Introduction
Pancreatic cancer develops from cancerous cells in
the tissues of the pancreas, a gland organ that lies inferior to
the stomach. It is one of the most lethal cancers. According to the
American Cancer Society, pancreatic cancer is expected to cause
48,960 new cases and 40,560 deaths in the United States in 2015
alone (1). Only 20% of patients
live for more than a year after diagnosis and fewer than 6% survive
past five years (2). Recent
evidence suggests the existence of a small population of
tumorigenic stem cells responsible for tumor initiation, resistance
to chemotherapy and radiation, and metastasis. These cancer stem
cells have the ability to self-renew, driving tumorigenicity,
recurrence, and metastasis. They also have the capacity to
differentiate aberrantly which gives rise to a heterogeneous
subpopulation of cancer cells that constitute the bulk of the
tumor. Surface markers cluster of differentiation 24 (CD24) and
cluster of differentiation 44 (CD44), along with aldehyde
dehydrogenase (ALDH1), a detoxifying enzyme responsible for the
oxidation of intracellular aldehydes, have been identified as
markers of stem cells of pancreatic adenocarcinomas (3,4). The
role of signal transducers and activators of transcription 3
(STAT3) in pancreatic cancer stem cells, however, is still unknown.
Hence, it is important to identify the regulatory mechanisms and
signaling pathways involved in pancreatic cancer stem cells and
develop novel agents to target pancreatic cancer stem cell
populations.
The signal transducers and activators of
transcription (STAT) protein family represents a group of
transcription factors that play a role in relaying extracellular
signals initiated by cytokines and growth factors from the
cytoplasm to the nucleus (5–7). In
response to extracellular signals, phosphorylated STAT proteins
dimerize and translocate to the nucleus where they regulate the
expression of numerous critical genes involved in cell cycle
progression, proliferation, invasion, and survival. STAT3
activation is dependent upon the phosphorylation of a conserved
tyrosine residue (Y705) which promotes the dimerization of STAT3
monomers via their Src-homology 2 (SH2) domains (8,9). The
constitutive activation of STAT3 is frequently detected in primary
human cancer cells including pancreatic cancer cells (10–12).
Blockade of STAT3 signaling has been shown to effectively inhibit
cell growth and induce apoptosis of pancreatic cancer cells in both
in vitro (13) and in
vivo studies (14,15). Although the role of STAT3 signaling
in stem cell-like pancreatic cancer cells is unknown, this pathway
may represent an attractive therapeutic target. Thus, it is
important to determine the role of STAT3 activation in pancreatic
stem cell-like cancer cells. We demonstrate for the first time that
the ALDH+ and CD44+/CD24+
subpopulations of pancreatic cancer cells express higher levels of
phosphorylated STAT3 (tyrosine 705) (P-STAT3, Y705) than
subpopulations that do not express these markers. In addition,
novel STAT3 inhibitors, LLL12, FLLL32, and Sttatic, inhibited STAT3
phosphorylation, cell viability, tumorsphere formation, and reduced
STAT3 downstream target gene expression in ALDH+ and
CD44+/CD24+ subpopulations. This report
indicates that constitutively activated STAT3 has an important role
in pancreatic stem cell-like cancer cell function and thus may
serve as an attractive therapeutic target for pancreatic
cancer.
Materials and methods
Pancreatic cancer cell lines
Human pancreatic cancer cell lines (Panc-1, BxPC3,
and HPAC) were purchased from the American Type Culture Collection
and maintained in Dulbecco's modified Eagle's medium supplemented
with 10% FBS, 4.5 g/l L-glutamine, sodium pyruvate, and 1%
penicillin/streptomycin. All cell lines were stored in a humidified
37°C incubator with 5% CO2. Cancer stem-like cells were
grown in a serum-free mammary epithelial basal medium (MEBM)
(Clonetics Division of Cambrex BioScience) supplemented with B27
(Invitrogen), 20 ng/ml EGF (BD Biosciences), 4 μg/ml gentamycin, 1
ng/ml hydrocortisone, 5 μg/ml insulin and 100 μM β-mercaptoethanol
(Sigma-Aldrich).
STAT3 inhibitors, LLL12, FLLL32 and
Stattic
Small molecules, LLL12 (16) and FLLL32 (17) that selectively target STAT3, were
synthesized by Dr Pui-Kai Li's laboratory at the Ohio State
University College of Pharmacy. Stattic, a previously reported
STAT3 inhibitor (18), was
purchased from Calbiochem (San Diego, CA, USA).
MTT cell viability assay
Pancreatic cancer stem-like cells (3,000/well in
96-well plates) were incubated with desired concentrations of
compounds in triplicate at 37°C for 72 h.
3-(4,5-Dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT)
viability assays were done and the absorbance was read at 595
nm.
Isolation of cancer stem cells
The AldeFluor kit (StemCell Technologies, Durham,
NC, USA) was used to isolate the population of cells with high ALDH
enzymatic activity as previously described (19–21).
Briefly, cells were trypsinized to single cells using 0.05% trypsin
and subsequently suspended in AldeFluor assay buffer containing
ALDH substrate (BAAA, 1 μmol/l per 1×106 cells) and then
incubated for 40 min at 37°C. For each sample, an aliquot of cells
was stained under identical conditions with 15 mmol/l
diethylaminobenzaldehyde (DEAB), a specific ALDH inhibitor, as a
negative control. In all experiments, the AldeFluor-stained cells
treated with DEAB served as ALDH-negative controls. Anti-human
PE-CD24 and PE/Cy5-CD44 antibodies (BioLegend) were used for
CD44/CD24 identification. ALDH+ and
CD44+/CD24+ subpopulations were separated
from Panc-1, BxPC3, and HPAC pancreatic cancer cells by a FACS
Wantage SE (Becton-Dickinson, Palo Alto, CA, USA) flow cytometer.
After sorting, ALDH+ and
CD44+/CD24+ cells were cultured in serum-free
stem cell medium (MEBM) to maintain cancer stem cell
characteristics. ALDH− and
CD44−/CD24− cells were cultured in regular
medium and replaced with stem cell medium for three days before
harvesting.
Western blot analysis
After treatment with LLL12 (5 μM), FLLL32 (5 μM),
Stattic (20 μM) or DMSO for 24 h, ALDH+ and
CD44+/CD24+ Panc-1 and HPAC pancreatic cancer
cells were lysed in cold RIPA lysis buffer containing protease
inhibitors and subjected to SDS-PAGE. Proteins were transferred on
to PVDF membrane and probed with antibodies (Cell Signaling
Technology). Membranes were probed with a 1:1,000 dilution of
antibodies (Cell Signaling Technology) against phospho-specific
STAT3 (tyrosine 705), phospho-independent STAT3, phospho-specific
ERK1/2 (threonine 202/tyrosine 204), and GAPDH. Membranes were
analyzed using enhanced chemiluminescence Plus reagents and scanned
with the Storm Scanner (Amersham Pharmacia Biotech Inc.,
Piscataway, NJ, USA).
Reverse transcriptase-polymerase chain
reaction (RT-PCR)
ALDH+ and
CD44+/CD24+ subpopulations of Panc-1 and HPAC
pancreatic cancer cells were treated with LLL12 (5 μM), FLLL32 (5
μM), or DMSO for 24 h. RNA was then collected using RNeasy kits
(Qiagen, Valencia, CA, USA). PCR amplification was done under the
following conditions: 5 min at 94°C followed by 25 cycles of 30 sec
at 94°C, 30 sec at 55°C, and 30 sec at 72°C with a final extension
of 5 min at 72°C. Primer sequences and source information of STAT3
downstream target genes can be found in Table I.
 | Table IPrimer sequences and source
information of STAT3 downstream target genes. |
Table I
Primer sequences and source
information of STAT3 downstream target genes.
| Gene | Primers | Size | Source |
|---|
| Cyclin D1 | Forward:
5′-GCTGGAGCCCGTGAAAAAGA-3′
Reverse: 5′-CTCCGCCTCTGGCATTTTG-3′ | 247 | (25) |
| Survivin | Forward:
5′-ACCAGGTGAGAAGTGAGGGA-3′
Reverse: 5′-AACAGTAGAGGAGCCAGGGA-3′ | 309 | (26) |
| Bcl-Xl | Forward:
5′-TTGGACAATGGACTGGTTGA-3′
Reverse: 5′-GTAGAGTGGATGGTCAGTG-3′ | 765 | (27) |
| Notch1 | Forward:
5′-CAACATCCAGGACAACATGG-3′
Reverse: 5′-GGACTTGCCCAGGTCATCTA-3′ | 229 | a |
| Notch3 | Forward:
5′-TGTCTTGCTGCTGGTCATTC-3′
Reverse: 5′-CATCTGGGCCACGCACATT-3′ | 413 | a |
Tumorsphere culture
The ALDH+ and
CD44+/CD24+ subpopulations of Panc-1, BxPC3,
and HPAC pancreatic cancer cells were plated as single cells in
ultra-low attachment 6-well plates (Corning, Lowell, MA, USA) at a
density of 25,000 viable cells/well. Cells were grown in a
serum-free mammary epithelial basal medium (MEBM) in a humidified
incubator (5% CO2) at 37°C. On the second day after
seeding, the ALDH+ cells were treated with 2.5–5 μM of
LLL12, 2.5–5 μM of FLLL32, or 5–10 μM of Stattic. Tumorspheres were
observed under microscope 21 to 28 days later. In order to compare
tumorsphere-forming ability, ALDH+,
CD44+/CD24+, ALDH− and
CD44−/CD24− cells were plated as single cells
in ultra-low attachment six-well plates at a density of 1,000 or
2,500 viable cells/well in triplicate in MEBM. Three to four weeks
later, the content of all wells was collected, pooled, and
transferred onto a collagen-coated 6-well dish in differentiating
medium (DMEM+10% FBS). Tumorspheres adhered in these conditions in
~24 h, after which they were stained with crystal violet and
counted under low magnification.
Results
ALDH+ and
CD44+/CD24+ subpopulations of pancreatic
cancer cells expressed higher levels of phosphorylated STAT3 and
greater tumorsphere-forming ability than ALDH− and
CD44−/CD24− subpopulations
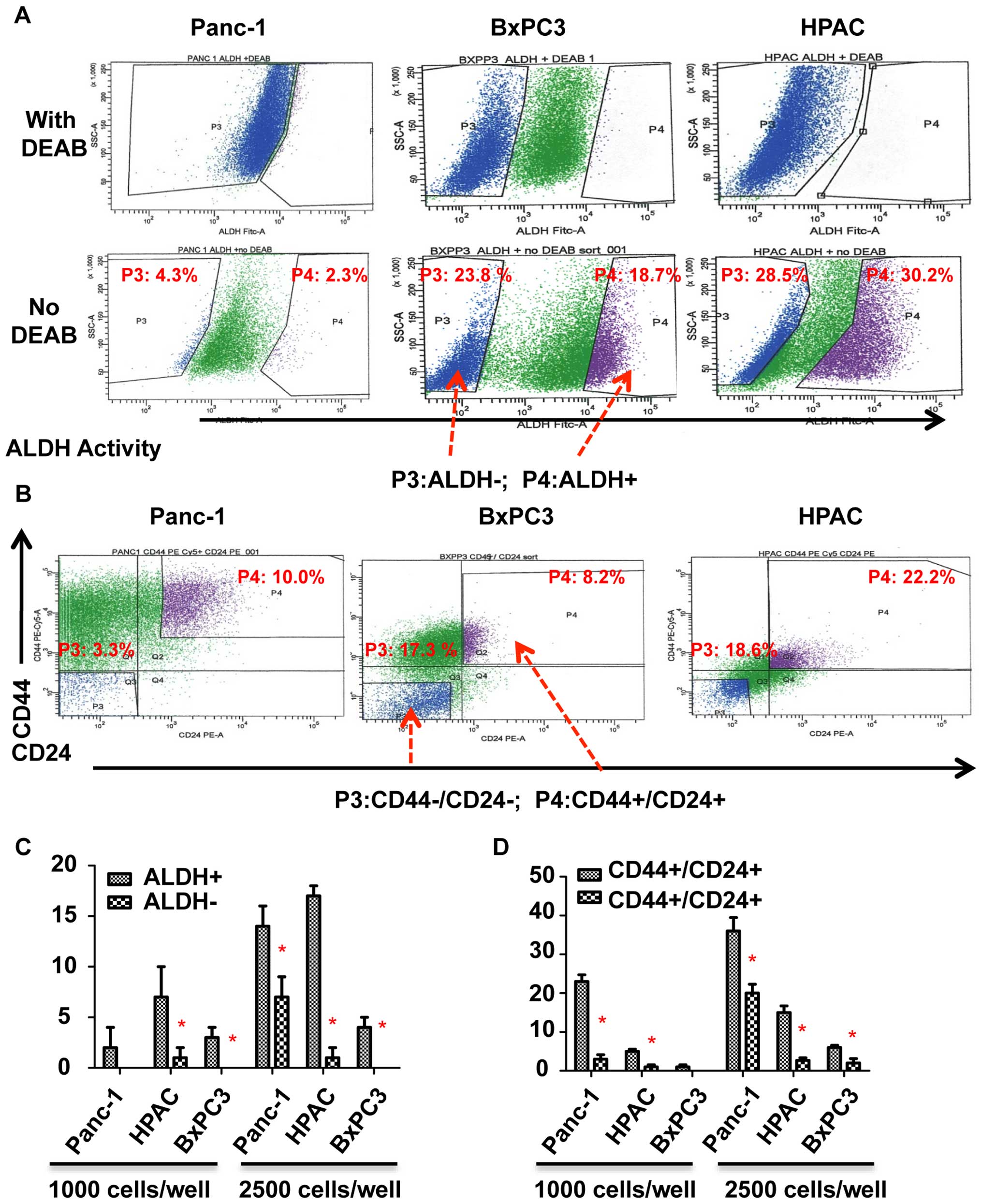
To determine whether STAT3 is activated in
pancreatic stem-like cancer cells, we separated ALDH+,
ALDH−, CD44+/CD24+ and
CD44−/CD24− subpopulations from Panc-1,
BxPC3, and HPAC pancreatic cancer cell lines by flow cytometry as
previously described (Fig. 1A and
B) (19). ALDH+,
CD44+, and CD24+ expressing subpopulations of
pancreatic cancer cells have been reported to exhibit cancer
stem-like cell properties (3,4). To
confirm the cancer stem-like cell properties of ALDH+
and CD44+/CD24+ subpopulations, we first
compared the tumorsphere-forming ability of ALDH+ and
CD44+/CD24+ subpopulations with
ALDH− and CD44−/CD24−
subpopulations. As shown in Fig. 1C
and D, ALDH+ and CD44+/CD24+
cells of Panc-1, BxPC3, and HPAC all generated more tumorspheres
than ALDH− and CD44−/CD24− cells.
We thus demonstrated that ALDH+ and
CD44+/CD24+ subpopulations of pancreatic
cancer cells have an increased ability to form tumorspheres than
ALDH− and CD44−/CD24− cells which
suggests that these markers are exhibited by pancreatic stem-like
cancer cells.
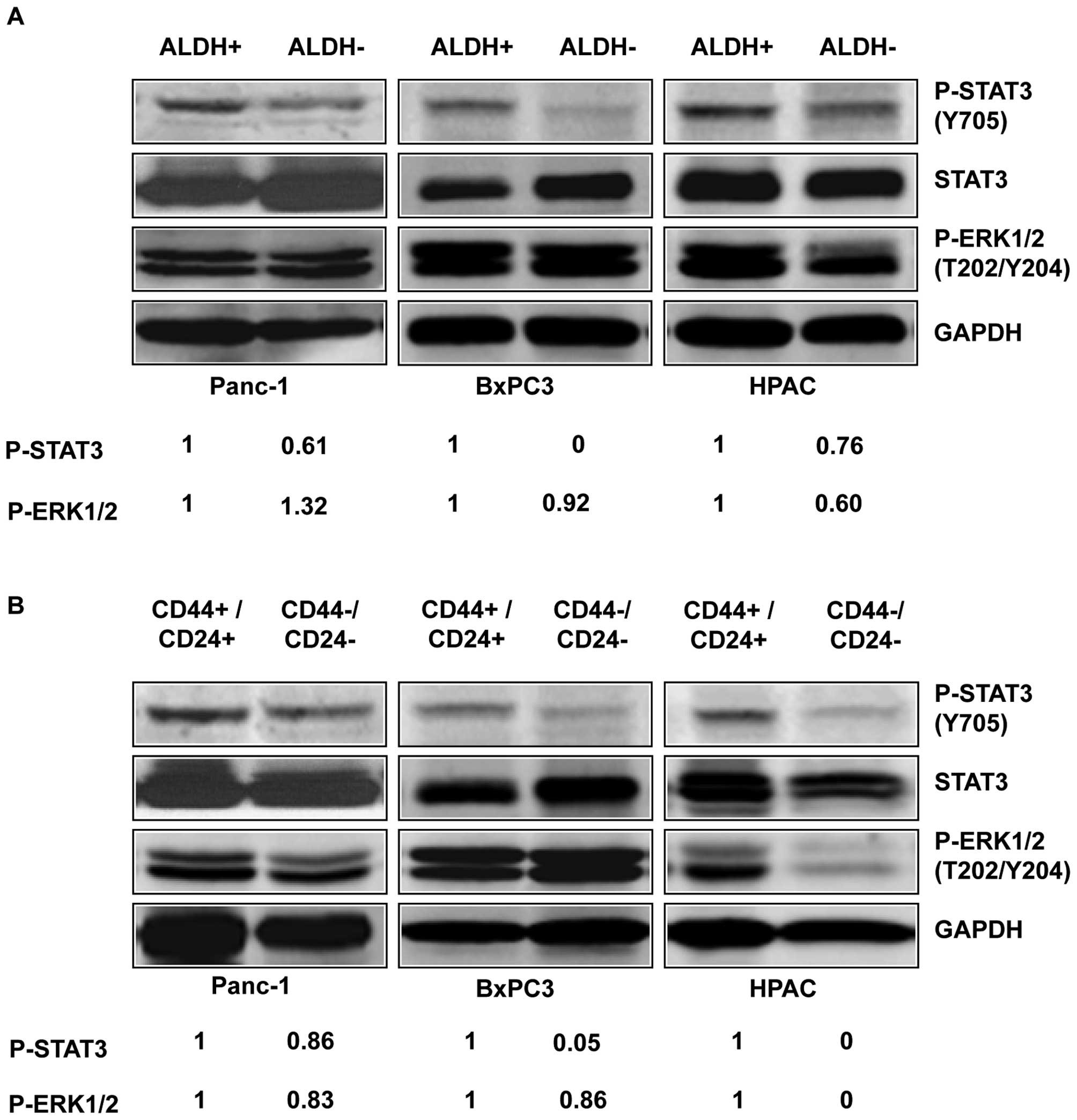
To determine the expression of activated P-STAT3 in
pancreatic stem-like cancer cells, we separated the
ALDH+ and CD44+/CD24+
subpopulations from the ALDH− and
CD44−/CD24− subpopulations of three
pancreatic cancer cell lines, Panc-1, BxPC3, and HPAC, and detected
the level of P-STAT3 by western blot analysis. Our results showed
that ALDH+ and CD44+/CD24+
subpopulations of pancreatic cancer cells expressed higher levels
of P-STAT3 (Y705) compared to ALDH− (Fig. 2A) and
CD44−/CD24− subpopulations (Fig. 2B). Phosphorylation of tyrosine
residue 705 (Y705) is important for the activation of STAT3
(22). In contrast to the
differences in STAT3 phosphorylation, the phosphorylation of
P-ERK1/2 (T202/Y204) in the ALDH+ and ALDH−
subpopulation were relatively similar in the three cell lines
(Fig. 2A). The phosphorylation of
P-ERK1/2 (T202/Y204) in the CD44+/CD24+ and
CD44−/CD24− subpopulations were also similar
in all three cell lines except HPAC (Fig. 2B). These results suggest that the
STAT3 pathway plays an important role in pancreatic cancer
stem-like cells and thus may serve as attractive pathway for
targeting stem cell-like pancreatic cancer populations.
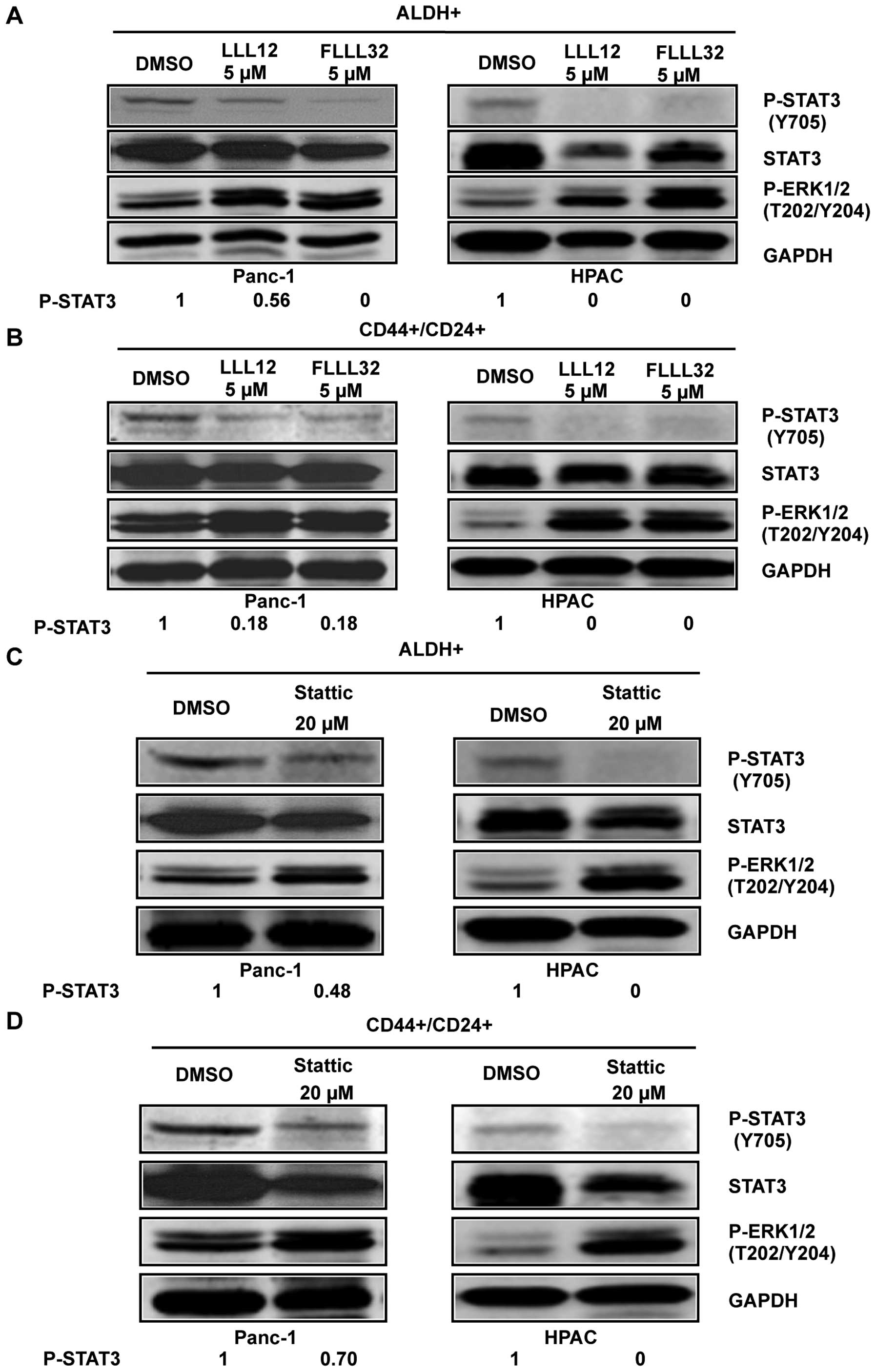
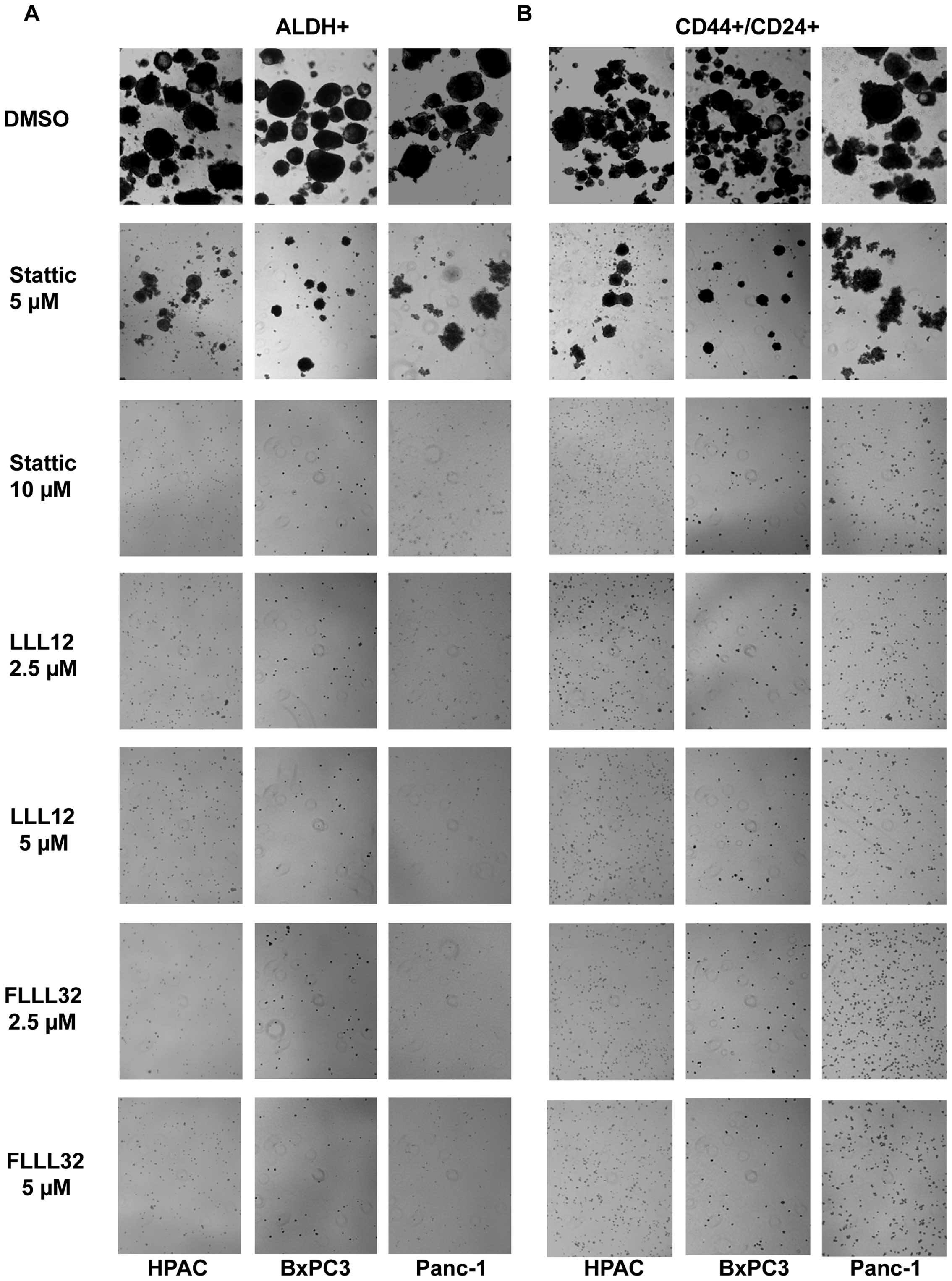
STAT3 inhibitors LLL12, FLLL32, and
Stattic selectively inhibited STAT3 phosphorylation in
ALDH+ and CD44+/CD24+
subpopulations of pancreatic cancer cells
To confirm the importance of STAT3 in pancreatic
cancer stem-like cells, STAT3 inhibitors, Stattic, LLL12, and
FLLL32 were used to target STAT3 in these cells. To confirm the
inhibition of phosphorylated STAT3 by Stattic, LLL12, and FLLL32 in
pancreatic cancer stem-like cells, we examined STAT3
phosphorylation at tyrosine residue 705 (Y705) in ALDH+
and CD44+/CD24+ pancreatic cancer cells using
phospho-STAT3 (tyrosine 705) antibody. Our results demonstrated
that LLL12 and FLLL32 significantly inhibited STAT3 phosphorylation
at tyrosine residue 705 (Y705) in Panc-1 and HPAC human pancreatic
cancer cell lines in both ALDH+ (Fig. 3A) and
CD44+/CD24+ subpopulations (Fig. 3B). Stattic also inhibited STAT3
phosphorylation (Y705) in ALDH+ (Fig. 3C) and
CD44+/CD24+ (Fig. 3D) subpopulations of Panc-1 and HPAC
pancreatic cancer cell lines at a higher concentration (20 μM).
LLL12, FLLL32, and Stattic selectively inhibited P-STAT3 as
demonstrated by the lack of inhibition of P-ERK1/2 in both
ALDH+ and CD44+/CD24+
subpopulations of Panc-1 and HPAC (Fig. 3).
STAT3 inhibitors LLL12, FLLL32, and
Stattic inhibited STAT3 downstream targets in ALDH+ and
CD44+/CD24+ subpopulations of pancreatic
cancer cells
The inhibition of STAT3 by LLL12 and FLLL32 also
downregulated the expression of many known STAT3-regulated genes in
ALDH+ (Fig. 4A) and
CD44+/CD24+ (Fig. 4B) pancreatic cancer stem-like cells
related to cancer cell proliferation, survival, and angiogenesis,
such as Cyclin D, Survivin, and Bcl-XL. Furthermore, LLL12 and
FLLL32 inhibited Notch1 and Notch3 expression in ALDH+
(Fig. 4A) and
CD44+/CD24+ cells (Fig. 4B), which have recently been
reported as putative STAT3 downstream target genes (23,24).
The Notch signaling pathway is known to be essential for normal
stem cell self-renewal and differentiation in a variety of tissues,
and is involved in the human cancer stem cell self-renewal capacity
and tumorigenicity.
STAT3 inhibitors, LLL12, FLLL32, and
Stattic, inhibited cell viability of ALDH+ and
CD44+/CD24+ subpopulations of pancreatic
cancer cells
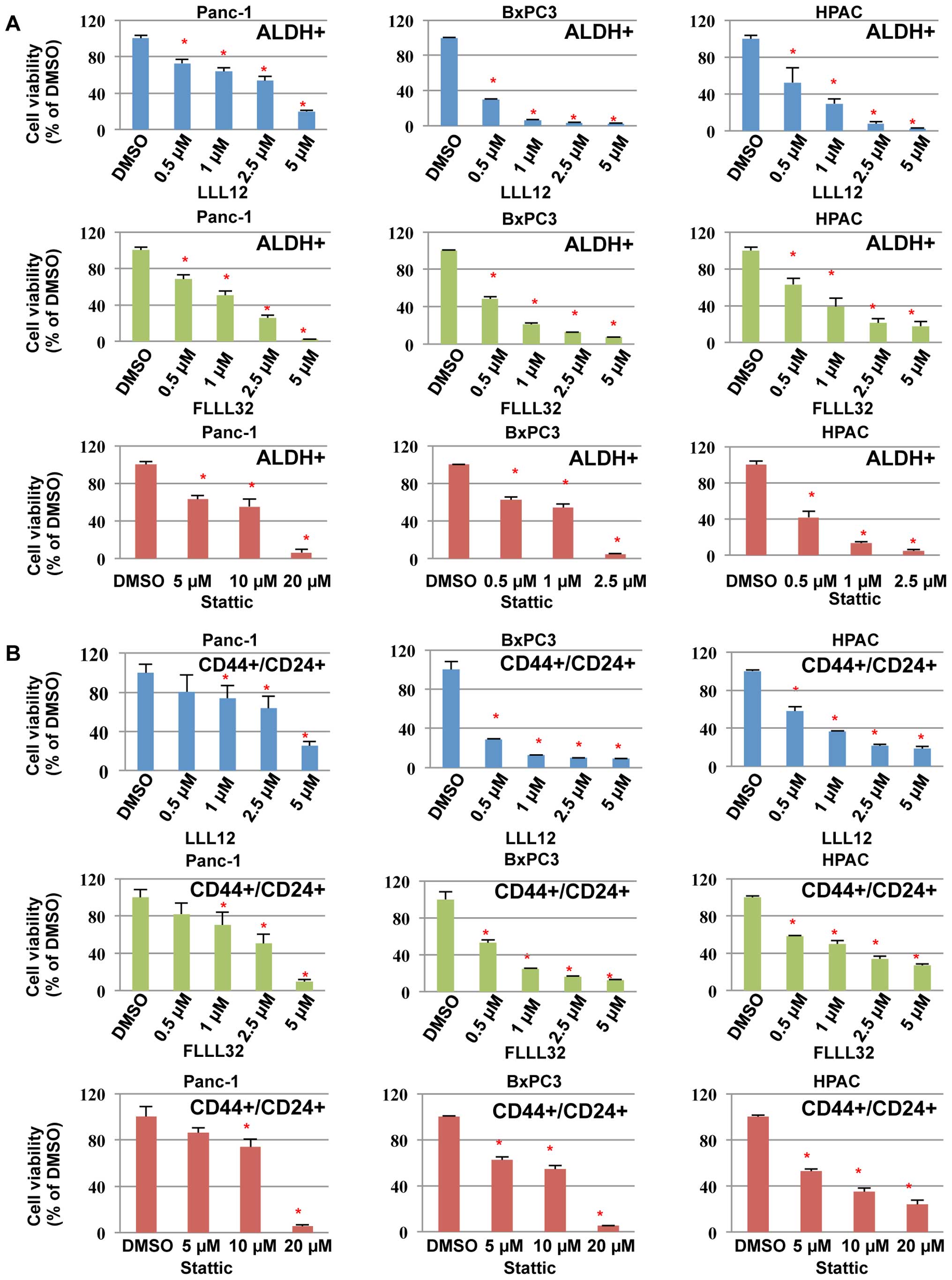
We next examined the inhibitory effects of LLL12,
FLLL32, and Stattic on cell viability in pancreatic cancer
stem-like cells. Our results demonstrated that LLL12, FLLL32, and
Stattic could inhibit cell viability of the ALDH+
(Fig. 5A) and
CD44+/CD24+ (Fig. 5B) subpopulations from Panc-1,
BxPC3, and HPAC pancreatic cancer stem-like cells, further
supporting the idea that these subpopulations of pancreatic cancer
cells are sensitive to STAT3 inhibitors, LLL12, FLLL32, and
Stattic. LLL12 and FLLL32 were more potent than Stattic in
inhibiting cell viability of the ALDH+ and
CD44+/CD24+ subpopulations from Panc-1,
BxPC3, and HPAC (Fig. 5).
STAT3 inhibitors, LLL12, FLLL32, and
Stattic, inhibited tumorsphere forming capacity of ALDH+
and CD44+/CD24+ subpopulations of pancreatic
cancer cells
We also examined the efficacy of LLL12, FLLL32 and
Stattic in inhibiting the survival and proliferation of pancreatic
cancer stem-like cells in anchorage-independent conditions and
ability to form tumor-spheres. Our results demonstrated that LLL12
and FLLL32 can significantly inhibit tumorsphere forming capacity
in the ALDH+ and CD44+/CD24+
subpopulations of Panc-1, BxPC3, and HPAC pancreatic cell lines
(Fig. 6A and B). As previously
demonstrated, Stattic was not as potent in inhibiting tumorsphere
forming capacity as LLL12 and FLLL32 in the ALDH+ and
CD44+/CD24+ subpopulations (Fig. 6). Thus, we demonstrated that
pancreatic cancer stem-like cells in the ALDH+ and
CD44+/CD24+ cells expressed an activated form
of STAT3 and are sensitive to inhibition by STAT3 inhibitors LLL12,
FLLL32 and Stattic. Our results also show that STAT3 inhibition by
LLL12, FLLL32, and Stattic decreases cell viability and tumorsphere
forming ability, which indicates that STAT3 is an important pathway
in pancreatic stem-like cancer cells and thus may serve as an
attractive target for therapeutic drugs.
Discussion
Presently, the STAT3 pathway has been characterized
in many cancers and the main effort to target constitutive STAT3
signaling is primarily on the bulk of cancer cells. No report has
been published on the role of STAT3 in pancreatic stem-like cancer
cells or on targeting the STAT3 pathway in these cells. Both CD44
and CD24, along with ALDH, have been used to isolate pancreatic
cancer stem cells (3,4). We utilized pancreatic stem cell
markers, ALDH, CD44, and CD24, to demonstrate that both
ALDH+ and CD44+/CD24+
subpopulations express higher levels of P-STAT3 (Y705) than
ALDH− and CD44−/CD24−
subpopulations suggesting that the phosphorylation of STAT3 plays a
role in their survival and proliferation. ALDH+ and
CD44+/CD24+ subpopulations also generated
more tumorspheres than ALDH− and
CD44−/CD24− subpopulations of pancreatic
cancer cells. Taken together, these data suggest that the STAT3
pathway may provide an attractive target for therapeutic treatment
in pancreatic stem-like cancer cells.
To explore the inhibition of STAT3 in pancreatic
cancer stem-like cells, we examined the inhibitory effects of three
STAT3 inhibitors, LLL12, FLLL32, and Stattic, on two subpopulations
of pancreatic stem-like cancer cells. All three molecules inhibited
the expression of P-STAT3 in ALDH+ and
CD44+/CD24+ subpopulations of pancreatic
cancer cells. In addition, LLL12, FLLL32, and Stattic were also
effective in inhibiting cell viability and tumorsphere formation in
both ALDH+ and CD44+/CD24+
subpopulations of cells. Although Stattic did inhibit cell
viability and tumorsphere formation in pancreatic stem-like cancer
cells, it was not as potent as either LLL12 or FLLL32. This
observation is consistent with the weaker predictive binding
affinity of Stattic to STAT3 than either LLL12 or FLLL32.
In conclusion, this study demonstrates that STAT3 is
activated in pancreatic cancer stem-like cells and that
constitutively activated STAT3 in these cells enhances
proliferation and survival. Our results show that STAT3 inhibition
by small molecules could inhibit tumor stem-like cell growth, cell
viability, and tumorsphere formation. Targeting STAT3 may provide
an effective method for depleting stem cell-like pancreatic cancer
cells and thus an effective treatment for pancreatic cancer. Our
study also demonstrated that both LLL12 and FLLL32 significantly
inhibited STAT3 in pancreatic cancer stem-like cells and thus are
promising drug candidates for targeting constitutive STAT3
expression in these cells.
Acknowledgements
This study was supported in part by the National
Natural Science Foundation of China (81001005, 81372402, 81570416),
the Outstanding Young Investigator Foundation of Tongji Hospital
(YXQN009) and the Fundamental Research Fund for the Central
Universities, HUST: 0118540019 to Li Lin, and supported by funding
from the AACR-Pancreatic Cancer Action Network to Jiayuh Lin.
Abbreviations:
|
STAT3
|
signal transducers and activators of
transcription 3
|
|
ALDH
|
aldehyde dehydrogenase
|
|
CD44
|
cluster of differentiation 44
|
|
MEBM
|
mammary epithelial basal medium
|
|
DEAB
|
diethylaminobenzaldehyde
|
References
|
1
|
Society AC. Cancer Factor & Figures.
American Cancer Society; 2015, http://www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf.
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li C, Heidt DG, Dalerba P, Burant CF,
Zhang L, Adsay V, Wicha M, Clarke MF and Simeone DM: Identification
of pancreatic cancer stem cells. Cancer Res. 67:1030–1037. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wei HJ, Yin T, Zhu Z, Shi PF, Tian Y and
Wang CY: Expression of CD44, CD24 and ESA in pancreatic
adenocarcinoma cell lines varies with local microenvironment.
Hepatobiliary Pancreat Dis Int. 10:428–434. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Calò V, Migliavacca M, Bazan V, Macaluso
M, Buscemi M, Gebbia N and Russo A: STAT proteins: From normal
control of cellular events to tumorigenesis. J Cell Physiol.
197:157–168. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Germain D and Frank DA: Targeting the
cytoplasmic and nuclear functions of signal transducers and
activators of transcription 3 for cancer therapy. Clin Cancer Res.
13:5665–5669. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Turkson J and Jove R: STAT proteins: Novel
molecular targets for cancer drug discovery. Oncogene.
19:6613–6626. 2000. View Article : Google Scholar
|
|
8
|
Zhong Z, Wen Z and Darnell JE Jr: Stat3: A
STAT family member activated by tyrosine phosphorylation in
response to epidermal growth factor and interleukin-6. Science.
264:95–98. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sasse J, Hemmann U, Schwartz C,
Schniertshauer U, Heesel B, Landgraf C, Schneider-Mergener J,
Heinrich PC and Horn F: Mutational analysis of acute-phase response
factor/Stat3 activation and dimerization. Mol Cell Biol.
17:4677–4686. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Corvinus FM, Orth C, Moriggl R, Tsareva
SA, Wagner S, Pfitzner EB, Baus D, Kaufmann R, Huber LA, Zatloukal
K, et al: Persistent STAT3 activation in colon cancer is associated
with enhanced cell proliferation and tumor growth. Neoplasia.
7:545–555. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kusaba T, Nakayama T, Yamazumi K, Yakata
Y, Yoshizaki A, Nagayasu T and Sekine I: Expression of p-STAT3 in
human colorectal adenocarcinoma and adenoma; correlation with
clinicopathological factors. J Clin Pathol. 58:833–838. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma XT, Wang S, Ye YJ, Du RY, Cui ZR and
Somsouk M: Constitutive activation of Stat3 signaling pathway in
human colorectal carcinoma. World J Gastroenterol. 10:1569–1573.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao S, Venkatasubbarao K, Lazor JW,
Sperry J, Jin C, Cao L and Freeman JW: Inhibition of STAT3 Tyr705
phosphorylation by Smad4 suppresses transforming growth factor
beta-mediated invasion and metastasis in pancreatic cancer cells.
Cancer Res. 68:4221–4228. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qiu Z, Huang C, Sun J, Qiu W, Zhang J, Li
H, Jiang T, Huang K and Cao J: RNA interference-mediated signal
transducers and activators of transcription 3 gene silencing
inhibits invasion and metastasis of human pancreatic cancer cells.
Cancer Sci. 98:1099–1106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sahu RP and Srivastava SK: The role of
STAT-3 in the induction of apoptosis in pancreatic cancer cells by
benzyl isothiocyanate. J Natl Cancer Inst. 101:176–193. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin L, Hutzen B, Li PK, Ball S, Zuo M,
DeAngelis S, Foust E, Sobo M, Friedman L, Bhasin D, et al: A novel
small molecule, LLL12, inhibits STAT3 phosphorylation and
activities and exhibits potent growth-suppressive activity in human
cancer cells. Neoplasia. 12:39–50. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin L, Hutzen B, Zuo M, Ball S, Deangelis
S, Foust E, Pandit B, Ihnat MA, Shenoy SS, Kulp S, et al: Novel
STAT3 phosphorylation inhibitors exhibit potent growth-suppressive
activity in pancreatic and breast cancer cells. Cancer Res.
70:2445–2454. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schust J, Sperl B, Hollis A, Mayer TU and
Berg T: Stattic: A small-molecule inhibitor of STAT3 activation and
dimerization. Chem Biol. 13:1235–1242. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ginestier C, Hur MH, Charafe-Jauffret E,
Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG,
Liu S, et al: ALDH1 is a marker of normal and malignant human
mammary stem cells and a predictor of poor clinical outcome. Cell
Stem Cell. 1:555–567. 2007. View Article : Google Scholar
|
|
20
|
Lin L, Liu A, Peng Z, Lin HJ, Li PK, Li C
and Lin J: STAT3 is necessary for proliferation and survival in
colon cancer-initiating cells. Cancer Res. 71:7226–7237. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tanei T, Morimoto K, Shimazu K, Kim SJ,
Tanji Y, Taguchi T, Tamaki Y and Noguchi S: Association of breast
cancer stem cells identified by aldehyde dehydrogenase 1 expression
with resistance to sequential paclitaxel and epirubicin-based
chemotherapy for breast cancers. Clin Cancer Res. 15:4234–4241.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kaptein A, Paillard V and Saunders M:
Dominant negative stat3 mutant inhibits interleukin-6-induced
Jak-STAT signal transduction. J Biol Chem. 271:5961–5964. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Grivennikov S and Karin M: Autocrine IL-6
signaling: A key event in tumorigenesis? Cancer Cell. 13:7–9. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dontu G, Jackson KW, McNicholas E,
Kawamura MJ, Abdallah WM and Wicha MS: Role of Notch signaling in
cell-fate determination of human mammary stem/progenitor cells.
Breast Cancer Res. 6:R605–R615. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tian X, Li J, Ma ZM, Zhao C, Wan DF and
Wen YM: Role of hepatitis B surface antigen in the development of
hepatocellular carcinoma: Regulation of lymphoid enhancer-binding
factor 1. J Exp Clin Cancer Res. 28:582009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Paydas S, Tanriverdi K, Yavuz S, Disel U,
Sahin B and Burgut R: Survivin and aven: Two distinct antiapoptotic
signals in acute leukemias. Ann Oncol. 14:1045–1050. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yamaguchi H, Inokuchi K, Tarusawa M and
Dan K: Mutation of bcl-x gene in non-Hodgkin's lymphoma. Am J
Hematol. 69:74–76. 2002. View Article : Google Scholar : PubMed/NCBI
|