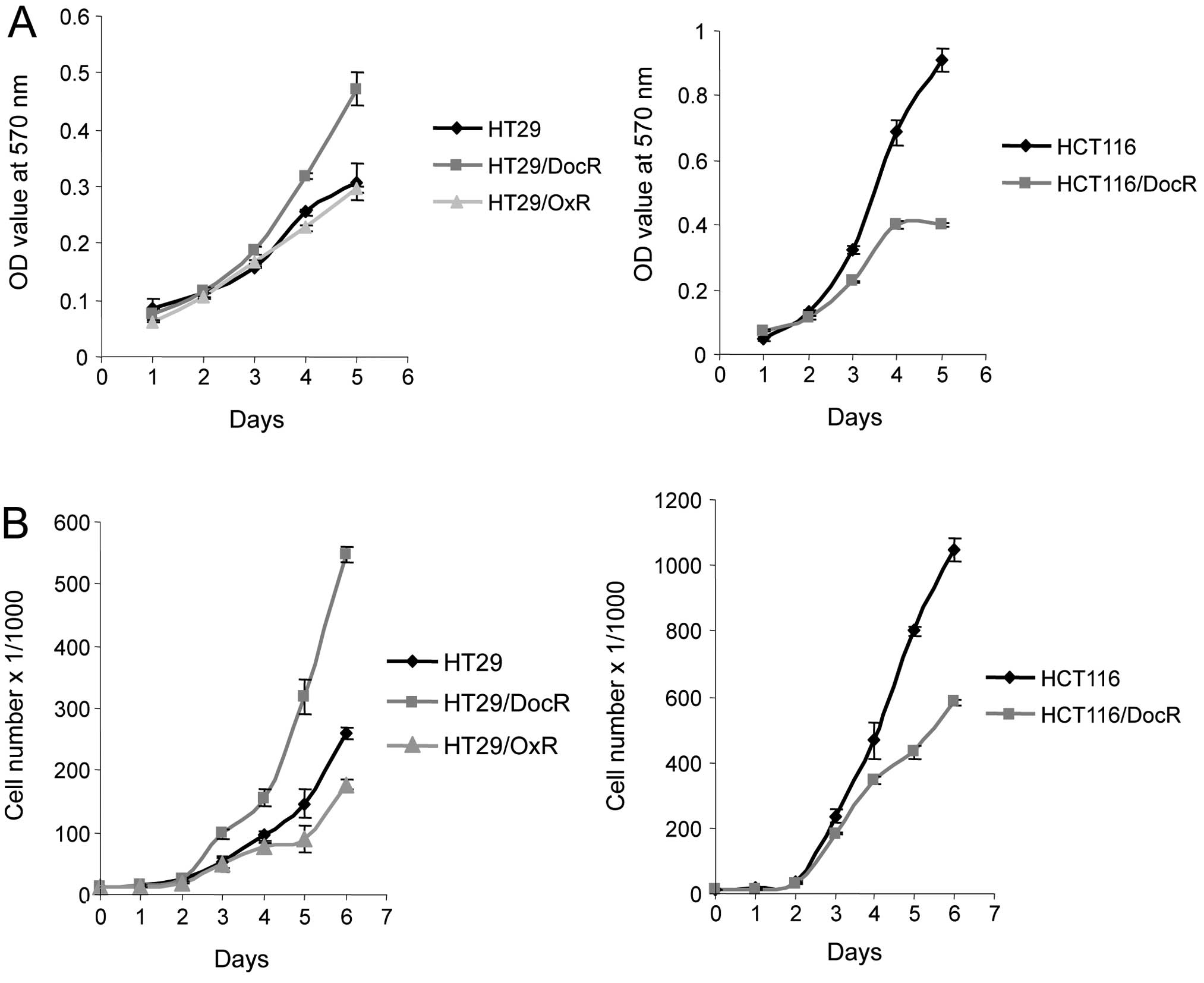
Growth of resistant cells
Growth of the drug-resistant cells was analyzed over
5- or 6-day periods. We first measured the in vitro
proliferation rate of parental and resistant cells using the MTT
cell viability assay. In parallel, the proliferation rate of the
cells was assessed using the cell counting feature of the BD accury
C6 flow cytometer. Both methods gave similar results for the
general trend of the growth curves (Fig. 2). The proliferation rate of the
HT29/DocR cells was higher than that of the parental cells. By
contrast, HCT116/DocR cells grew more slowly than HCT116 parental
cells. In addition, HT29/OxR cells grew somewhat more slowly than
HT29 parental cells.
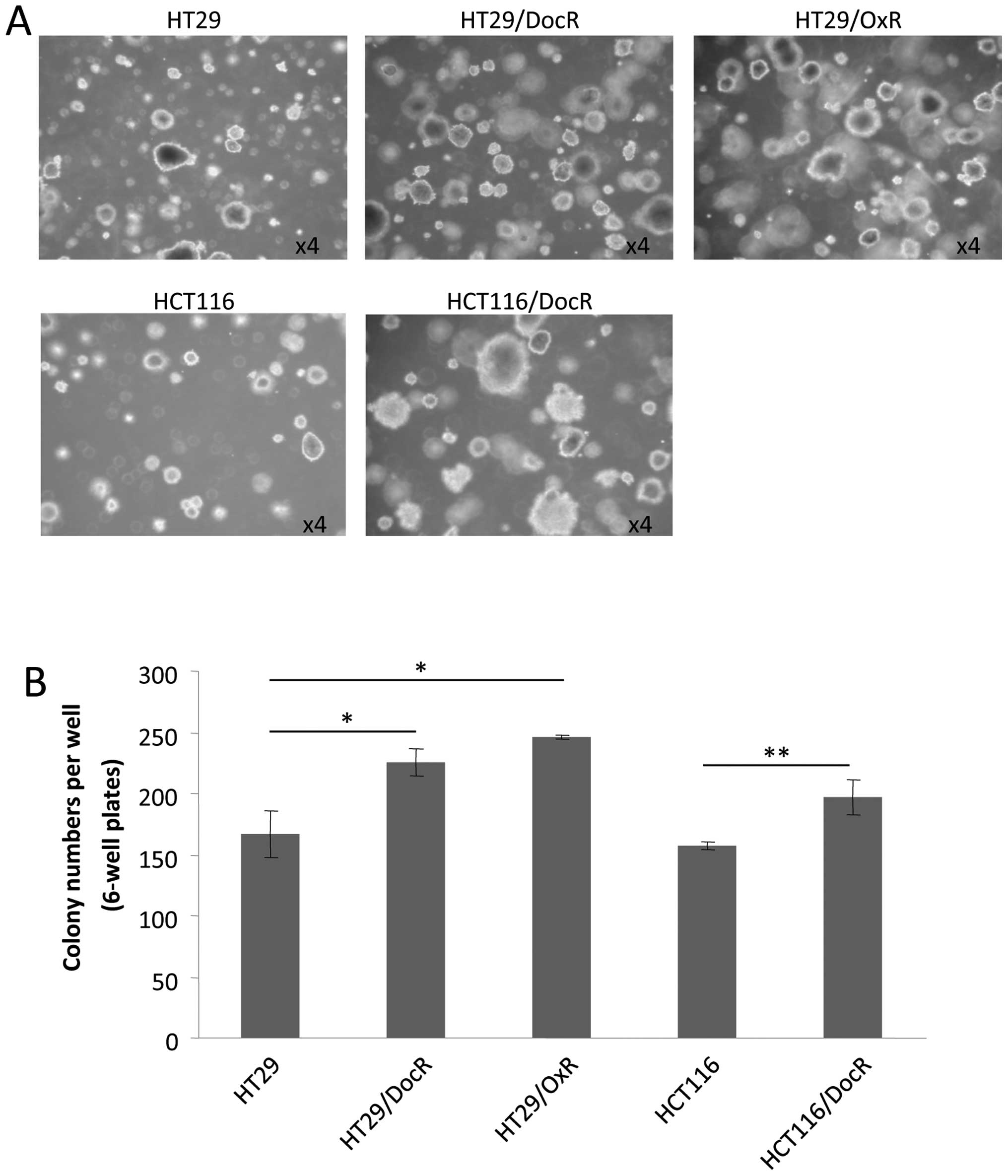
Colony formation of chemoresistant
cells
A phenotypic characteristic of CSC is their ability
to grow as colonospheres or spheroids, which can be evaluated in
presence of agarose, i.e., when no adhesion to the plate is
possible. Both the parental and the three resistant cell lines
generated spheres after 18 days of incubation. However,
chemoresistant cells produced essentially larger spheroids
(Fig. 3A). Moreover, the number of
spheroids was significantly higher for the resistant cells
(Fig. 3B). HT29 cells formed on
average 177 spheroids, versus 220 (124%) and 246 (139%) spheroids,
respectively for HT29/DocR and HT29/OxR cells (p<0.05). For
HCT116 cells, an average 156 spheres were counted compared to 190
(122%) spheres for HCT116/DocR cells (p<0.01).
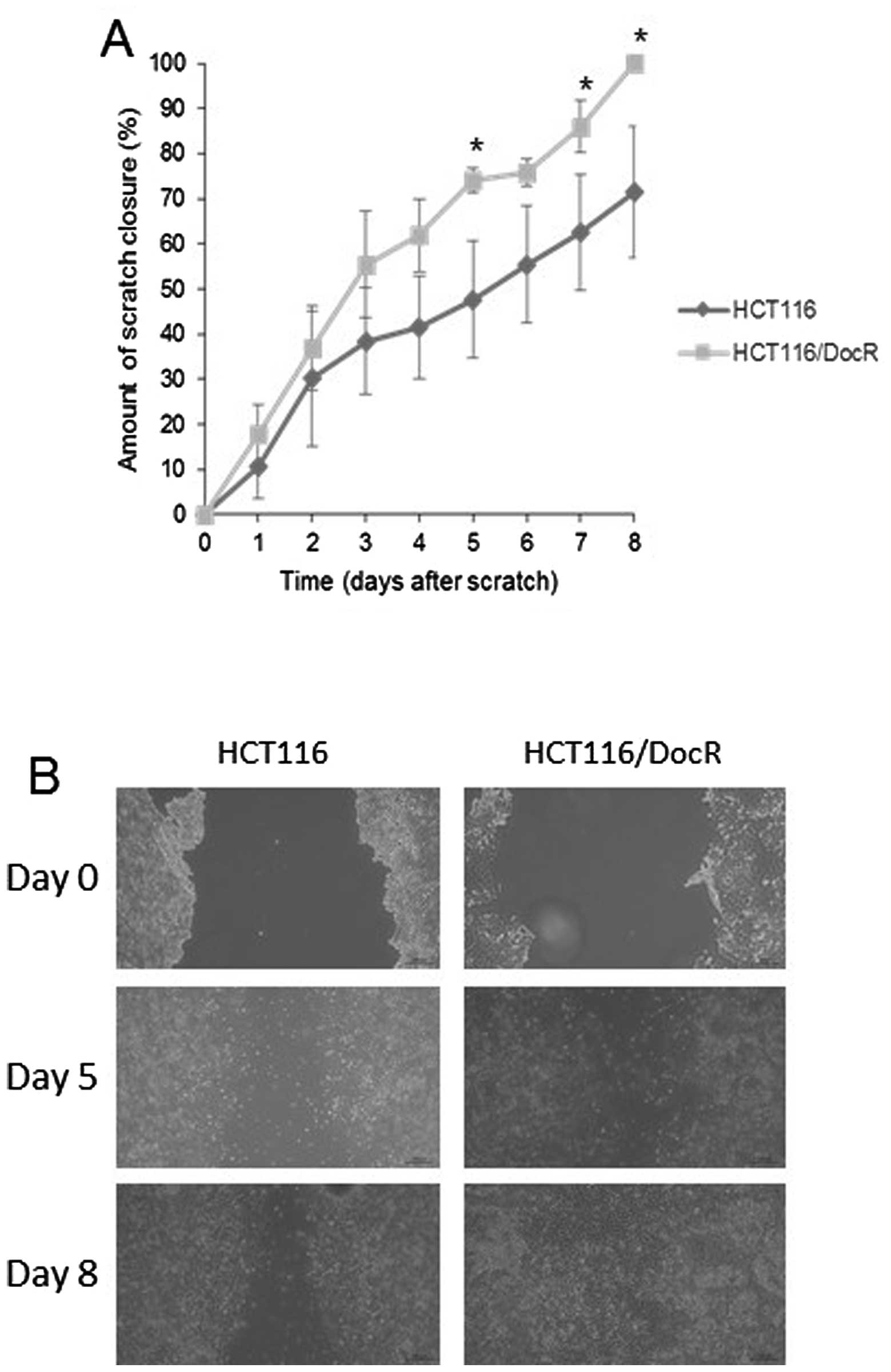
Wound-healing assay
In order to assay the migration ability of resistant
cells, an indicator of the stemness-like phenotype, we used the
wound-healing assay method (29).
Parental and resistant cells were cultured in 6-well plates, and a
gap was made upon scratching the plate surface after cells had
reached confluency. Images were captured daily and the distance (in
μm) between the two edges of the scratch was measured. For the HT29
cells, no significant difference was recorded in the percentage (%)
of scratch closure between the parental (49.9%) and resistant cells
(48 and 49.8% for HT29/DocR and HT29/OxR cells, respectively - data
not shown). By contrast, the HCT116/DocR cells were able to achieve
a complete closure of the scratch (100%) at day 8 post-scratch
while the HCT116 parental cells had closed only 71% of the gap on
the same day (Fig. 4).
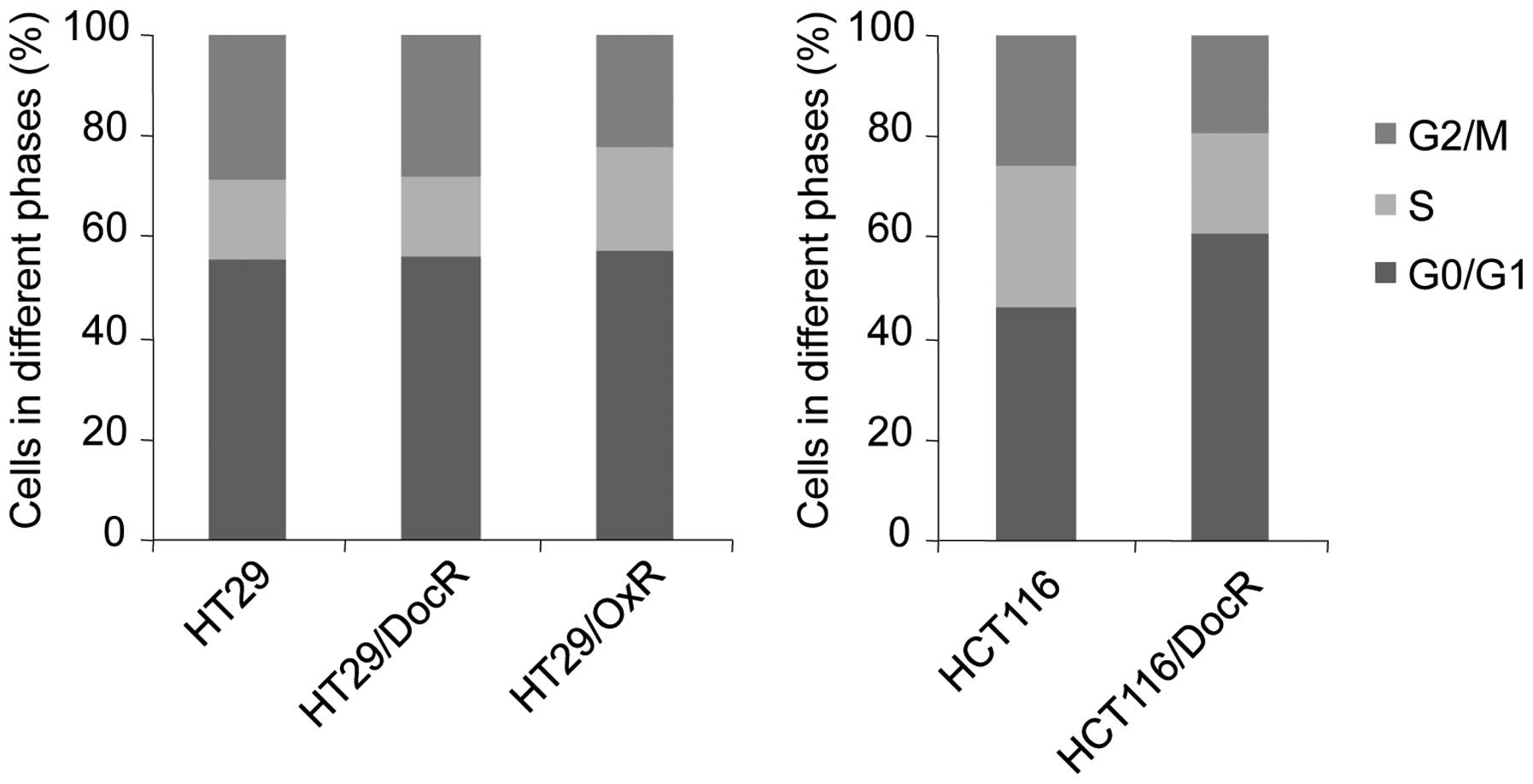
Cell cycle distribution
The effects of chemoresistance on the cell cycle
were evaluated using flow cytometry analysis. Genes implicated in
cell cycle or apoptosis control were studied by RTqPCR. No change
in cell cycle distribution was observed in HT29/DocR cells
(Fig. 5). However, the proportion
of HCT116/DocR cells (61%) in the G0/G1 phase was higher than that
of the parental cells (46%), and fewer cells were in the S (20 vs
28% in parental cells) and G2/M phases (19 vs 26%). Accordingly,
the level of cyclin B1, which controls the G2/M transition, was
decreased in HCT116/DocR cells, which correlated with the cell
cycle distribution of the cells (Table
I) and the slowest growth. For the HT29/OxR cells, the fraction
of cells in G2/M was decreased (from 29 to 22%) and that in S phase
was increased (from 15 to 21%) (Fig.
5). Surprisingly, cyclin B1 was increased while the fraction of
HT29/OxR cells in G2/M was decreased, suggesting that some other
mechanism may be involved in downregulating HT29/OxR cells growth.
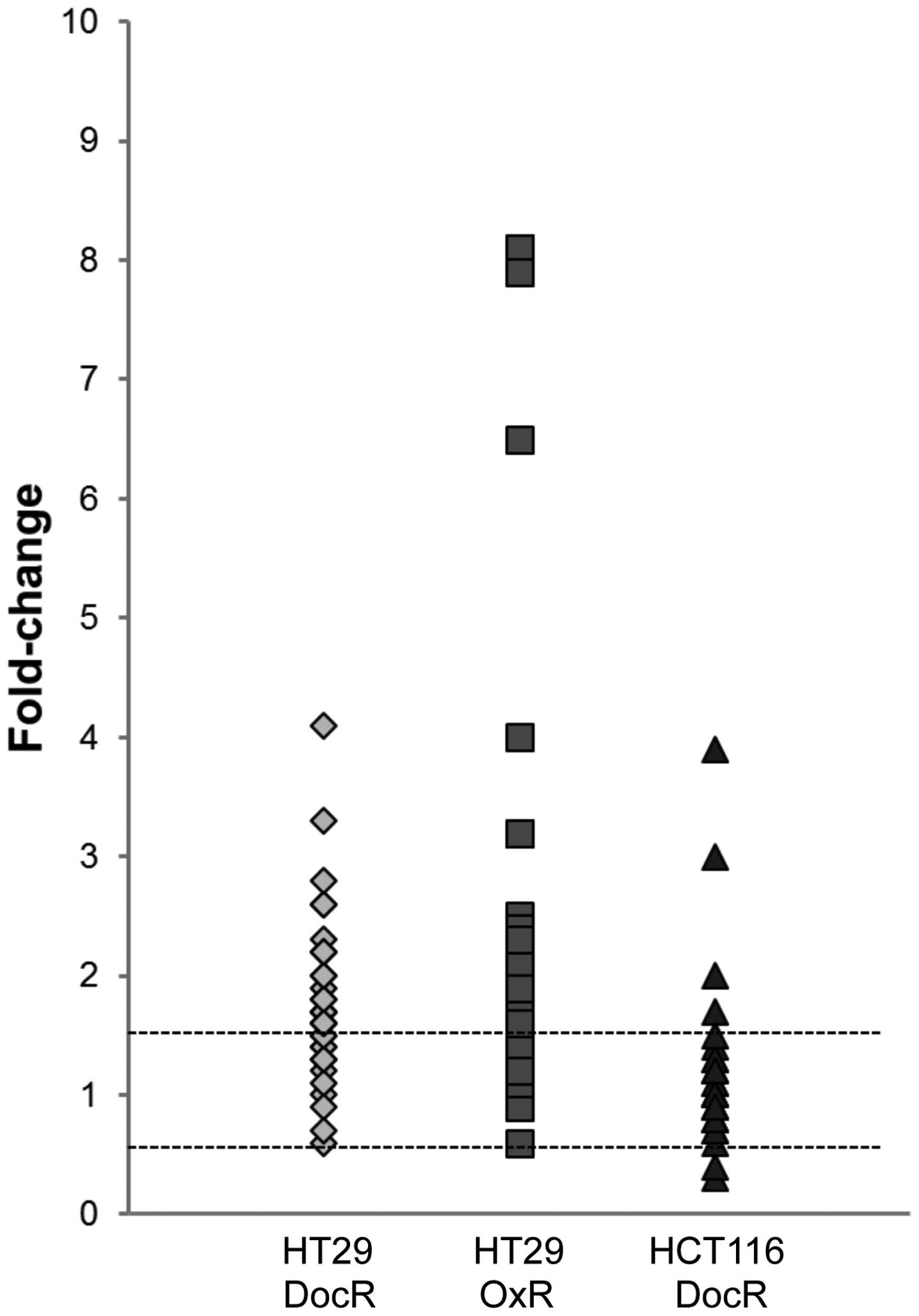
Cyclin D1, which controls progression through the G1 phase, showed
a 1.7–1.8-fold increase in the three resistant cell lines. The
cyclin-dependent kinase inhibitor 1 (p21), which acts as a negative
regulator of cell cycle progression during the G1 and S phases, was
overexpressed in the resistant cells with a 2.6-, an 8.1- and a
3-fold change in the HT29/DocR, HT29/OxR and HCT116/DocR cells,
respectively (Table I). The
cyclin-dependent kinase inhibitor 1B (p27) was marginally increased
(1.6- and 1.4-fold in HT29/DocR and OxR cells, respectively).
Survivin, which opposes apoptosis, was overexpressed in HT29/OxR
cells (3.2-fold).
 | Table IRT-qPCR analyses of marker genes in
drug-resistant cells, cell cycle and apoptosis-related genes,
differentiation markers and the CSC potential marker
expression.a |
Table I
RT-qPCR analyses of marker genes in
drug-resistant cells, cell cycle and apoptosis-related genes,
differentiation markers and the CSC potential marker
expression.a
| Gene | Fold change versus
levels in parental cell lines |
|---|
|
|---|
| HT29 DocR | HT29 OxR | HCT116 DocR |
|---|
| Drug
resistance | | | |
| MDR1 |
3.3±1.4b |
8.1±2.3b |
22±5.8b |
| ABCG2 |
0.6±0.1c | 0.6±0.2 |
3.9±0.6c |
| MRP2 | 1.9±0.7 |
4±1c |
0.3±0.1c |
| OATP2B1 | 1.6±0.5 | 0.9±0.2 | 0.9±0.3 |
| ATP7A |
1.7±0.2c |
1.6±0.3b |
0.7±0.1b |
| ATP7B |
2.2±0.5c | 1.1±0.3 | 1.4±0.5 |
| hCTR1 | 1±0.1 | 1.5±0.6 | 1.1±0.1 |
| MT2A | 1.2±0.4 |
2.4±0.4c |
0.96±0.01c |
| GSTπ |
1.1±0.1b |
1.2±0.1b | 1.3±0.4 |
| CYP 3A5 |
1.4±0.2c | 1.1±0.2 | 1±0.4 |
| XPD |
1.5±0.05c |
1.2±0.03c | 0.9±0.2 |
| CSB |
1.8±0.1c |
1.8±0.1c | 1.2±0.4 |
| XPF |
1.7±0.3c |
1.9±0.1c | 1±0.2 |
| SRPK1 | 0.9±0.1 |
1.6±0.4b | 1±0.2 |
| NFκB1 |
1.4±0.1c |
1.5±0.1c | 1.1±0.2 |
| Cell cycle | | | |
| Cyclin B1 | 1.3±0.3 |
2.5±0.2c |
0.6±0.1c |
| Cyclin D1 |
1.7±0.2c |
1.8±0.3b | 1.7±0.6 |
| p21 |
2.6±0.7b |
8.1±1.2c |
3±0.9b |
| p27 | 1.6±0.5 |
1.4±0.2b | 0.8±0.2 |
| Survivin | 1.3±0.3 | 3.2±1.3 |
0.8±0.1b |
|
Differentiation | | | |
| E-cadherin |
2±0.5b |
1.8±0.3c | 1.2±0.2 |
| TGFβ1 |
2.6±0.4c |
2±0.4c |
1.2±0.1c |
| TGFβ2 | 0.7±0.2 | 1.2±0.2 |
0.4±0.2b |
| Villin |
1.5±0.3b |
1.2±0.1c | Undetected |
| β1 catenin |
1.7±0.3c |
2.4±0.4c |
0.7±0.2b |
| α1 catenin |
1.6±0.3b |
2.1±0.4b |
0.8±0.1b |
| CSC potential
markers | | | |
| CD26 |
2.3±0.4c | 2±0.6 |
2±0.3c |
| CD133 |
2.8±0.7c |
2.1±0.6b | 1±0.3 |
| CD166 |
4.1±0.9c |
7.9±1.9c |
1.5±0.3b |
| EpCAM |
1.8±0.1c |
2.3±0.4c | 0.9±0.1 |
| EPHB2 | 2.2±0.8 |
6.5±0.8c | 1.3±0.2 |
| Oct4 |
2±0.6b | 1.6±0.1 | 1±0.3 |
| Myc |
2.2±0.4b |
1.9±0.4b |
0.9±0.03b |
| ITGB1 | 1.6±0.4 |
3.2±0.04c | 1.2±0.2 |
| ALDH1A1 |
0.8±0.01c | 0.6±0.2 | Undetected |
To follow the characterization of the resistant cell
phenotypes, we selected a number of specific markers associated
with drug resistance, colon cancer differentiation and cancer
stemness. A global survey of these markers showed that
drug-resistant HT29 cells expressed most of them at higher levels
than parental cells (24 and 25 overexpressed out of 34 markers for
HT29/DocR and HT29/OxR, respectively). By contrast, most of these
markers remained unchanged in HCT116/DocR in comparison with
parental cells (24 unchanged out of 34 markers). CD26, CD166,
Cyclin D1, p21 and MDR1 were the five genes commonly upregulated
between all three drug-resistant cell populations (Table I and Fig. 6).
Drug resistance and expression of
chemo-resistance-related markers
The expression level of genes potentially involved
in oxaliplatin or docetaxel resistance was analyzed by RT-PCR,
RT-qPCR and/or western blot analyses. The three resistant cell
populations exhibited higher levels of MDR1, a member of the
superfamily of ATP-binding cassette (ABC) transporters responsible
for decreased drug accumulation in multidrug-resistant cells. A
3.3-fold-change for the HT29/DocR cells, an 8.1-fold-change in the
HT29/OxR cells and a 22-fold-change increase for the HCT116/DocR
cells were obtained (Table I).
However, the western blot analysis showed overexpression of the
MDR1 protein in HCT116/DocR cells, but not in HT29-resistant cells,
possibly because of a very low basal expression level (Fig. 7). The level of ATP-binding cassette
sub-family G member 2 (ABCG2) mRNA was increased in HCT116/DocR
cells, but declined in HT29/DocR and HT29/OxR cells. The multidrug
resistance-associated protein 2 (MRP2), which was reported to be
involved in the response to oxaliplatin (30), was increased in HT29/OxR cells but
decreased in HCT116/DocR cells. The level of the organic anion
transporting polypeptide 2B1 (OATP2B1), which contribution to
docetaxel uptake and clearance is not well established (31), was evaluated. No change was
registered for the HT29/OxR cells or the HCT116/DocR cells, while a
1.6-fold increase was observed in HT29/DocR cells. The copper
transporter ATP7A that pumps out platinum compounds (32), was increased in some tumors,
including CRC, and slightly increased in the HT29/DocR and HT29/OxR
cells. ATP7B levels were increased only in HT29/DocR. Drug uptake
was evaluated with the copper transporter hCTR1, but no significant
difference was observed in the resistant cells. We looked also at
expression of drug detoxification and drug-metabolizing enzymes.
Metallothionein 2A (MT2A) was increased (2.4-fold) in HT29/OxR
cells. Glutathione-S-transferase π (GSTπ and cytochrome CYP3A5
levels showed no change in the resistant cells. Since DNA repair is
one of the predominant events that occur in resistance to platinum
agents, we analyzed expression of xeroderma pigmentosum group D and
group F (XPD and XPF) and cocaine syndrome group B (CSB), members
of the nucleotide excision repair (NER) pathway (33). No change was observed in
HCT116/DocR mRNA levels of these genes, whereas CSB and XPF and, to
a lower extent, XPD, were increased (1.5- to 1.9-fold) in HT29/DocR
and HT29/OxR cells, although docetaxel does not trigger DNA damage.
Finally the nuclear factor NFκB1 that plays a role in the
inhibition of apoptosis and the induction of resistance to various
chemotherapeutic agents (34), was
slightly overexpressed in HT29 (1.4- to 1.5-fold), but not in
HCT116-resistant cells.
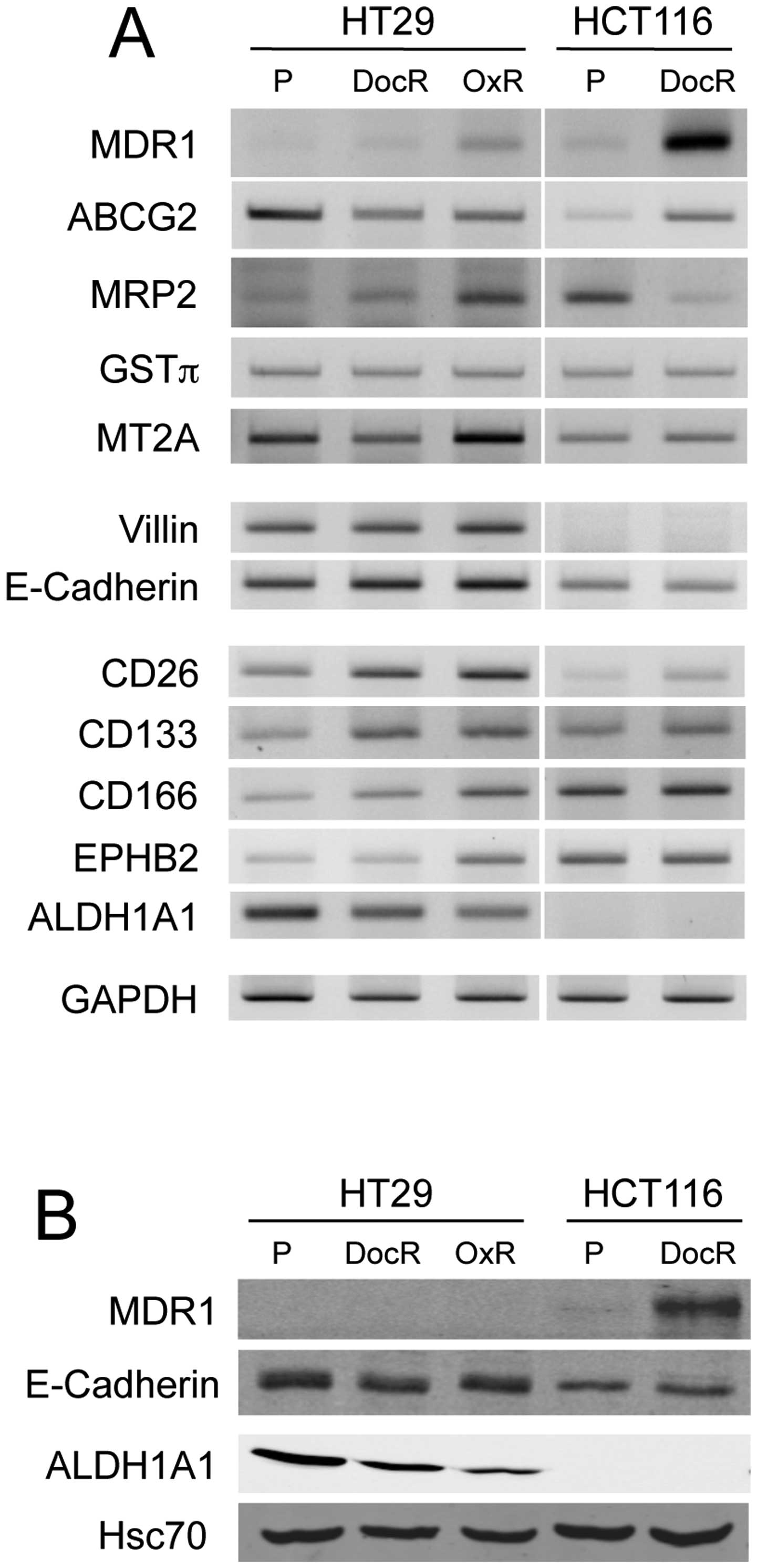 | Figure 7Comparative expression of
drug-resistance-related markers, differentiation and CSC potential
markers in parental and chemo-resistant cells. (A) RT-PCR analyses
of MDR1, ABCG2, MRP2, GSTπ, MT2A, villin, E-cadherin, CD26, CD133,
CD166, EPHB2 and ALDH1A1 expression. GAPDH was used as control. (B)
Western blot analysis of MDR1, E-cadherin, ALDH1A1 expression.
Hsc70 served as protein loading control. P, parental;
drug-sensitive cells; DocR, docetaxel (5 nM)-resistant cells; OxR,
oxaliplatin (10 μM)-resistant cells. |
Effect of chemo-resistance on the
expression of differentiation markers
Solid cancer resistance to therapy has been linked
to the existence of CSC that may be mostly resistant to anti-cancer
treatments. We used RT-PCR, RT-qPCR and western blot analyses to
survey differentiation markers. E-cadherin, which plays a role in
cell-cell adhesion, is reportedly associated with the epithelial to
mesenchymal transition (EMT), a characteristic feature of
cancer-associated aggressiveness, when underexpressed (35). However, E-cadherin mRNA increased
by 2- and 1.8-fold in HT29/DocR and OxR cells (Table I), while no change was detected at
the protein level (Fig. 7), and
remained unchanged in HCT116/DocR cells. In addition, expression of
TGFβ1, which is involved in metastasis and invasion, was increased
in HT29/DocR and HT29/OxR cells, but not in HCT116/DocR cells.
TGFβ2 was reduced in HCT116/DocR cells. The tissue-specific
actin-binding protein villin, which is associated with invasion and
aggressiveness in cancers, was slightly overexpressed in HT29/DocR
cells. β1 catenin and α1 catenin that are implicated in EMT in
cancer, were overexpressed in both HT29/DocR and HT29/OxR cells,
but no change in expression occurred in HCT116/DocR cells.
Chemo-resistance and expression of CSC
markers
We next analyzed the expression of several CSC
markers (Table I). Dipeptidyl
peptidase-4 (DPP4), also known as CD26 (cluster of differentiation
26) (36) is a stem cell-inducing
gene; it was overexpressed in HT29/DocR with a 2.3-fold-change,
likewise in HT29/OxR cells and HCT116/DocR cells with a
2-fold-change. We next studied the expression of CD133 and CD166,
trans-membrane glycoproteins implicated in cell adhesion and
migration (37,38). CD133 mRNA levels increased by 2.8-
and 2.1-fold in HT29/DocR and HT29/OxR cells, respectively, but did
not change in HCT116/DocR cells. CD166 expression was strongly
increased in HT29/DocR (4.1-fold) and HT29/OxR (7.9-fold) cells,
whereas it was increased only 1.5-fold in HCT116/DocR cells. The
epithelial cell adhesion molecule (EpCAM) was overexpressed (1.8-
to 2.3-fold) in the HT29-resistant cells, but not in
HCT116-resistant cells. The ephrin type-B receptor 2 (EPHB2) gene,
which encodes a receptor with tyrosine kinase activity that
characterizes colorectal cancer stem cells, was also overexpressed
in HT29/DocR (2.2-fold) and HT29/OxR (6.5-fold) cells, but not in
HCT116/DocR cells (Table I and
Fig. 7). The octamer-binding
transcription factor 4 (Oct-4) is a transcription factor essential
for maintenance of the self-renewal, or pluripotency of
undifferentiated embryonic stem cells (37). Its level was increased in the
HT29/DocR (2-fold) and HT29/OxR (1.6-fold) cells, but not in
HCT116/DocR cells. The Myc oncogene, considered as an essential
factor in the generation and maintenance of the stemness status
(39), was also overexpressed in
both HT29/DocR (2.2-fold) and HT29/OxR (1.9-fold) cells, but not in
HCT116/DocR cells. Integrin β-1 (ITGβ-1) mRNA, which has been shown
to play a role in the resistance of hepatic and esophageal
carcinoma cells to docetaxel (40,41),
showed an increased expression in HT29/DocR (1.6-fold) and HT29/OxR
(3.2-fold) cells, again with no noticeable change in HCT116/DocR
cells. The serine/arginine protein kinase 1 (SRPK1), involved in
phosphorylation-dependent activation of serine/arginine-rich RNA
binding proteins, in platinum resistance (42) and in the acquisition of an EMT
phenotype (43) showed a 1.6-fold
increase in HT29/OxR.
Discussion
In this study, we set out to analyze the overall
phenotype of human colon cancer cells selected for their acquired
resistance to the anticancer drugs oxaliplatin and docetaxel. We
were successful in isolating drug-resistant HT29 and HCT116 cells
following 2-month continuous exposure to the drugs. Short-term drug
treatments showed that HCT116 cells were much more sensitive than
HT29 cells, possibly due, at least in part, to their higher
multiplication rate. Camptothecin was the most toxic for both cell
lines after 48 or 72 h. However, we could not isolate
camptothecin-resistant cells, presumably because DNA topoisomerase
I activity is mandatory. The drug-resistant cells were capable of
forming more and larger colonies in agarose-containing medium than
naïve cells. This capacity fits well with stem cell properties
whereby a cell can expand and give rise to a heterogeneous colony
in vitro, under anchorage-independent conditions, resembling
that happening during EMT and invasion. In addition, the ability of
scratch repair was strongly magnified in HCT116/DocR cells as
compared to parental cells, although this was not the case for
HT29-resistant cells, making this another clear distinction among
the HT29 or HCT116-resistant cells.
Drug metabolism and disposition
Several drug-metabolizing enzymes have been
implicated in the metabolism of docetaxel and/or platinum salts
(6,44,45).
The expression levels of CYP3A5, GSTπ and MT2A were barely changed
in the resistant cells, except in HT29/OxR cells where a 2.4-fold
increase occurred for MT2A, which agreed with previous reports
(46,47) linking the overexpression of MT2A to
resistance to platinum drugs.
The multidrug resistance protein 1 (MDR1 or Pgp or
ABCB1) is an ATP-dependent efflux pump localized at the cell
membrane that plays a role in expulsing a variety of toxic agents.
MDR1 is primarily expressed in normal tissues such as the liver,
the brain and the gastrointestinal tract, before the initiation of
any therapy (48,49). However, it has been linked to a
general drug resistance phenotype, as its gene is often amplified
and/or overexpressed in tumors or in colon CSC (50,51).
Docetaxel is mainly eliminated by Pgp-mediated efflux (11). Hence, the resistant phenotype
observed here agrees well with an increased expulsion of docetaxel
due to overexpression of Pgp in HCT116/DocR. Strikingly, ABCG2 was
overexpressed in HCT116/DocR cells only. Although oxaliplatin is
not an ABCG2 substrate (21),
upregulation of its mRNA has been observed in some
oxaliplatin-resistant CRC cells (52). In addition, docetaxel may (53) or may not (21) be an ABCG2 substrate, depending on
the study. The MRP2 (encoded by the ABCC2 gene) increase was
associated with resistance to oxaliplatin of HT29/OxR, as reported
(54). Hence, the main genes
potentially involved in oxaliplatin resistance were MDR1 and MRP2.
OATP2B1, although reported to affect the intracellular
concentration of several anticancer drugs (31,45,55,56),
was only marginally increased in HT29/DocR. No consistent pattern
of regulation was observed for the ATP7A, ATP7B and hCTR1 copper
transporters, although a 2.2-fold increase in ATP7B levels occurred
in HT29/DocR.
DNA repair and signaling
The nucleotide excision repair (NER) pathway is
responsible for repairing DNA adducts produced by platinum agents
(57,58). Expression of CSB and XPF genes was
increased in HT29/OxR cells, in good agreement with the increase in
DNA-repair ability and the development of resistance to cisplatin
(59). Expression of these genes
was also increased in HT29/DocR but not in HCT116/DocR cells.
As splicing abnormalities frequently occur in cancer
(60), we looked at SRPK1, a
kinase that phosphorylates, and hence positively controls, the
activity of the SR-rich proteins. However, only HT29/OxR cells
showed a moderate 1.6-fold increase SRPK1 compared to HT29 parental
cells. The nuclear transcription factor NFκB was reported to
mediate tumor progression, metastasis and resistance to drugs
(61). NFκB is activated in
response to many stimuli such as tumor necrosis factor α, radiation
and chemotherapeutics, including docetaxel (62). This activation leads to inhibition
of apoptosis and to resistance to various chemotherapeutic agents
(34), including docetaxel.
However, only small changes in expression were observed in the DocR
cells.
Cell cycle and apoptosis regulation
Distribution of the cells in the different phases of
the cycle was measured by flow cytometry. Although only slight
modifications were observed for HT29-resistant cells, a higher
proportion of HCT116/DocR cells were recorded in the G0/G1 phase,
together with a decreased proportion of cells in the S phase and a
lower proportion in the G2/M phase, in agreement with previous
reports (63). Surprisingly,
Cyclin B1, which main role is to control the G2/M phase, was
increased in the HT29/OxR cells. Cyclin D1, which is required for
the progression through the G1 phase, was increased in the three
resistant cell populations. The cyclin-dependent kinase inhibitor 1
(p21) that mediates growth arrest was increased strongly in the
three resistant cell populations. p21 has also a well-known role in
protecting colon cancer cells against a variety of injuries,
including those caused by anticancer drugs (64). By contrast, the cyclin-dependent
inhibitor 1B (p27), a cell cycle inhibitor, was slightly
overexpressed in HT29-resistant cells. Survivin, that inhibits
caspase activation, thereby preventing apoptosis, was uniquely
increased in HT29/OxR cells.
EMT markers
Resistance to chemotherapeutics has been partly
correlated to an EMT phenotype (65). Three major factors associated with
EMT, E-cadherin, β-catenin and α-catenin were overexpressed in both
HT29-resistant cells, but β-catenin and α-catenin were decreased in
HCT116/DocR cells. TGFβ is a pluripotent cytokine expressed in the
colon that promotes invasion and metastasis during advanced stage
CRC. The increase of TGFβ1 was linked to the EMT in a number of
solid tumors, including CRC (66).
TGFβ1 level was increased in HT29-resistant cells, while TGFβ2 was
either suppressed or unchanged across all cells. The level of the
invasion-associated gene villin was barely changed, although
previous reports showed that overexpression of villin was
correlated to an aggressive cell phenotype and to EMT (67).
Stem cell markers
Many CSC markers, such as cell surface receptors or
transmembrane proteins involved in cell-cell or cell-matrix
adhesion, including CD133, EpCAM, CD166, the integrin family, or
stem cell inducing genes (Oct-4, CD26) have been reported (37,68).
Our data showed that all markers but ALDH1A1 were increased in
HT29-resistant cells. ALDH1A1 expression was undetected in HCT116
parental and drug-resistant cells, although, ALDH1A1 was proposed
to characterize a subpopulation of cells with tumor-initiating or
cancer stem cell properties in several malignancies (69,70).
By contrast with HT29-resistant cells, only CD26 and CD166 showed
some increase, but a very limited one, in HCT116/DocR cells.
Enhanced expression of CD26 correlated with the associated increase
in metastatic capacity and resistance to drugs in CRC (36), and that of CD166, a transmembrane
glycoprotein, was previously reported to result in an enhanced cell
adhesion and migration (38).
EpCAM, a transmembrane glycoprotein involved in cell signaling,
migration, proliferation and differentiation (71), was also increased in HT29
drug-resistant derivatives. Similarly, the ephrin type-B receptor
(EPHB2), which may be controlled by the Wnt pathway, an event
critically required for the progression of colorectal cancer, or
Oct-4, a pluripotent stem cell inducer and a self-renewal
regulatory factor, which may play a role in the physiology of CSC
(72), were also upregulated. Myc
plays a central role in regulating proliferation and survival of
normal cells and CSC and connects malignancy with stemness
(39), was also overexpressed in
HT29-resistant cells. Receptors of the β1 integrin family are
involved in many tumor-promoting activities (73), and overexpression of ITGβ-1 has
been reported in hepatocellular carcinoma (HCC) (41) and esophageal squamous cell
carcinoma (40). It has been
associated with resistance to apoptosis via the activation of a MAP
kinase-dependent pathway. For esophageal squamous cell carcinoma,
it was suggested that targeting ITGβ-1 could enhance the effect of
chemotherapy, particularly in using docetaxel (40).
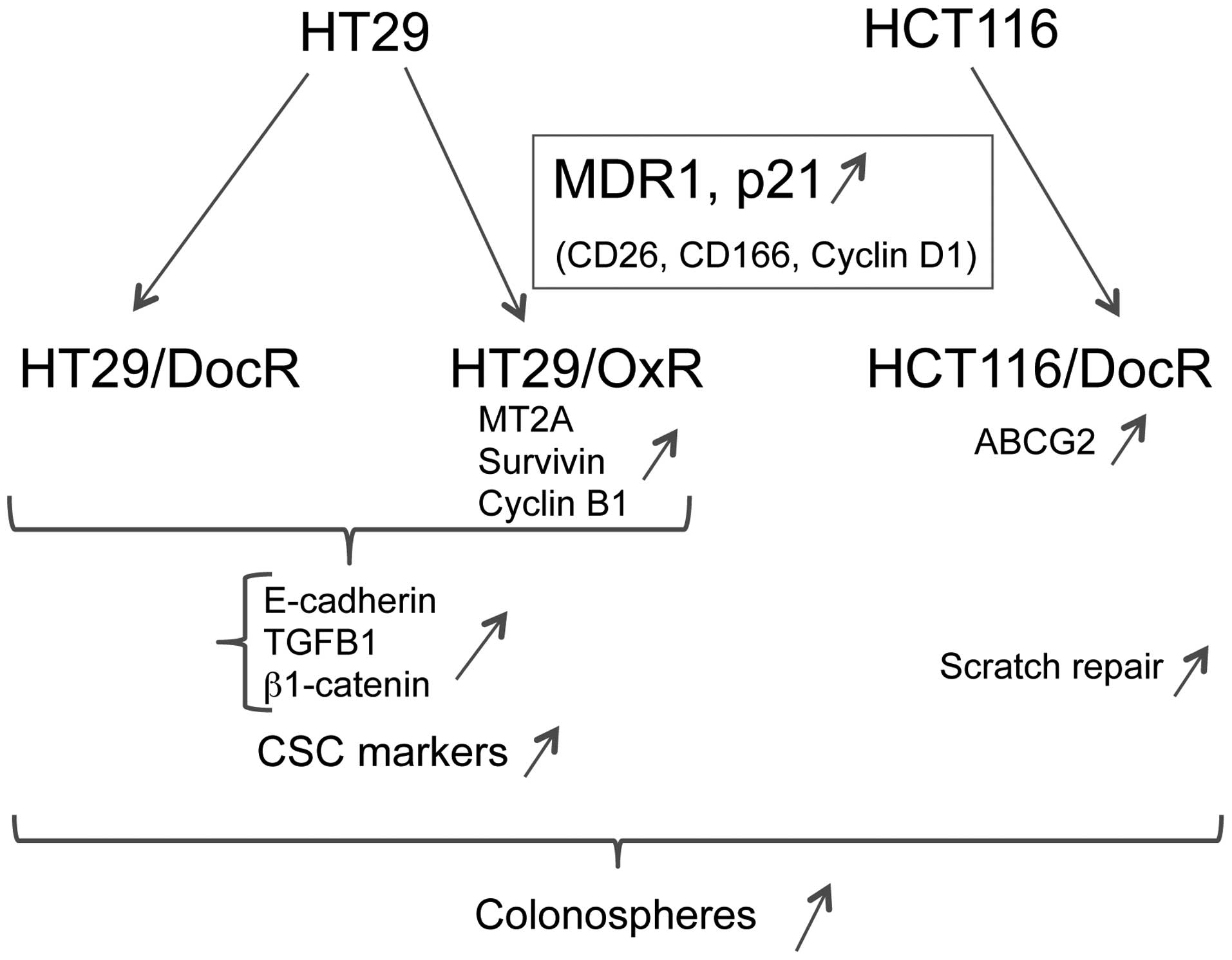
In conclusion, the isolation of oxaliplatin and/or
docetaxel-resistant cell populations from 2 different human colon
cancer cell lines revealed a number of shared phenotypic traits.
Increased ability to form colonospheres was common to HT29 and
HCT116 drug-resistant cells, and all cell populations showed
increased expression of the CD26, CD166, Cyclin D1, p21 and MDR1
genes. However, other markers showed cell line-restricted
regulations. Strikingly, differentiation and EMT markers were
increased in HT29, but not in HCT116 drug-resistant cells. By
contrast, a marked increase in wound repair activity was specific
for HCT116/DocR cells, indicating a higher ability to engage into
active migration. Our results suggest that better chances to
overcome colon cancer resistance, intrinsic or acquired, might be
obtained by lowering tumor expressed MDR1 and, possibly, CD26,
CD166, Cyclin D1 and p21. In conclusion, the selection of
drug-resistant derivatives of HT29 and HCT116 cells led to the
isolation of cell populations displaying a few shared but, mostly,
unshared phenotypic properties (Fig.
8). This suggests that, in the case of oxaliplatin and
docetaxel, the establishment of a drug-resistant phenotype may use
different routes, depending on the cancer cell of origin. Taken
together, the results presented here indicate that the acquisition
of drug resistance by established colon cancer cell lines is only
partly associated with the expression of a strict colon cancer stem
cell phenotype.
Acknowledgements
F.E.K. was supported by a fellowship from the French
Ministry of Education and Research; this study was supported by the
INSERM, Brest University (UBO) and the Ligue Regionale Contre le
Cancer (comité du Finistère).
References
|
1
|
Bray F, Ren JS, Masuyer E and Ferlay J:
Global estimates of cancer prevalence for 27 sites in the adult
population in 2008. Int J Cancer. 132:1133–1145. 2013. View Article : Google Scholar
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
3
|
Wang D and Lippard SJ: Cellular processing
of platinum anti-cancer drugs. Nat Rev Drug Discov. 4:307–320.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barker N, van Es JH, Kuipers J, Kujala P,
van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H,
Peters PJ, et al: Identification of stem cells in small intestine
and colon by marker gene Lgr5. Nature. 449:1003–1007. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kanwar SS, Yu Y, Nautiyal J, Patel BB and
Majumdar AP: The Wnt/beta-catenin pathway regulates growth and
maintenance of colonospheres. Mol Cancer. 9:2122010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arnould S, Hennebelle I, Canal P, Bugat R
and Guichard S: Cellular determinants of oxaliplatin sensitivity in
colon cancer cell lines. Eur J Cancer. 39:112–119. 2003. View Article : Google Scholar
|
|
7
|
Ringel I and Horwitz SB: Studies with RP
56976 (taxotere): A semisynthetic analogue of taxol. J Natl Cancer
Inst. 83:288–291. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sternberg CN, ten Bokkel Huinink WW, Smyth
JF, Bruntsch V, Dirix LY, Pavlidis NA, Franklin H, Wanders S, Le
Bail N and Kaye SB: Docetaxel (Taxotere), a novel taxoid, in the
treatment of advanced colorectal carcinoma: An EORTC Early Clinical
Trials Group Study. Br J Cancer. 70:376–379. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee HH, Leake BF, Teft W, Tirona RG, Kim
RB and Ho RH: Contribution of hepatic organic anion-transporting
polypeptides to docetaxel uptake and clearance. Mol Cancer Ther.
14:994–1003. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
van Herwaarden AE, Wagenaar E, van der
Kruijssen CM, van Waterschoot RA, Smit JW, Song JY, van der Valk
MA, van Tellingen O, van der Hoorn JW, Rosing H, et al: Knockout of
cytochrome P450 3A yields new mouse models for understanding
xenobiotic metabolism. J Clin Invest. 117:3583–3592. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wils P, Phung-Ba V, Warnery A, Lechardeur
D, Raeissi S, Hidalgo IJ and Scherman D: Polarized transport of
docetaxel and vinblastine mediated by P-glycoprotein in human
intestinal epithelial cell monolayers. Biochem Pharmacol.
48:1528–1530. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsai SM, Lin CY, Wu SH, Hou LA, Ma H, Tsai
LY and Hou MF: Side effects after docetaxel treatment in Taiwanese
breast cancer patients with CYP3A4, CYP3A5, and ABCB1 gene
polymorphisms. Clin Chim Acta. 404:160–165. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Murray S, Briasoulis E, Linardou H,
Bafaloukos D and Papadimitriou C: Taxane resistance in breast
cancer: Mechanisms, predictive biomarkers and circumvention
strategies. Cancer Treat Rev. 38:890–903. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wen K, Fu Z, Wu X, Feng J, Chen W and Qian
J: Oct-4 is required for an antiapoptotic behavior of
chemoresistant colorectal cancer cells enriched for cancer stem
cells: Effects associated with STAT3/Survivin. Cancer Lett.
333:56–65. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li C, Heidt DG, Dalerba P, Burant CF,
Zhang L, Adsay V, Wicha M, Clarke MF and Simeone DM: Identification
of pancreatic cancer stem cells. Cancer Res. 67:1030–1037. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Singh SK, Hawkins C, Clarke ID, Squire JA,
Bayani J, Hide T, Henkelman RM, Cusimano MD and Dirks PB:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
O’Brien CA, Pollett A, Gallinger S and
Dick JE: A human colon cancer cell capable of initiating tumour
growth in immunodeficient mice. Nature. 445:106–110. 2007.
View Article : Google Scholar
|
|
19
|
Ricci-Vitiani L, Lombardi DG, Pilozzi E,
Biffoni M, Todaro M, Peschle C and De Maria R: Identification and
expansion of human colon-cancer-initiating cells. Nature.
445:111–115. 2007. View Article : Google Scholar
|
|
20
|
Jordan CT: Cancer stem cell biology: From
leukemia to solid tumors. Curr Opin Cell Biol. 16:708–712. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
An Y and Ongkeko WM: ABCG2: The key to
chemoresistance in cancer stem cells? Expert Opin Drug Metab
Toxicol. 5:1529–1542. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
D’Arcangelo M, Todaro M, Salvini J,
Benfante A, Colorito ML, D’Incecco A, Landi L, Apuzzo T, Rossi E,
Sani S, et al: Cancer Stem Cells Sensitivity Assay (STELLA) in
patients with advanced lung and colorectal cancer: A feasibility
study. PLoS One. 10:e01250372015. View Article : Google Scholar :
|
|
23
|
Maugeri-Saccà M, Vigneri P and De Maria R:
Cancer stem cells and chemosensitivity. Clin Cancer Res.
17:4942–4947. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chakrabarty S: Regulation of human
colon-carcinoma cell adhesion to extracellular matrix by
transforming growth factor beta 1. Int J Cancer. 50:968–973. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Whissell G, Montagni E, Martinelli P,
Hernando-Momblona X, Sevillano M, Jung P, Cortina C, Calon A, Abuli
A, Castells A, et al: The transcription factor GATA6 enables
self-renewal of colon adenoma stem cells by repressing BMP gene
expression. Nat Cell Biol. 16:695–707. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang R, Wang G, Song Y, Tang Q, You Q,
Liu Z, Chen Y, Zhang Q, Li J, Muhammand S, et al: Colorectal cancer
stem cell and chemoresistant colorectal cancer cell phenotypes and
increased sensitivity to Notch pathway inhibitor. Mol Med Rep.
12:2417–2424. 2015.PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Liang CC, Park AY and Guan JL: In vitro
scratch assay: A convenient and inexpensive method for analysis of
cell migration in vitro. Nat Protoc. 2:329–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Saha A, Shree Padhi S, Roy S and Banerjee
B: Method of detecting new cancer stem cell-like enrichment in
development front assay (DFA). J Stem Cells. 9:235–242. 2014.
|
|
30
|
Myint K, Li Y, Paxton J and McKeage M:
Multidrug resistance-associated protein 2 (MRP2) mediated transport
of oxaliplatin-derived platinum in membrane vesicles. PLoS One.
10:e01307272015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Buxhofer-Ausch V, Secky L, Wlcek K,
Svoboda M, Kounnis V, Briasoulis E, Tzakos AG, Jaeger W and
Thalhammer T: Tumor-specific expression of organic
anion-transporting polypeptides: Transporters as novel targets for
cancer therapy. J Drug Deliv. 2013:8635392013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rabik CA, Maryon EB, Kasza K, Shafer JT,
Bartnik CM and Dolan ME: Role of copper transporters in resistance
to platinating agents. Cancer Chemother Pharmacol. 64:133–142.
2009. View Article : Google Scholar :
|
|
33
|
Furuta T, Ueda T, Aune G, Sarasin A,
Kraemer KH and Pommier Y: Transcription-coupled nucleotide excision
repair as a determinant of cisplatin sensitivity of human cells.
Cancer Res. 62:4899–4902. 2002.PubMed/NCBI
|
|
34
|
Garg A and Aggarwal BB: Nuclear
transcription factor-kappaB as a target for cancer drug
development. Leukemia. 16:1053–1068. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yilmaz M and Christofori G: EMT, the
cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev.
28:15–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pang R, Law WL, Chu AC, Poon JT, Lam CS,
Chow AK, Ng L, Cheung LW, Lan XR, Lan HY, et al: A subpopulation of
CD26+ cancer stem cells with metastatic capacity in
human colorectal cancer. Cell Stem Cell. 6:603–615. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Papailiou J, Bramis KJ, Gazouli M and
Theodoropoulos G: Stem cells in colon cancer. A new era in cancer
theory begins. Int J Colorectal Dis. 26:1–11. 2011. View Article : Google Scholar
|
|
38
|
Hermann PC, Bhaskar S, Cioffi M and
Heeschen C: Cancer stem cells in solid tumors. Semin Cancer Biol.
20:77–84. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang J, Wang H, Li Z, Wu Q, Lathia JD,
McLendon RE, Hjelmeland AB and Rich JN: c-Myc is required for
maintenance of glioma cancer stem cells. PLoS One. 3:e37692008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mori R, Ishiguro H, Kuwabara Y, Kimura M,
Mitsui A, Tomoda K, Mori Y, Ogawa R, Katada T, Harata K, et al:
Targeting beta1 integrin restores sensitivity to docetaxel of
esophageal squamous cell carcinoma. Oncol Rep. 20:1345–1351.
2008.PubMed/NCBI
|
|
41
|
Zhang H, Ozaki I, Mizuta T, Matsuhashi S,
Yoshimura T, Hisatomi A, Tadano J, Sakai T and Yamamoto K: Beta
1-integrin protects hepatoma cells from chemotherapy induced
apoptosis via a mitogen-activated protein kinase dependent pathway.
Cancer. 95:896–906. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schenk PW, Stoop H, Bokemeyer C, Mayer F,
Stoter G, Oosterhuis JW, Wiemer E, Looijenga LH and Nooter K:
Resistance to platinum-containing chemotherapy in testicular germ
cell tumors is associated with downregulation of the protein kinase
SRPK1. Neoplasia. 6:297–301. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gout S, Brambilla E, Boudria A, Drissi R,
Lantuejoul S, Gazzeri S and Eymin B: Abnormal expression of the
pre-mRNA splicing regulators SRSF1, SRSF2, SRPK1 and SRPK2 in non
small cell lung carcinoma. PLoS One. 7:e465392012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
el-Akawi Z, Abu-Hadid M, Perez R, Glavy J,
Zdanowicz J, Creaven PJ and Pendyala L: Altered glutathione
metabolism in oxaliplatin resistant ovarian carcinoma cells. Cancer
Lett. 105:5–14. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hall MD, Okabe M, Shen DW, Liang XJ and
Gottesman MM: The role of cellular accumulation in determining
sensitivity to platinum-based chemotherapy. Annu Rev Pharmacol
Toxicol. 48:495–535. 2008. View Article : Google Scholar
|
|
46
|
Habel N, Hamidouche Z, Girault I,
Patiño-García A, Lecanda F, Marie PJ and Fromigué O: Zinc
chelation: A metallothionein 2A’s mechanism of action involved in
osteosarcoma cell death and chemotherapy resistance. Cell Death
Dis. 4:e8742013. View Article : Google Scholar
|
|
47
|
L’Espérance S, Popa I, Bachvarova M,
Plante M, Patten N, Wu L, Têtu B and Bachvarov D: Gene expression
profiling of paired ovarian tumors obtained prior to and following
adjuvant chemotherapy: Molecular signatures of chemoresistant
tumors. Int J Oncol. 29:5–24. 2006.
|
|
48
|
Gottesman MM and Pastan I: Biochemistry of
multidrug resistance mediated by the multidrug transporter. Annu
Rev Biochem. 62:385–427. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Levêque D and Jehl F: P-glycoprotein and
pharmacokinetics. Anticancer Res. 15:331–336. 1995.PubMed/NCBI
|
|
50
|
Roninson IB: Molecular mechanism of
multidrug resistance in tumor cells. Clin Physiol Biochem.
5:140–151. 1987.PubMed/NCBI
|
|
51
|
She JJ, Zhang PG, Wang X, Che XM and Wang
ZM: Side population cells isolated from KATO III human gastric
cancer cell line have cancer stem cell-like characteristics. World
J Gastroenterol. 18:4610–4617. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ceckova M, Vackova Z, Radilova H, Libra A,
Buncek M and Staud F: Effect of ABCG2 on cytotoxicity of platinum
drugs: Interference of EGFP. Toxicol In Vitro. 22:1846–1852. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Qu H, Fang L, Duan L and Long X:
Expression of ABCG2 and p-glycoprotein in residual breast cancer
tissue after chemotherapy and their correlation with
epithelial-mesenchymal transition. Zhonghua Bing Li Xue Za Zhi.
43:236–240. 2014.(In Chinese). PubMed/NCBI
|
|
54
|
Hinoshita E, Uchiumi T, Taguchi K,
Kinukawa N, Tsuneyoshi M, Maehara Y, Sugimachi K and Kuwano M:
Increased expression of an ATP-binding cassette superfamily
transporter, multidrug resistance protein 2, in human colorectal
carcinomas. Clin Cancer Res. 6:2401–2407. 2000.PubMed/NCBI
|
|
55
|
Tamai I: Oral drug delivery utilizing
intestinal OATP transporters. Adv Drug Deliv Rev. 64:508–514. 2012.
View Article : Google Scholar
|
|
56
|
Plasencia C, Martínez-Balibrea E,
Martinez-Cardús A, Quinn DI, Abad A and Neamati N: Expression
analysis of genes involved in oxaliplatin response and development
of oxaliplatin-resistant HT29 colon cancer cells. Int J Oncol.
29:225–235. 2006.PubMed/NCBI
|
|
57
|
Damia G, Guidi G and D’Incalci M:
Expression of genes involved in nucleotide excision repair and
sensitivity to cisplatin and melphalan in human cancer cell lines.
Eur J Cancer. 34:1783–1788. 1998. View Article : Google Scholar
|
|
58
|
Selvakumaran M, Pisarcik DA, Bao R, Yeung
AT and Hamilton TC: Enhanced cisplatin cytotoxicity by disturbing
the nucleotide excision repair pathway in ovarian cancer cell
lines. Cancer Res. 63:1311–1316. 2003.PubMed/NCBI
|
|
59
|
Rosell R, Taron M, Barnadas A, Scagliotti
G, Sarries C and Roig B: Nucleotide excision repair pathways
involved in Cisplatin resistance in non-small-cell lung cancer.
Cancer Control. 10:297–305. 2003.PubMed/NCBI
|
|
60
|
David CJ and Manley JL: Alternative
pre-mRNA splicing regulation in cancer: Pathways and programs
unhinged. Genes Dev. 24:2343–2364. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kim SM, Lee SY, Yuk DY, Moon DC, Choi SS,
Kim Y, Han SB, Oh KW and Hong JT: Inhibition of NF-kappaB by
ginsenoside Rg3 enhances the susceptibility of colon cancer cells
to docetaxel. Arch Pharm Res. 32:755–765. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wang CY, Cusack JC Jr, Liu R and Baldwin
AS Jr: Control of inducible chemoresistance: Enhanced anti-tumor
therapy through increased apoptosis by inhibition of NF-kappaB. Nat
Med. 5:412–417. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
63
|
Shen W, Pang H, Liu J, Zhou J, Zhang F and
Liu L, Ma N, Zhang N, Zhang H and Liu L: Epithelial-mesenchymal
transition contributes to docetaxel resistance in human non-small
cell lung cancer. Oncol Res. 22:47–55. 2014. View Article : Google Scholar
|
|
64
|
Mahyar-Roemer M and Roemer K: p21
Waf1/Cip1 can protect human colon carcinoma cells against
p53-dependent and p53-independent apoptosis induced by natural
chemopreventive and therapeutic agents. Oncogene. 20:3387–3398.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yang AD, Fan F, Camp ER, van Buren G, Liu
W, Somcio R, Gray MJ, Cheng H, Hoff PM and Ellis LM: Chronic
oxaliplatin resistance induces epithelial-to-mesenchymal transition
in colorectal cancer cell lines. Clin Cancer Res. 12:4147–4153.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Bates RC, DeLeo MJ III and Mercurio AM:
The epithelial-mesenchymal transition of colon carcinoma involves
expression of IL-8 and CXCR-1-mediated chemotaxis. Exp Cell Res.
299:315–324. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Na DC, Lee JE, Yoo JE, Oh BK, Choi GH and
Park YN: Invasion and EMT-associated genes are up-regulated in B
viral hepatocellular carcinoma with high expression of CD133-human
and cell culture study. Exp Mol Pathol. 90:66–73. 2011. View Article : Google Scholar
|
|
68
|
Dalerba P, Dylla SJ, Park IK, Liu R, Wang
X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, et al:
Phenotypic characterization of human colorectal cancer stem cells.
Proc Natl Acad Sci USA. 104:10158–10163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Leinung M, Ernst B, Döring C, Wagenblast
J, Tahtali A, Diensthuber M, Stöver T and Geissler C: Expression of
ALDH1A1 and CD44 in primary head and neck squamous cell carcinoma
and their value for carcinogenesis, tumor progression and cancer
stem cell identification. Oncol Lett. 10:2289–2294. 2015.PubMed/NCBI
|
|
70
|
Sullivan JP, Spinola M, Dodge M, Raso MG,
Behrens C, Gao B, Schuster K, Shao C, Larsen JE, Sullivan LA, et
al: Aldehyde dehydrogenase activity selects for lung adenocarcinoma
stem cells dependent on notch signaling. Cancer Res. 70:9937–9948.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Maetzel D, Denzel S, Mack B, Canis M, Went
P, Benk M, Kieu C, Papior P, Baeuerle PA, Munz M, et al: Nuclear
signalling by tumour-associated antigen EpCAM. Nat Cell Biol.
11:162–171. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Amini S, Fathi F, Mobalegi J,
Sofimajidpour H and Ghadimi T: The expressions of stem cell
markers: Oct4, Nanog, Sox2, nucleostemin, Bmi, Zfx, Tcl1, Tbx3,
Dppa4, and Esrrb in bladder, colon, and prostate cancer, and
certain cancer cell lines. Anat Cell Biol. 47:1–11. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wiktorska M, Sacewicz-Hofman I,
Stasikowska-Kanicka O, Danilewicz M and Niewiarowska J: Distinct
inhibitory efficiency of siRNAs and DNAzymes to β1 integrin subunit
in blocking tumor growth. Acta Biochim Pol. 60:77–82. 2013.
|