Introduction
Lung cancer is the leading cause of cancer-related
mortality worldwide, and ~90% of lung cancers are classified as
non-small-cell lung cancer (NSCLC) (1). Although the prognosis of advanced
NSCLC is very poor, the identification of epidermal growth factor
receptor gene mutations (EGFR mutations) as oncogenic driver
mutations in a subset of patients with NSCLC, coupled with the
development of EGFR tyrosine kinase inhibitors (EGFR-TKIs), has
opened the door to a new era in the treatment of this disease
(2–7). Receptor tyrosine kinases (RTKs),
including EGFR, have been shown to act not only as key regulators
of normal cellular processes, but also to play a critical role in
the development and progression of many cancers, and several RTKs
including ALK, ROS1, RET, and MET have also been identified as
therapeutic targets in NSCLC (8–10).
RTKs generally undergo autophosphorylation, which in
turn promotes the recruitment of downstream effector proteins
leading to the activation of multiple signal cascades, including
the mitogen-activated protein kinase (MAPK), PI3K/AKT, and STAT
pathways (11,12). Among these pathways, the MAPK
pathway is particularly important for cancer cell proliferation,
differentiation and survival (13,14).
The three-tiered kinase cascade consisting of RAF,
mitogen-activated protein kinase kinase (MEK), and extracellular
signal-regulated kinase (ERK) is frequently dysregulated in many
malignancies including NSCLC (13). In this study, we tested the effects
of MEK inhibitors (trametinib and PD0325901) in several NSCLC cell
lines with driver gene alterations, especially RTK genes, in
vitro and found a wide range of sensitivities. Among them, MEK
inhibitors were effective against all MET-amplified cell
lines but were not effective against any EGFR-mutated NSCLC
cell line. Next, the mechanism and synergistic effect of a MET
inhibitor and a MEK inhibitor against MET-amplified NSCLC
cell lines were also investigated.
Materials and methods
Cell cultures and reagents
The A549 cell line (KRAS mutation) was
maintained in DMEM medium (Sigma-Aldrich, St. Louis, MO, USA)
supplemented with 10% FBS (Gibco BRL, Grand Island, NY, USA) in a
humidified atmosphere of 5% CO2 at 37°C. The H358
(KRAS mutation), H1299 (NRAS mutation), PC-9, HCC827,
Ma-1, 11_18, PC-9/ZD, H1975 (EGFR mutation), EBC-1, H1993
(MET amplification), H2228, and H3122 (ALK fusion)
cell lines were maintained in RPMI medium (Sigma-Aldrich)
supplemented with 10% FBS in a humidified atmosphere of 5%
CO2 at 37°C. The HCC827CNXR cell line whose driver gene
shifts from an EGFR mutation to a MET amplification
(oncogene swap) was established as described previously and was
maintained in RPMI medium supplemented with 10% FBS (15). Trametinib, PD0325901 (MEK
inhibitors), LY294002 (a PI3K inhibitor), and crizotinib (a MET
inhibitor) were purchased from Selleck Chemicals (Houston, TX,
USA).
Growth inhibition assay in vitro
The growth-inhibitory effects of drugs were examined
using a 3,4,-5-dimethyl-2H-tetrazolium bromide assay (MTT;
Sigma-Aldrich) (16). The
experiment was performed in triplicate.
Bioinformatics technique
The mutational profiles of known driver oncogenes
such as KRAS mutation, EGFR mutation, BRAF
mutation, ALK, RET or ROS1 fusion, MAP2K1,
NRAS or HRAS mutation, MET exon 14 skipping
mutation, MET amplification, ERBB2 mutation,
ERBB2 amplification, RIT1 mutation, and NF1
loss, for 230 lung adenocarcinomas were based on the previously
published report (17) and the
gene expression profiles of these samples were extracted from the
Cancer Genome Atlas (TCGA) data portal (https://tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp). The
average expression profile for the 12 mutational subclasses
(17) was calculated and used for
further analysis. In total, 2,320 genes with a median gene
expression (log2 value) of >7.0- and a 4-fold change among the
12 subclasses were extracted and used for clustering analysis.
Cluster 3.0 was used for hierarchical clustering analysis, and JAVA
TreeView was used for display. For the pathway analyses, genes
involved in each specific pathway were extracted from the REACTOME
database (http://www.reactome.org/). The
Z-scores for each gene in the 12 subclasses were calculated and
summarized for each subclass. A positive value indicated that the
genes in the pathway were relatively activated.
Antibodies
Rabbit antibodies specific for EGFR, phospho-EGFR,
phospho-MET, AKT, phospho-AKT, ERK1/2, phospho-ERK1/2, poly
(ADP-ribose) polymerase (PARP), caspase-3, cleaved PARP, cleaved
caspase-3, and β-actin, and a mouse antibody specific for MET were
obtained from Cell Signaling (Beverly, MA, USA).
Western blot analysis
A western blot analysis was performed as described
previously (16). Briefly,
subconfluent cells were washed with cold phosphate-buffered saline
(PBS) and harvested with lysis A buffer containing 1% Triton X-100,
20 mM Tris-HCl (pH 7.0), 5 mM EDTA, 50 mM sodium chloride, 10 mM
sodium pyrophosphate, 50 mM sodium fluoride, 1 mM sodium
orthovanadate, and a protease inhibitor mix, Complete™ (Roche
Diagnostics). Whole-cell lyses were separated using SDS-PAGE and
were blotted onto a polyvinylidene fluoride membrane. After
blocking with 3% bovine serum albumin in a TBS buffer (pH 8.0) with
0.1% Tween-20, the membrane was probed with the primary antibody.
After rinsing twice with TBS buffer, the membrane was incubated
with a horseradish peroxidase-conjugated secondary antibody and
washed, followed by visualization using an ECL detection system and
LAS-4000 (GE Healthcare, Buckinghamshire, UK). When the
phosphorylation and apoptosis were examined after the drug
exposure, the samples were collected 3 and 24 h after the exposure,
respectively.
Statistical analysis
Continuous variables were analyzed using the Student
t-test, and the results were expressed as the average and standard
deviation (SD). The statistical analyses were two-tailed and were
performed using Microsoft Excel (Microsoft, Redmond, WA, USA). A
P-value of <0.05 was considered statistically significant.
Results
Sensitivity to MEK inhibitors in several
NSCLC cell lines with driver gene alterations
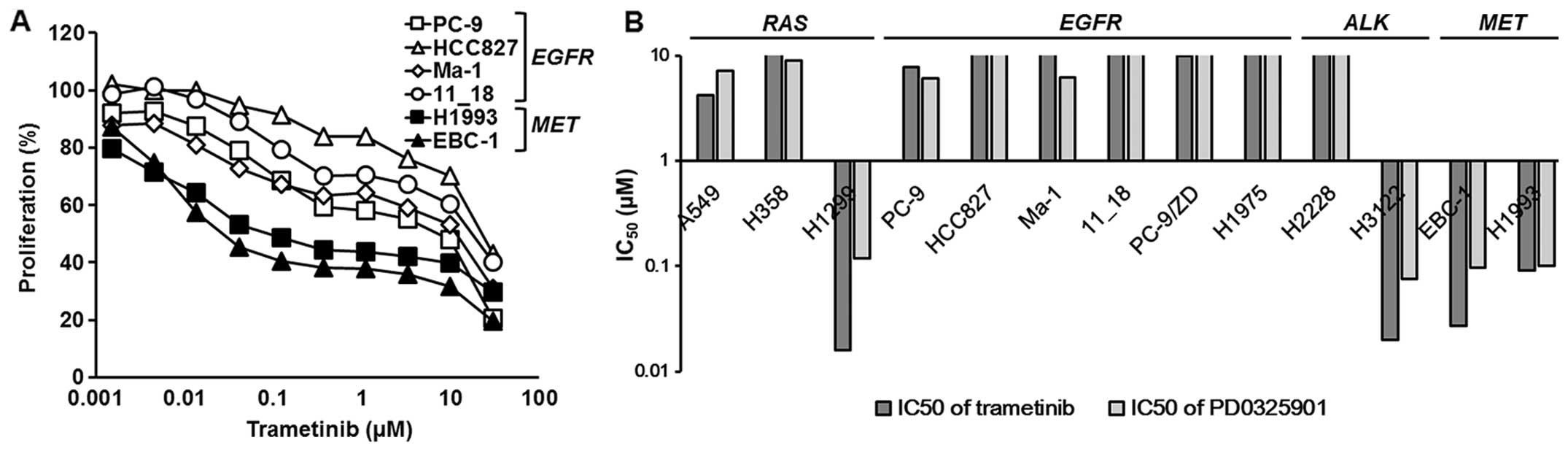
To investigate the effects of MEK inhibitors, we
used two MEK inhibitors (trametinib and PD0325901), and the growth
inhibitory assay was performed using an MTT assay. The inhibitory
curves for trametinib in the EGFR-mutated or
MET-amplified NSCLC cell lines are shown in Fig. 1A, and the 50% inhibitory
concentrations (IC50) are summarized in Fig. 1B and Table I. Although the MAPK pathway is the
same downstream pathway associated with cellular growth and
survival among RAS-mutated, EGFR-mutated,
ALK-fused, and MET-amplified NSCLC cell lines, a wide
range of sensitivities to MEK inhibitors was observed (Fig. 1B and Table I). Among them, all the
EGFR-mutated cell lines were resistant to MEK inhibitors
(IC50 ≥1 μM), whereas all the MET-amplified cell
lines were sensitive (IC50 ~0.1 μM) (Fig. 1 and Table I).
 | Table IThe 50% inhibitory concentrations
(IC50) of MEK inhibitors in non-small cell lung cancer
cell lines with driver gene alterations. |
Table I
The 50% inhibitory concentrations
(IC50) of MEK inhibitors in non-small cell lung cancer
cell lines with driver gene alterations.
| Driver gene | Cell line | Driver gene
alterations |
IC50 |
|---|
|
|---|
| Trametinib
(μM) | PD0325901 (μM) |
|---|
| RAS | A549 | KRAS
G12S | 4.17 | 7.23 |
| H358 | KRAS
G12C | 11.3 | 8.90 |
| H1299 | NRAS
Q61K | 0.016 | 0.12 |
| EGFR | PC-9 | EGFR exon 19
deletion | 7.82 | 6.02 |
| HCC827 | EGFR exon 19
deletion | 22.7 | >30 |
| Ma-1 | EGFR exon 19
deletion | 11.7 | 6.24 |
| 11_18 | EGFR exon 21
L858R | 17.6 | 16.5 |
| PC-9/ZD | EGFR exon 19
deletion and exon 20 T790M | 10.0 | 15.5 |
| H1975 | EGFR exon 21
L858R and exon 20 T790M | 12.4 | 25.6 |
| ALK | H2228 |
EML4-ALK | >30 | >30 |
| H3122 |
EML4-ALK | 0.020 | 0.076 |
| MET | EBC-1 | MET
amplification | 0.027 | 0.097 |
| H1993 | MET
amplification | 0.092 | 0.10 |
| HCC827CNXR | MET
amplification | 0.14 | 0.55 |
PI3K/AKT pathway is more activated in
EGFR-mutated NSCLC than in MET-amplified NSCLC
To investigate the mechanism responsible for the
difference in sensitivities, bioinformatics techniques were used
based on the TCGA dataset for lung adenocarcinomas. Overall, 230
lung adenocarcinomas were clustered into 12 subclasses based on the
mutational profiles of known driver oncogenes (17). The average expression profile for
each subclass was calculated and used for further analysis. The
average gene expression profiles of samples with MET
amplification (MET-amp subclass) were clustered next to those of
samples with mutations in MAP2K1, NRAS and HRAS
(MAP2K1 subclass). These two expression profiles were clustered in
a different branch from the other types of expression profiles,
suggesting their similarity compared with the other profiles. In
addition, the expression profile of the EGFR-mutated samples
(EGFR subclass) was clustered in a notably different branch from
that of the MET-amp subclass (Fig.
2A). These findings suggest that the expression profile of the
MET-amp subclass is similar to that of the MAP2K1 subclass and
notably different from that of the EGFR subclass. Next, the PI3K
pathway was analyzed, since several reports have shown the
activation of the PI3K/AKT pathway in EGFR-mutated NSCLC
(18,19). Genes involved in the ‘PI3K-cascade’
were extracted from the REACTOME database. The Z-scores for each
gene in the 12 subclasses were calculated and were summarized for
each subclass. A positive value indicated that the genes in this
pathway were relatively activated. As shown in Fig. 2A, the MAP2K1 subclass exhibited a
particularly negative value, suggesting that the genes involved in
the PI3K/AKT pathway play a less important role in this cluster.
The MET-amp subclass, which was similar to the MAP2K1 subclass,
also had a negative value, whereas the EGFR subclass had a positive
value. These findings indicate that the PI3K/AKT pathway is
activated in EGFR-mutated NSCLC, compared with
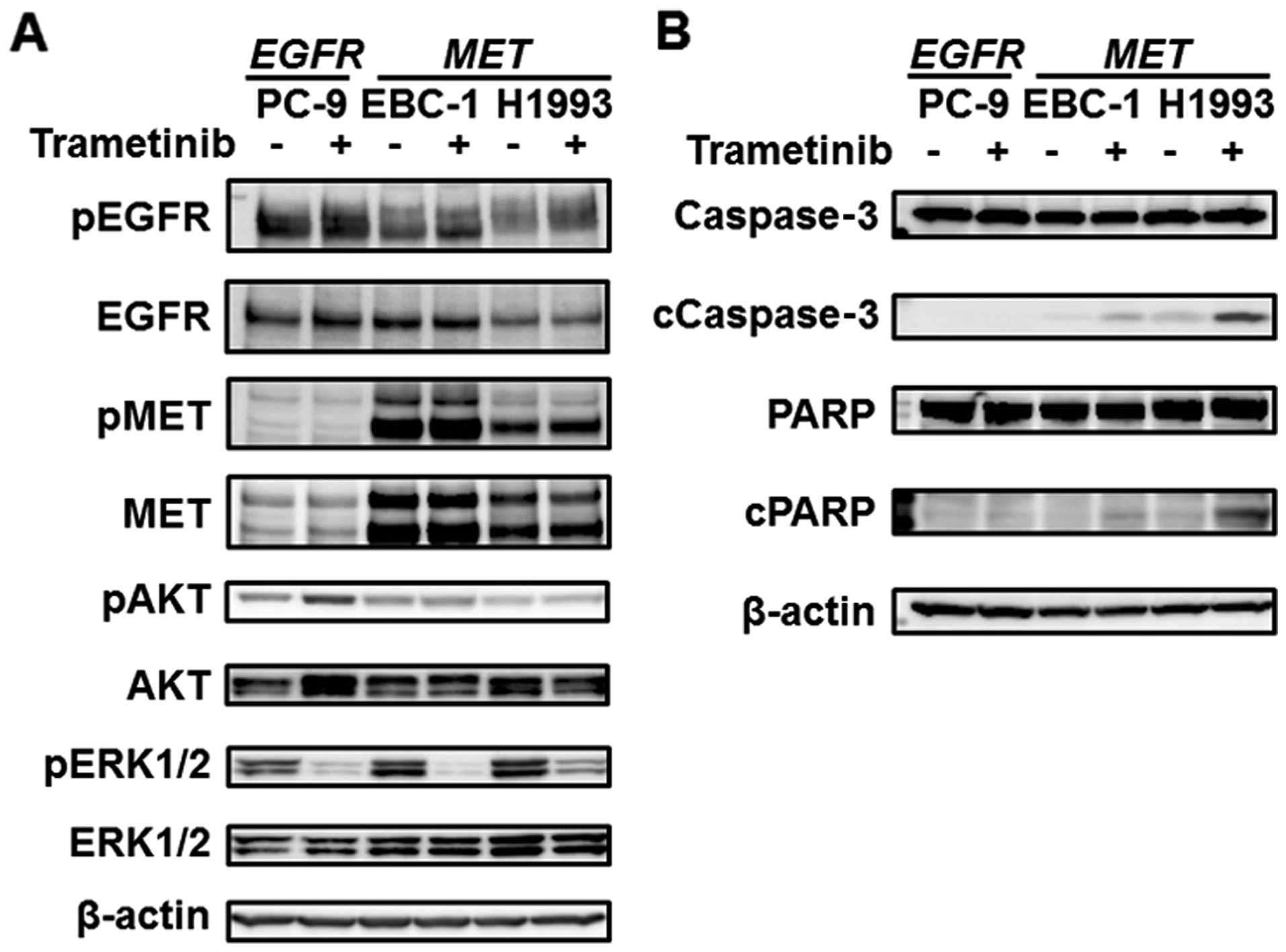
MET-amplified NSCLC. The higher expression of phospho-AKT in
the EGFR-mutated NSCLC cell lines than that in the
MET-amplified NSCLC cell lines was observed in western blot
analyses, indicating the activated PI3K/AKT pathway in
EGFR-mutated NSCLC (Fig.
2B). Furthermore, a western blot analysis also showed that the
A549 cell line (KRAS mutation; KRAS subclass) had a
significantly higher expression of phospho-AKT, compared with the
H1299 cell line (NRAS mutation; MAP2K1 subclass) (Fig. 2B). These findings are consistent
with our database analyses revealing that the KRAS subclass had a
positive value and the MAP2K1 subclass had a negative value
(Fig. 2A). In addition, the A549
cell line was resistant to MEK inhibitors, while the H1299 cell
line was sensitive (Fig. 1B and
Table I). Altogether, these
findings suggest that the activated PI3K/AKT pathway might be
associated with resistance to MEK inhibitors.
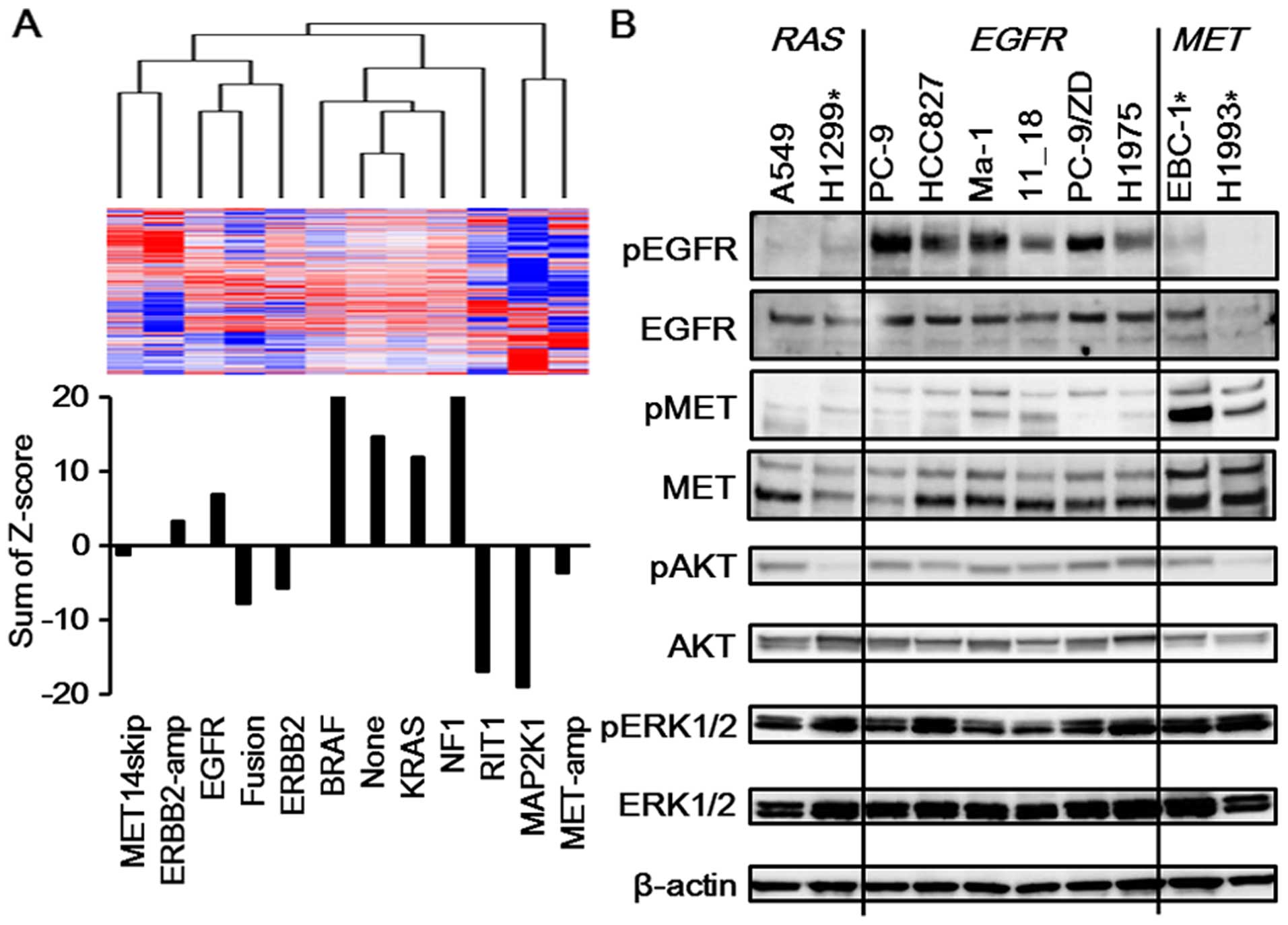 | Figure 2Activated PI3K/AKT pathway in
EGFR-mutated NSCLC. (A) PI3K/AKT pathway in each NSCLC
cluster. The mutational profiles of known driver oncogenes for 230
lung adenocarcinomas were based on The Cancer Genome Atlas (TCGA)
dataset, and the expression profiles of those samples were
extracted from the TCGA data portal. The average expression profile
for each subclass was calculated and was used for further analysis.
The average gene expression profile of the MET-amp subclass (n=5)
was clustered next to that of the MAP2K1 subclass (n=4). These two
expression profiles were clustered together in a different branch
from those of the other types of expression profiles. In addition,
the expression profile of the EGFR subclass was clustered in a
notably different branch from that of the MET-amp subclass. Next,
genes involved in the ‘PI3K-cascade’ from the REACTOME database
were extracted. The Z-scores for each gene in the 12 subclasses
were calculated and were summarized for each subclass. A positive
value indicated that the genes in the pathway were relatively
activated. The MAP2K1 subclass had a particularly negative value.
The MET-amp subclass, which is similar to the MAP2K1 subclass, also
had a negative value, whereas the EGFR and KRAS subclasses had
positive values. MET14skip, subclass of samples with MET
exon 14 skipping mutations; ERBB2-amp, subclass of samples with
ERBB2 amplification; EGFR, subclass of samples with
EGFR mutations; fusion, subclass of samples with ALK,
RET or ROS1 fusions; ERBB2, subclass of samples with
ERBB2 mutations; BRAF, subclass of samples with BRAF
mutations; KRAS, subclass of samples with KRAS mutations;
NF1, subclass of samples with NF1 loss; RIT1, subclass of
samples with RIT1 mutations; MAP2K1, subclass of samples
with MAP2K1, NRAS or HRAS mutations; MET-amp,
subclass of samples with MET amplification; none, subclass
of samples without any of the above driver gene alterations. (B)
Western blotting for related pathways. To confirm the
bioinformatics analyses, western blot analyses were performed using
the RAS-mutated, EGFR-mutated or MET-amplified
NSCLC cell lines. The phosphorylation levels of AKT were
significantly elevated in the A549 (KRAS mutation) and
EGFR-mutated cell lines (PC-9, HCC827, Ma-1, 11_18, PC-9/ZD,
and H1975), compared with those in the H1299 (NRAS mutation)
and MET-amplified cell lines (EBC-1 and H1993). The
phosphorylation level of EGFR was elevated in the
EGFR-mutated cell lines and that of MET was elevated in the
MET-amplified cell lines. β-actin was used as an internal
control. *These cell lines were sensitive to MEK
inhibitors. |
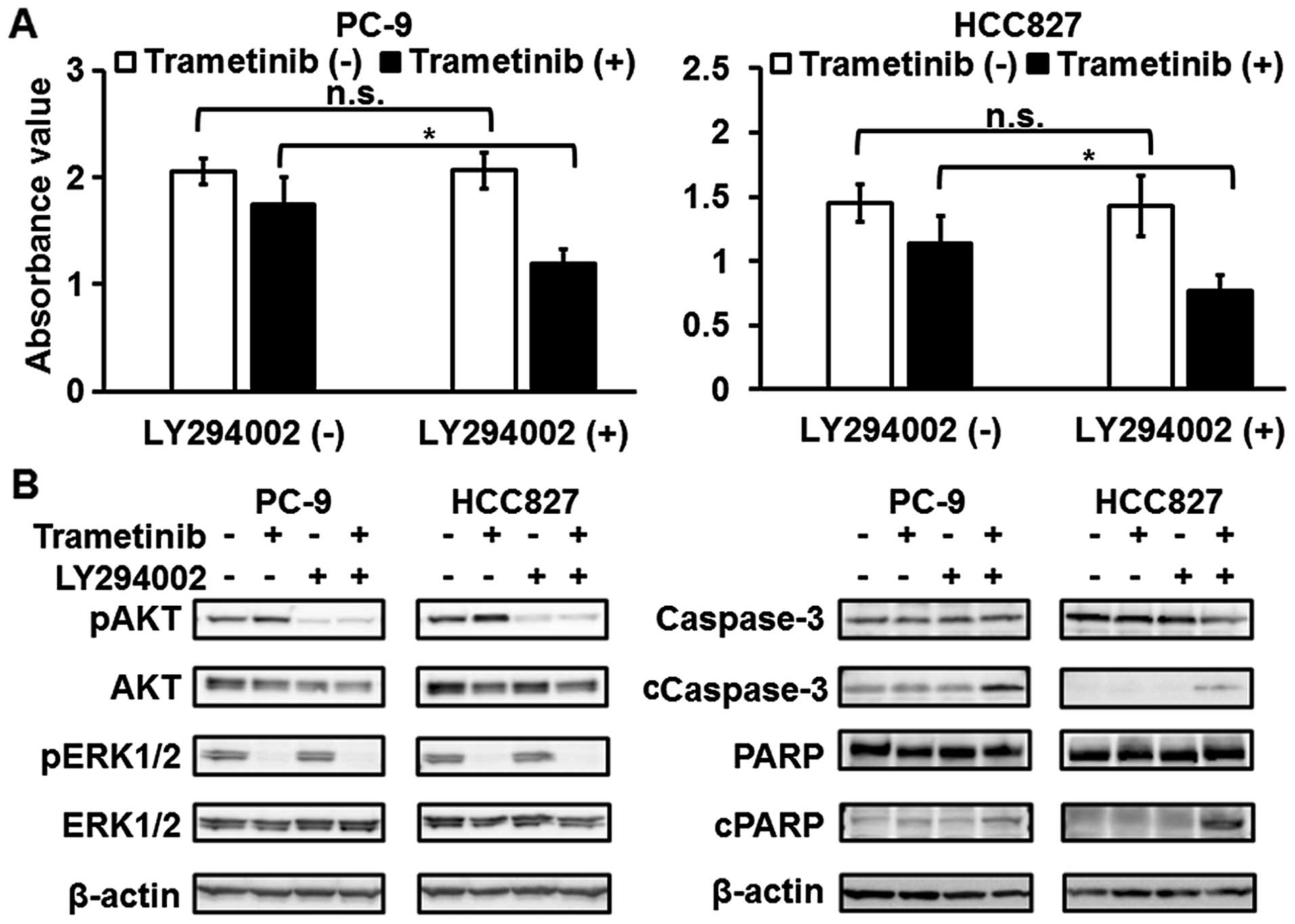
PI3K/AKT pathway is associated with
resistance to MEK inhibitors in EGFR-mutated NSCLC cell lines
To elucidate the association between the PI3K/AKT
pathway and resistance to MEK inhibitors, western blot analyses
after treatment with trametinib were performed. The phosphorylation
level of ERK1/2 was reduced after the treatment (Figs. 3A and 4B). When the PC-9 and HCC827 cell lines
(EGFR mutation) were exposed to trametinib, the
phosphorylation level of AKT was elevated with no change in EGFR or
MET phosphorylation (Figs. 3A and
4B). In contrast, the
phosphorylation of AKT was not changed in the EBC-1 and H1993 cell
lines (MET amplification) (Fig.
3A). In addition, the expression levels of apoptosis-related
molecules (cleaved caspase-3 and cleaved PARP) were elevated after
treatment in the EBC-1 and H1993 cell lines, whereas these levels
were not elevated in the PC-9 and HCC827 cell lines (Figs. 3B and 4B). Next, combination treatment with
trametinib and a PI3K inhibitor (LY294002) was tested. The
sensitivities to trametinib in the PC-9 and HCC827 cell lines were
enhanced using LY294002 (Fig. 4A).
The trametinib-induced elevation in the phosphorylation level of
AKT was reduced and apoptosis was also significantly induced by the
combined treatment with trametinib and LY294002 in these cell lines
(Fig. 4B). These findings support
the hypothesis that the activated PI3K/AKT pathway is associated
with resistance to MEK inhibitors in EGFR-mutated NSCLC cell
lines.
MEK inhibitors are effective for the
HCC827CNXR cell line, in which the driver gene has shifted from
EGFR to MET
We previously created a CNX-2006 (third-generation
EGFR-TKI)-resistant HCC827 cell line (HCC827CNXR) (15). This HCC827CNXR cell line lost the
EGFR mutations and instead harbored MET amplification
(Fig. 5A) (15). HCC827CNXR was sensitive to MET
inhibitors, meaning that the driver gene had shifted from the
EGFR mutation to MET amplification (oncogene swap)
(15). As is seen in other
MET-amplified NSCLC cell lines, the phosphorylation level of AKT
was reduced in the HCC827CNXR cell line, compared with that in the
HCC827 cell line (Fig. 5A). In
addition, the HCC827CNXR cell line was sensitive to MEK inhibitors,
while the HCC827 cell line was resistant (Fig. 5B and Table I), meaning that the downstream RTK
pathway had shifted to the MAPK pathway along with the oncogene
swap. In the HCC827 cell line, the phosphorylation level of AKT was
elevated after treatment with trametinib, whereas a similar
elevation was not observed in the HCC827CNXR cell line (Fig. 5C). The expression levels of
apoptosis-related molecules were also elevated in the HCC827CNXR
cell line after treatment with trametinib (Fig. 5C). These findings suggest that the
MAPK pathway is biologically important for MET-amplified
NSCLC.
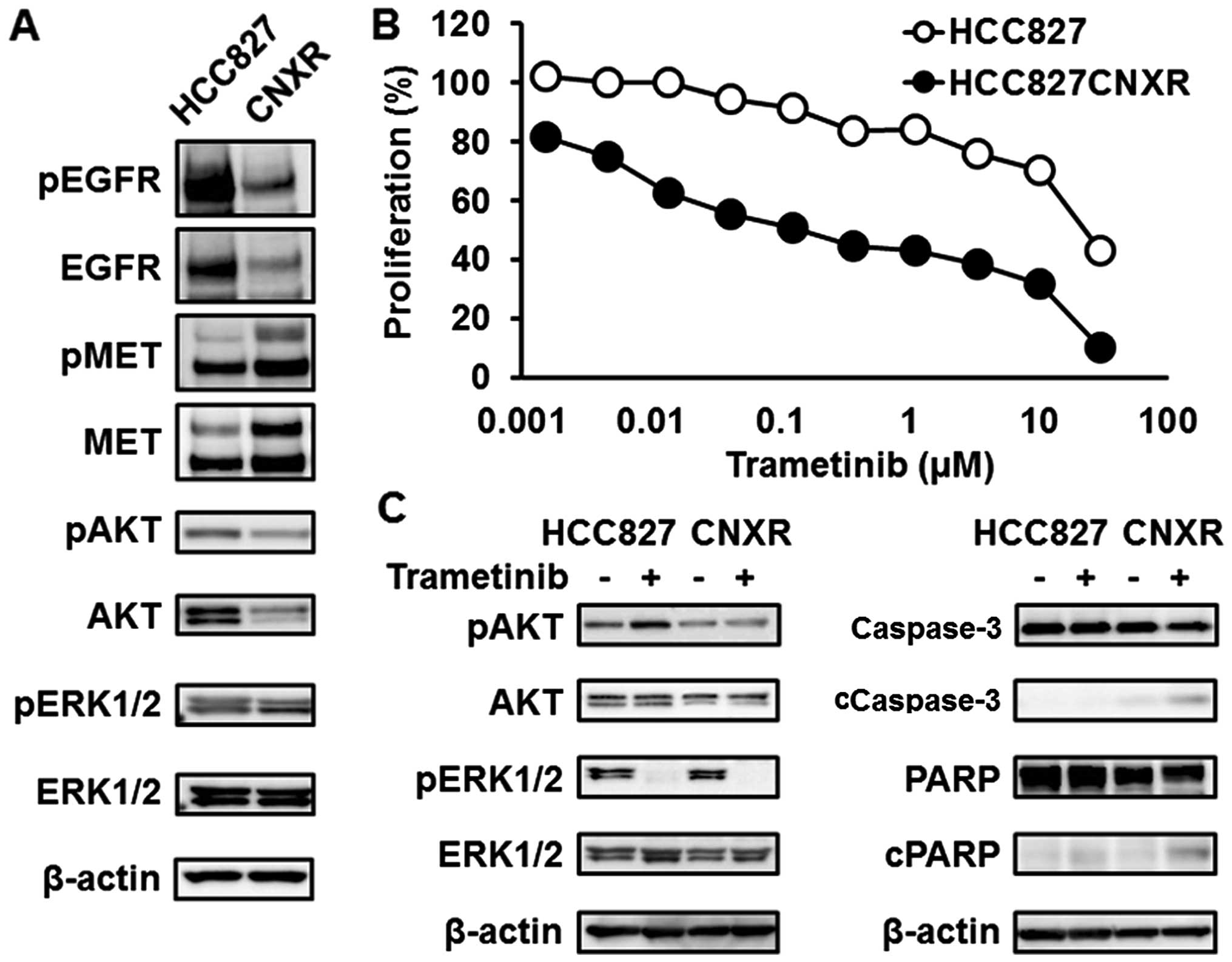 | Figure 5Trametinib against HCC827
(EGFR mutation) or HCC827CNXR (MET amplification)
cell lines. (A) Western blotting for related pathways. The
expression levels of EGFR and phospho-EGFR were elevated in the
HCC827 cell line. In contrast, the expression levels of MET and
phospho-MET were elevated, instead of a decrease in EGFR, in the
HCC827CNXR cell line (oncogene swap). The expression levels of AKT
and phospho-AKT were reduced along with the oncogene swap. β-actin
was used as an internal control. (B) Inhibitory curves for
trametinib in the HCC827 and HCC827CNXR cell lines. To investigate
the efficacy of trametinib against the HCC827CNXR cell line, a
growth inhibitory assay was performed using an MTT assay. The
HCC827CNXR cell line was sensitive to trametinib, while the HCC827
cell line was resistant. Line, mean of independent triplicate
experiments. (C) Western blotting for related pathways and
apoptosis-related molecules. Western blot analyses for related
pathways and apoptosis-related molecules were performed 3 and 24 h
after treatment with trametinib (0.1 μM), respectively. The
phosphorylation level of AKT was elevated after treatment with
trametinib in the HCC827 cell line, whereas a similar elevation was
not observed in the HCC827CNXR cell line. The expression levels of
cleaved caspase-3 and cleaved PARP were also significantly elevated
by treatment in the HCC827CNXR cell line. β-actin was used as an
internal control. cCaspase-3, cleaved caspase-3; cPARP, cleaved
PARP. |
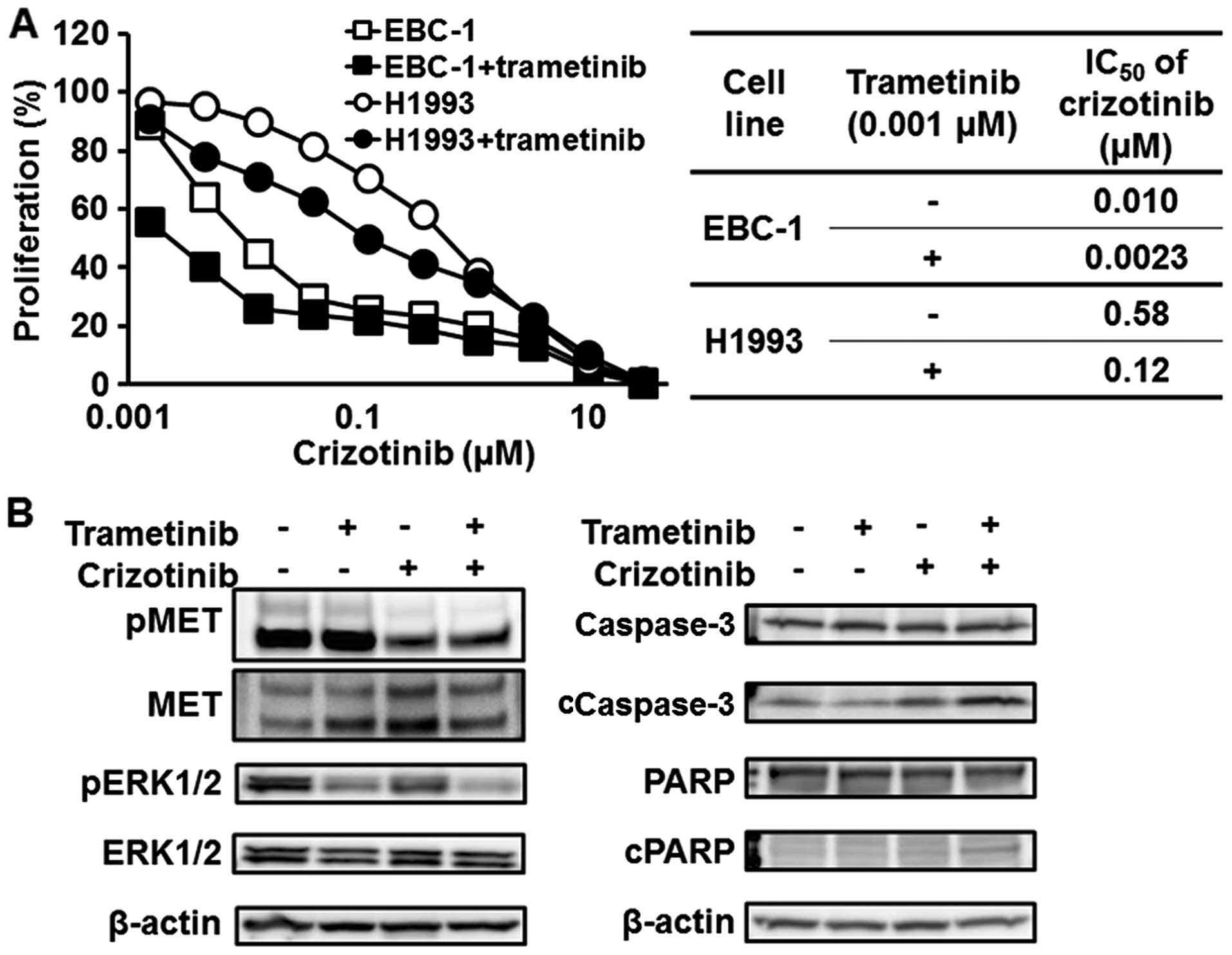
Synergistic effect of a MET inhibitor and
a MEK inhibitor against MET-amplified NSCLC cell lines
Since our experimental findings suggest that the
MAPK pathway is biologically important for MET-amplified
NSCLC, the synergistic effect of a MET inhibitor (crizotinib) and
trametinib was investigated. As shown in Fig. 6A, trametinib enhanced the efficacy
of crizotinib against MET-amplified NSCLC cell lines (EBC-1
and H1993). The IC50 of crizotinib in the EBC-1 and
H1993 cell lines decreased to 0.0023 and 0.12 μM from 0.010 and
0.58 μM, respectively (Fig. 6A).
The combination with trametinib significantly reduced the
phosphorylation level of ERK1/2 and induced apoptosis, compared
with crizotinib monotherapy (Fig.
6B).
Discussion
In this study, the sensitivities of several NSCLC
cell lines with driver gene alterations to MEK inhibitors were
investigated, and interesting results were obtained. MEK inhibitors
were not effective against any of the EGFR-mutated cell
lines but were effective against all the MET-amplified cell
lines. Furthermore, combination therapy with a MET inhibitor and a
MEK inhibitor was particularly effective against
MET-amplified NSCLC cell lines. To the best of our
knowledge, this is the first study to demonstrate such a difference
in the sensitivity to MEK inhibitors among NSCLC cell lines, the
biological importance of the MAPK pathway in MET-amplified
NSCLC, and the synergistic effect of a MET inhibitor and a MEK
inhibitor against MET-amplified NSCLC.
The MAPK pathway includes RAS, RAF, MEK, and ERK.
The constitutive activation of this pathway can lead to
uncontrolled cell growth and survival, ultimately resulting in
oncogenic transformation and progression (13,14).
Reflecting the central role of the MAPK pathway in cell
proliferation, activated mutants of RAS family members are
among the oncoproteins most frequently detected in human
malignancies (20). In addition,
among the downstream targets of RTK pathways, the MAPK pathway is
particularly important for cancer cell proliferation and survival
(11–14). Therefore, MEK inhibitors are
expected to be effective against NSCLC cell lines in which cell
growth or survival is dependent on oncogenic driver RTK gene
alterations, i.e., EGFR-mutated, ALK-fused or
MET-amplified NSCLC cell lines. However, a wide range of
sensitivities to MEK inhibitors was observed. Especially, MEK
inhibitors were effective against all the MET-amplified
NSCLC cell lines, whereas they were not effective against any of
the EGFR-mutated cell lines. Furthermore, the established
HCC827CNXR cell line, in which the driver gene had shifted from
EGFR to MET (oncogene swap), exhibited enhanced
sensitivity to MEK inhibitors. These findings suggest that the MAPK
pathway is biologically important for MET-amplified NSCLC.
The mechanism responsible for this difference was investigated
using a bioinformatics technique, which showed an association with
the activated PI3K/AKT pathway in EGFR-mutated NSCLC.
Indeed, the phosphorylation level of AKT in the EGFR-mutated
cell lines was significantly elevated, compared with that in the
MET-amplified cell lines, and this phosphorylation increased
after treatment with trametinib, suggesting that the PI3K/AKT
pathway plays a salvage role in EGFR-mutated NSCLC treated
with MEK inhibitors. In addition, a PI3K inhibitor enhanced the
sensitivity to a MEK inhibitor in the EGFR-mutated NSCLC
cell lines. These experimental findings support the association
between the activated PI3K/AKT pathway and resistance to MEK
inhibitors in EGFR-mutated NSCLC. Although further
mechanisms remain unclear, similar reports showing the activated
PI3K/AKT pathway in EGFR-mutated NSCLC have been previously
reported (18,19). Differences in adopter protein
binding might exist between EGFR and MET, and further research is
needed.
MET is overexpressed in 22–67% of NSCLCs, and one
type of MET abnormality is amplification (21–24).
The prevalence of MET amplification is ~5% in untreated patients
and 20% in those with acquired resistance to EGFR-TKIs (21–31).
MET positivity via IHC and an increased MET gene copy number are
both significantly associated with poor overall survival (21–24,32).
Although some preclinical studies, phase I/II trials, and subgroup
analyses of phase III trials of small molecules targeting MET (used
either singly or in combination) for MET-amplified NSCLC
have yielded favorable results, the efficacy of such agents has not
yet been confirmed in large clinical trials (10,33–35).
Furthermore, two large phase III trials using anti-MET drugs in
combination with EGFR-TKIs have failed (36,37).
A previous report showed the synergistic effect of a MET inhibitor
and a MEK inhibitor in RAS-mutated cell lines and the role
of RAS mutations in resistance to a MET inhibitor (38). These findings indicate the
importance of the MAPK pathway in treatment with a MET inhibitor,
and our present study also showed the importance of this pathway
for MET-amplified NSCLC and the efficacy of MEK inhibitors
against such NSCLC. In addition, our experimental findings revealed
the synergistic effect of a MET inhibitor and a MEK inhibitor
against MET-amplified NSCLC, suggesting that combination
therapy with a MET inhibitor and a MEK inhibitor might be a
promising treatment strategy for MET-amplified NSCLC.
In conclusion, we observed that MEK inhibitors were
not effective against any of the EGFR-mutated NSCLC cell
lines that were examined, but were effective against all the
MET-amplified NSCLC cell lines. Although this difference
seems to have been caused by the activation of the PI3K/AKT pathway
in EGFR-mutated NSCLC, the detailed molecular mechanism
remains unclear and further research is needed. In addition, the
importance of the MAPK pathway for MET-amplified NSCLC has
been shown, and combination therapy consisting of a MET inhibitor
and a MEK inhibitor was particularly effective against
MET-amplified NSCLC cell lines. These experimental findings
strongly encourage the development of this combination therapy
against MET-amplified NSCLC.
Acknowledgements
We thank Ms. Tomoko Kitayama and Ms. Ayaka
Kurumatani for their technical assistance. This study was supported
in part by a Grant-in-Aid for Research Activity start-up (Y.
Togashi), The Uehara Memorial Foundation, and The Takeda Science
Foundation (T. Mitsudomi). T. Mitsudomi received a lecture fee from
Pfizer Pharmaceuticals.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pao W, Miller V, Zakowski M, Doherty J,
Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, et al:
EGF receptor gene mutations are common in lung cancers from ‘never
smokers’ and are associated with sensitivity of tumors to gefitinib
and erlotinib. Proc Natl Acad Sci USA. 101:13306–13311. 2004.
View Article : Google Scholar
|
|
5
|
Mok TS, Wu YL, Thongprasert S, Yang CH,
Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et
al: Gefitinib or carboplatin-paclitaxel in pulmonary
adenocarcinoma. N Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mitsudomi T, Morita S, Yatabe Y, Negoro S,
Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, et
al; West Japan Oncology Group. Gefitinib versus cisplatin plus
docetaxel in patients with non-small-cell lung cancer harbouring
mutations of the epidermal growth factor receptor (WJTOG3405): An
open label, randomised phase 3 trial. Lancet Oncol. 11:121–128.
2010. View Article : Google Scholar
|
|
7
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I,
et al; North-East Japan Study Group. Gefitinib or chemotherapy for
non-small-cell lung cancer with mutated EGFR. N Engl J Med.
362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moreira AL and Eng J: Personalized therapy
for lung cancer. Chest. 146:1649–1657. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morgensztern D, Campo MJ, Dahlberg SE,
Doebele RC, Garon E, Gerber DE, Goldberg SB, Hammerman PS, Heist
RS, Hensing T, et al: Molecularly targeted therapies in
non-small-cell lung cancer annual update 2014. J Thorac Oncol.
10:S1–S63. 2015. View Article : Google Scholar :
|
|
10
|
Califano R, Abidin A, Tariq NU,
Economopoulou P, Metro G and Mountzios G: Beyond EGFR and ALK
inhibition: Unravelling and exploiting novel genetic alterations in
advanced non small-cell lung cancer. Cancer Treat Rev. 41:401–411.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Regad T: Targeting RTK signaling pathways
in cancer. Cancers (Basel). 7:1758–1784. 2015. View Article : Google Scholar :
|
|
12
|
Trusolino L, Bertotti A and Comoglio PM:
MET signalling: Principles and functions in development, organ
regeneration and cancer. Nat Rev Mol Cell Biol. 11:834–848. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
De Luca A, Maiello MR, D'Alessio A,
Pergameno M and Normanno N: The RAS/RAF/MEK/ERK and the PI3K/AKT
signalling pathways: Role in cancer pathogenesis and implications
for therapeutic approaches. Expert Opin Ther Targets. 16(Suppl 2):
S17–S27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mizuuchi H, Suda K, Murakami I, Sakai K,
Sato K, Kobayashi Y, Shimoji M, Chiba M, Sesumi Y, Tomizawa K, et
al: Oncogene swap as a novel mechanism of acquired resistance to
epidermal growth factor receptor-tyrosine kinase inhibitor in lung
cancer. Cancer Sci. 107:461–468. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Togashi Y, Hayashi H, Terashima M, de
Velasco MA, Sakai K, Fujita Y, Tomida S, Nakagawa K and Nishio K:
Inhibition of β-catenin enhances the anticancer effect of
irreversible EGFR-TKI in EGFR-mutated non-small-cell lung cancer
with a T790M mutation. J Thorac Oncol. 10:93–101. 2015. View Article : Google Scholar
|
|
17
|
Network CGAR; Cancer Genome Atlas Research
Network. Comprehensive molecular profiling of lung adenocarcinoma.
Nature. 511:543–550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Conde E, Angulo B, Tang M, Morente M,
Torres-Lanzas J, Lopez-Encuentra A, Lopez-Rios F and
Sanchez-Cespedes M: Molecular context of the EGFR mutations:
evidence for the activation of mTOR/S6K signaling. Clin Cancer Res.
12:710–717. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou Y, Rideout WM III, Zi T, Bressel A,
Reddypalli S, Rancourt R, Woo JK, Horner JW, Chin L, Chiu MI, et
al: Chimeric mouse tumor models reveal differences in pathway
activation between ERBB family- and KRAS-dependent lung
adenocarcinomas. Nat Biotechnol. 28:71–78. 2010. View Article : Google Scholar
|
|
20
|
Karnoub AE and Weinberg RA: Ras oncogenes:
Split personalities. Nat Rev Mol Cell Biol. 9:517–531. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tsuta K, Kozu Y, Mimae T, Yoshida A, Kohno
T, Sekine I, Tamura T, Asamura H, Furuta K and Tsuda H:
c-MET/phospho-MET protein expression and MET gene copy number in
non-small cell lung carcinomas. J Thorac Oncol. 7:331–339. 2012.
View Article : Google Scholar
|
|
22
|
Tachibana K, Minami Y, Shiba-Ishii A, Kano
J, Nakazato Y, Sato Y, Goya T and Noguchi M: Abnormality of the
hepatocyte growth factor/MET pathway in pulmonary
adenocarcinogenesis. Lung Cancer. 75:181–188. 2012. View Article : Google Scholar
|
|
23
|
Sun W, Song L, Ai T, Zhang Y, Gao Y and
Cui J: Prognostic value of MET, cyclin D1 and MET gene copy number
in non-small cell lung cancer. J Biomed Res. 27:220–230. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang L, An SJ, Chen ZH, Su J, Yan HH and
Wu YL: MET expression plays differing roles in non-small-cell lung
cancer patients with or without EGFR mutation. J Thorac Oncol.
9:725–728. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bean J, Brennan C, Shih JY, Riely G, Viale
A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, et al: MET
amplification occurs with or without T790M mutations in EGFR mutant
lung tumors with acquired resistance to gefitinib or erlotinib.
Proc Natl Acad Sci USA. 104:20932–20937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Beau-Faller M, Ruppert AM, Voegeli AC,
Neuville A, Meyer N, Guerin E, Legrain M, Mennecier B, Wihlm JM,
Massard G, et al: MET gene copy number in non-small cell lung
cancer: Molecular analysis in a targeted tyrosine kinase inhibitor
naive cohort. J Thorac Oncol. 3:331–339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Onozato R, Kosaka T, Kuwano H, Sekido Y,
Yatabe Y and Mitsudomi T: Activation of MET by gene amplification
or by splice mutations deleting the juxtamembrane domain in primary
resected lung cancers. J Thorac Oncol. 4:5–11. 2009. View Article : Google Scholar
|
|
29
|
Cappuzzo F, Janne PA, Skokan M,
Finocchiaro G, Rossi E, Ligorio C, Zucali PA, Terracciano L, Toschi
L, Roncalli M, et al: MET increased gene copy number and primary
resistance to gefitinib therapy in non-small-cell lung cancer
patients. Ann Oncol. 20:298–304. 2009. View Article : Google Scholar :
|
|
30
|
Kubo T, Yamamoto H, Lockwood WW, Valencia
I, Soh J, Peyton M, Jida M, Otani H, Fujii T, Ouchida M, et al: MET
gene amplification or EGFR mutation activate MET in lung cancers
untreated with EGFR tyrosine kinase inhibitors. Int J Cancer.
124:1778–1784. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sequist LV, von Pawel J, Garmey EG,
Akerley WL, Brugger W, Ferrari D, Chen Y, Costa DB, Gerber DE,
Orlov S, et al: Randomized phase II study of erlotinib plus
tivantinib versus erlotinib plus placebo in previously treated
non-small-cell lung cancer. J Clin Oncol. 29:3307–3315. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dimou A, Non L, Chae YK, Tester WJ and
Syrigos KN: MET gene copy number predicts worse overall survival in
patients with non-small cell lung cancer (NSCLC); a systematic
review and meta-analysis. PLoS One. 9:e1076772014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lutterbach B, Zeng Q, Davis LJ, Hatch H,
Hang G, Kohl NE, Gibbs JB and Pan BS: Lung cancer cell lines
harboring MET gene amplification are dependent on Met for growth
and survival. Cancer Res. 67:2081–2088. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Spigel DR, Edelman MJ, O'Byrne K, Paz-Ares
L, Shames DS, Yu W, Paton VE and Mok T: Onartuzumab plus erlotinib
versus erlotinib in previously treated stage IIIb or IV NSCLC:
Results from the pivotal phase III randomized, multicenter,
placebo-controlled METLung (OAM4971g) global trial. J Clin Oncol.
32(Suppl 15): 80002014.
|
|
35
|
Li A, Gao HF and Wu YL: Targeting the MET
pathway for potential treatment of NSCLC. Expert Opin Ther Targets.
19:663–674. 2015. View Article : Google Scholar
|
|
36
|
Scagliotti G, von Pawel J, Novello S,
Ramlau R, Favaretto A, Barlesi F, Akerley W, Orlov S, Santoro A,
Spigel D, et al: Phase III multinational, randomized, double-blind,
placebo-controlled study of Tivantinib (ARQ 197) Plus Erlotinib
Versus Erlotinib alone in previously treated patients with locally
advanced or metastatic nonsquamous non-small-cell lung cancer. J
Clin Oncol. 33:2667–2674. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Camidge DS, Ou S-HI, Shapiro Geoffrey,
Otterson GA, Villaruz LC, Villalona-Calero MA, Iafrate AJ,
Varella-Garcia M, Dacic S, Cardarella S, et al: Efficacy and safety
of crizotinib in patients with advanced c-MET-amplified non-small
cell lung cancer (NSCLC). J Clin Oncol. 32(Suppl 15): 80012014.
|
|
38
|
Leiser D, Medová M, Mikami K, Nisa L,
Stroka D, Blaukat A, Bladt F, Aebersold DM and Zimmer Y: KRAS and
HRAS mutations confer resistance to MET targeting in preclinical
models of MET-expressing tumor cells. Mol Oncol. 9:1434–1446. 2015.
View Article : Google Scholar : PubMed/NCBI
|