Introduction
Neuroblastoma (NB) is the most commonly occurring
extracranial solid neoplasm diagnosed in young children that shows
aggressive behaviour. NB is a childhood malignancy, which may
originate from multipotent crest cells of the sympathetic nervous
system and accounts for 8 to 10% of all childhood cancers, yet
disproportionately responsible for 15% of all childhood cancer
deaths (1,2). Approximately 40% of the children with
NB have an aggressive form with a 5-year event-free survival rate
<50% (3). Despite intensive
treatment protocols including myeloablative cytotoxic therapy and
13-cis-retinoic acid (4) or
anti-GD2 immunotherapy (5) that
were applied, the majority of the children diagnosed with high-risk
NB experience a recurrence and the long-term survival <40%
(6,7). Although the initial tumor response is
very good in children with high-risk NB, >60% of the treated
patients experience relapse and still die of this disease (8,9).
Currently, in the clinic relapsed/refractory patients still lack
effective treatment strategies which has been recognized as a
critical problem, and the poor outcome remains.
Initially, responsiveness to chemotherapy in NB
tumors is high (10), but
eventually, these patients often relapse and develop a multiple
drug resistant (MDR) phenotype. Acquired MDR is a major
contributing factor to chemotherapeutic treatment failure (11,12).
MDR is a phenomenon in which tumor cells become resistant to
diverse chemical structures and functionally unrelated
chemotherapeutic agents (13).
Once NB develops MDR, as a consequence, the long-term survival rate
of NB patients is poor (11).
Strategies to improve the outcomes in high-risk NB include
13-cis-retinoic acid, anti-GD2 immunotherapy and bone marrow
transplantation. Still, the high-risk NB patients eventually
develop progressive disease.
The mechanism of NB developing MDR is complicated
and several factors are involved, including loss of function of p53
(14), overexpression of the
membrane bound ATP-binding cassette transporters (ABC proteins)
(13), inactivation of the
detoxification system, changing drug targets and significant
downregulation of apoptotic cell death-related genes (12). Especially, the most often observed
the main mechanism for MDR includes loss of function of p53, high
levels of P-glycoprotein (P-gp) and MRP1 (multidrug
resistance-related protein). When overexpressed in neoplastic
tissue, P-gp and MRP1 effectively extrude various cytotoxic agents
and eliminate chemotherapeutics, may contribute to their
refractoriness to large group of cytotoxic drugs, such as
anthracycline, epipodophyllotoxin, vinca alkaloids, taxanes and
methotrexate (15,16).
A number of conventional anticancer agents show
cytotoxic effects by interfering with DNA or RNA replication and
protein synthesis in cancer cells, as well as activation of the
expression of p53 gene, but unfortunately, most of chemotherapy
drugs can also increase gene mutations and change tumor suppressor
gene expression. In some highly malignant tumor cells, apoptotic
mechanisms of secondary damage due to chemotherapy, such as
inactivation of p53 gene containing cysteine aspartic protease
(caspase) fail, limiting their application. These include increased
DNA repair, altered target sensitivity, decreased apoptotic
response, and numerous aberrant signal transduction pathways.
Commonly used chemotherapy drugs such as Dox and DDP induce tumor
cells formed MDR in that they can increase the leukemia cells or NB
the expression of P-gp. While increasing the dose of
chemotherapeutic agents may successfully eliminate MDR to some
extent, but high-dose chemotherapy can cause inevitable severe
side-effects, such as heart, liver, kidney and hearing damage, thus
patients can not tolerate and abandon treatment (17). This prompted us to investigate new
treatment strategies against MDR NB caused by diverse
mechanisms.
It is necessary to develop potential
chemotherapeutic strategies to enhance chemosensitivity and to
overcome the MDR. While in the last few years, arsenic was also
seen as a carcinogen, but recent reports have indicated that
arsenic trioxide (As2O3) has been found to
inhibit cell growth and/or induce apoptosis in acute promyelocytic
leukemia (APL) (18,19) and successfully employed as a highly
effective agent for other malignancies. As2O3
has been associated with complicated anticancer mechanisms,
including induction of tumor cell differentiation, inhibition of
tumor cell growth and induce apoptosis (20), although the efficacy of
As2O3 on different tumors remains unsure
(21). In the APL cells,
As2O3 exert antitumor effect by acting on the
PML/RARA as target (22); in other
tumor cells, the antitumor effect was associated with activation of
caspase cascade leading to apoptotic cell death and reduced
expression of Bcl-2 gene (23).
Many studies have confirmed that
As2O3 has the capacity to kill NB cells in
vitro and in vitro (24–27).
Despite that invasive tumor in the body often is in a low oxygen
environment, Karlsson et al (26) show that the cytotoxic effect of
As2O3 is retained under hypoxic conditions,
even in MDR NB cells. In contrast, the cell death induced by the
conventionally used drug etoposide (Vp16) was significantly
impaired in Vp16-sensitive NB cell lines cultivated at hypoxia.
These results support the idea of using As2O3
as an efficient treatment strategy also in solid tumors with
hypoxic areas like NB. Memorial Sloan-Kettering Cancer Center has
completed phase II clinical trials of treatment of NB and other
malignancies, preliminarily showing that
As2O3 treatment of MDR NB has better safety,
the primary efficacy outcome are not yet available.
Some investigators have shown that
As2O3-induced NB apoptosis is caused by
downregulation of Bcl-2 protein, activate caspase, the generation
of reactive oxygen species, loss of the mitochondrial transmembrane
potential, and cell cycle arrest in G1 or G2/M phase (23). Little is known about
As2O3 impact on the expression of multidrug
resistant-related P-gp in NB, however, further studies are
required. We examined the effect of As2O3 and
conventionally used drugs on P-glycoprotein by western blotting;
flow cytometric assay was used to examine the impact of various
antitumor drugs on cell cycle and cell apoptosis in SK-N-SH
cells.
Overall, we compared and contrasted the effects of
As2O3 and conventional chemotherapeutic drugs
at varying concentrations on the expression of P-gp and the impact
of cell cycle in NB SK-N-SH cells. In this study, our results
indicated that compared to conventional chemotherapeutic drugs,
As2O3 mediates this effect by inducing
apoptosis and provides a suggestion that
As2O3 is a potential chemotherapeutic reagent
for the treatment of NB including MDR NB.
Materials and methods
Cell line
Human NB cell line, SK-N-SH, was purchased from Cell
Bank of Sun-Yet Sen Medical School. SK-N-SH cells were grown in
DMEM supplemented with 12% fetal calf serum (FCS), 100 U/ml
penicillin and 100 μg/ml streptomycin. Cells were maintained at
37°C in a humidified incubator of 5% CO2/95%.
Reagents
As2O3, VP16 and DDP was
purchased from Sun Yat-Sen Memorial Hospital.
As2O3 was dissolved in phosphate-buffered
saline as 10 mM stock and kept at 4°C. MDR1/ABCB1 (D3H1Q) rabbit
mAb and anti-GAPDH antibody rabbit antibody was obtained from Cell
Signaling Technology. HRP affinipure goat anti-rabbit IgG for
western blotting was purchased from Sigma Chemical Co. Immobilon
western chemiluminescent HRP substrate was purchased from
Millipore, and DMEM medium and trypsin were purchased from Hyclone.
PVDF membrane was purchased from Millipore, and FBS was purchased
from NBQQ.
Flow cytometry with Annexin V-PI staining
for apoptosis
Logarithmically growing cells were exposed to
various concentrations and exposure intervals of
As2O3, VP16 and DDP. For the experiments,
cells were seeded in 6-well plates at a density of
~3×105 cells per milliliter at 37°C in a humidified
incubator of 5% for 24 h. For determination of cell growth
inhibition, cells were exposed to varying concentrations of
As2O3, VP16 and DDP at 37°C for the indicated
time. After treatment, the cells at 1–5×105/ml harvested
from culture and washed by PBS, and then resuspended in 200 μl
staining solution that contained 5 μl of Annexin V-FITC and 10 μl
propidium iodide, according to the protocol of the Annexin V
Staining kit. The proportion of cell apoptosis was analyzed by flow
cytometry within one hour. The IC50 value resulting from
50% inhibition of cell growth was calculated graphically as a
comparison with the control. All treatments were done in
triplicates and the experiments were repeated three times.
Flow cytometry for cell cycle
Logarithmically growing cells were exposed to
various concentrations and exposure intervals of
As2O3, VP16 and DDP. For the experiments,
cells were seeded in 6-well plates at a density of approximately
3×105 cells per milliliter at 37°C in a humidified
incubator of 5% for 24 h. IC50 of
As2O3 was added to the experimental group.
According to the literature (28)
and our result of IC50 of As2O3 on
SK-N-SH cells, we used 3 μM As2O3-treated
SK-N-SH cells for 48 and 72 h. IC50 of DDP and Vp16 were
added to the drug control groups. According to our prior research,
we chose 8 μg/ml DDP-treated cells for 12 and 24 h, 100 μg/ml
Vp16-treated cells for 24 and 48 h. Each group was set in three
parallel wells. Untreated or drug-treated cells were centrifuged,
washed with PBS and fixed with ice-cold 75% ethanol overnight.
Then, the cells were centrifuged and the cell pellet was
resuspended in 50 μl of PBS. The fixed cells were incubated with 50
μl of RNase for 30 min in 37°C water and 450 μl of propidium iodide
incubated for 30 min in the dark. The percentage of cells at a
particular phase was calculated by flow cytometry.
Western blot analysis was performed to
investigate the expression of P-gp
Logarithmically growing cells were exposed to
various concentrations and exposure intervals of
As2O3, VP16 and DDP. For the experiments,
cells were seeded in 6-well plates at a density of
~3×105 cells per milliliter at 37°C in a humidified
incubator of 5% for 24 h. The concentrations were based on our
results of IC50 of As2O3, DDP and
Vp16 (the concentration gradients based on the results of
preliminary experiments). Each group was set in three parallel
wells. As2O3 2, 3 and 4 μM (incubation 48, 72
and 96 h); DDP 5, 8 and 10 μg/ml (incubation 12 and 24 h); Vp16 50,
100 and 150 μg/ml (incubation 24 and 48 h). After treatment with
appropriate drug concentration, protein lysate was prepared in
lysis buffer and the protein concentration was determined by
bicinchonic acid (BCA) protein assay. Equal amount of proteins were
loaded on 8% SDS-polyacrylamide gel. Voltage 80 V was prepared for
the stacking gel; 100 V was prepared for separating gel in the end.
The proteins were then transferred to a polyvinylidene fluoride
(PVDF) membrane by semidry transfer system. After blocking the
non-specific space of the membrane with 5% non-fat milk in TBST
(TBS with 0.1% Tween-20) for one hour, membranes were first
incubated with monoclonal antibody against P-glycoprotein (1:1,000)
(Cell Signaling Technology) overnight at 4°C with constant shaking.
Then the membranes were incubated with horseradish
peroxidase-conjugated secondary antibody (1:1,000) (Sigma Chemical
Co.) for 1 h with constant shaking. After electrophoresis, proteins
were transferred to a nitrocellulose membrane and probed by
corresponding antibodies. The quantification of protein levels in
each sample was performed by ImageJ software. All experiments were
performed in triplicate unless otherwise noted.
Statistical analysis
Each apoptosis value represents the mean ± SD.
Subsequently, each experiment was performed at least three times.
Statistical analysis of data was carried out using a one-way ANOVA
and a probability value of <0.05 (P<0.05) was accepted as a
significant difference. Computations were performed using SPSS17
software.
Results
Effects of As2O3,
DDP and Vp16 on SK-N-SH cells
To characterize the effects of
As2O3, DDP and Vp16 on SK-N-SH cells, we
performed flow cytometry analyses of the viability of SK-N-SH
cells. It has previously been shown that
As2O3 efficiently induces apoptosis of human
NB cells in vitro. The initial experiments were conducted
for evaluation of As2O3, DDP and Vp16 induced
anti-proliferation in SK-N-SH cell lines. SK-N-SH cells were
treated with either PBS or drugs in each well. At first, according
to the literature review and the results of our previous study, we
determined the treatment conditions (drug concentration and
treatment duration). The growth inhibitory effects of various
concentrations of As2O3, DDP and Vp16 were
determined as described in Materials and methods. The viability of
the cells was measured at different therapy times. Each point
represents the mean ± SD of three independent experiments. The
IC50 values were determined by FACS.
As2O3 exhibited dose- and
time-dependent inhibitory effects on SK-N-SH cells with an
IC50 of 3 μM (Fig. 1A and
B). Furthermore, these results also indicated that
As2O3 exerted its cytotoxic effect in SK-N-SH
cells via induction of apoptosis. For example, without the
As2O3 treatment, there were only 6.9±1.3%
apoptotic cells in the SK-N-SH cell line, but the apoptotic cells
were gradually increased and reached 49.51±4.27% with a 3 μM
concentration of As2O3 at 72 h of exposure.
As shown in Fig. 1A and B, only
40.77% of cells were apoptotic at 2 μM As2O3
of exposure, 49.51% of cells were apoptotic at 3 μM
As2O3, and 54.21% were apoptotic at 4 μM
As2O3 after 72 h of exposure. Thus, the
concentrations of As2O3 (2–4 μM) were the
doses chosen for subsequent experiments. Moreover, flow cytometry
analysis of As2O3-treated cells revealed that
following treatment with As2O3 (3 μM) for 48
and 72 h, the percentage of apoptotic cells increased from
27.87±4.47 to 49.51±4.27%; As2O3 (4 μM) for
48 and 72 h, the percentage of apoptotic cells increased from
31.92±2.97 to 54.21±3.82%. These data suggest that
As2O3 may inhibit NB growth in
vitro.
The growth and survival of SK-N-SH cells were
markedly inhibited by the DDP and Vp16 treatment in a dose- and
time-dependent manner (Fig. 1C and
D). Fig. 1C and D demonstrate
the results of flow cytometry with Annex in V-PI staining on
SK-N-SH cells for IC50 of DDP and Vp16 found to be 8 and
100 μg/ml; consistent with our previous results. DDP significantly
reduced SK-N-SH cell viability at concentrations of 8 μg/ml or more
(Fig. 1C). By 24-h treatment high
levels of apoptosis were observed in SK-N-SH cells.
At 8 μg/ml, DDP strongly reduced the viability of
SK-N-SH cells compared with the effects at 5 μg/ml (Fig. 1C) for 12 and 24 h. Essentially the
same results were obtained in previous research results. After 5, 8
and 10 μg/ml DDP treatment of SK-N-SH cells for 12 h, the apoptosis
rates were (9.68±2.69)%, (15.38±4.36)% and (21.25±2.82)%; after 5,
8 and 10 μg/ml DDP treatment of SK-N-SH cells for 24 h, the
apoptosis rates were (26.37±4.6)%, (49.55±2.48)% and (53.89±3.31)%,
DDP cytotoxity to SK-N-SH cells in a concentration-dependent
manner. The apoptosis rates significantly increased after the same
concentration of DDP treatment of SK-N-SH cells for 24 h relative
to 12 h, DDP cytotoxity to SK-N-SH cells in a time-dependent
manner.
After 50, 100 and 150 μg/ml Vp16 (Fig. 1D) treatment of SK-N-SH cells for 48
h, the apoptosis rates were (14.86±2.86)%, (16.44±4.41)% and
(19.67±4.62)%; after 50, 100 and 150 μg/ml Vp16 treatent of SK-N-SH
cells for 72 h, the apoptosis rates were (40.54±3.53)%,
(49.12±3.96)% and (53.78±3.85)%, Vp16 cytotoxity to SK-N-SH cells
in a concentration-dependent manner. The apoptosis rates
significantly increased after the same concentration of Vp16
treatent of SK-N-SH cells for 48 h relative to 24 h, Vp16
cytotoxity to SK-N-SH cells in a time-dependent manner.
Effect of As2O3,
DDP and Vp16 on cell cycle arrest of SK-N-SH cells
Analysis of cell cycle phase distribution was
carried out to study the anti-proliferative mechanism of
As2O3 on SK-N-SH cells. IC50 of
As2O3 was added to the experimental group.
According to the literature (28)
and our result of IC50 of As2O3 on
SK-N-SH cells, we chose 3 μM As2O3-treated
SK-N-SH cells for 48 and 72 h. IC50 of DDP and Vp16 were
added to the drug control groups. According to our prior research,
we chose 8 μg/ml DDP-treated cells for 12 and 24 h, 100 μg/ml
Vp16-treated cells for 24 and 48 h. Each group was set in three
parallel wells. The result indicated that
As2O3 inhibited the cell proliferation of
SK-N-SH cells was partly due to the cell cycle arrest, not only
cytotoxic effect.
As2O3 treatment resulted in a
time-dependent accumulation of SK-N-SH cells in the G2/M phase
(Fig. 2A). At time 0, only
3.13±2.6% of cells were in the G2/M phase. After 48 h of treatment,
18.75±3.44% of cells were in the G2/M phase. The value of G2/M
phase cells peaked at 72 h (33.04%).
As shown in Fig.
2B, 21.01±0.35% of cells were at S-phase at time 0 and
33.86±0.39% of cells were at S-phase at 12 h. Treatment with DDP
for 24 h caused a small decrease in the S peak in SK-N-SH cells,
with no statistical difference. Treatment with Vp16 showed similar
results in SK-N-SH cells (Fig.
2C). S-phase cells reached peak value (77.49±9.08) at 48 h.
Effects of As2O3,
DDP and Vp16 on the expression changes of P-gp in SK-N-SH
cells
Overexpression of P-gp as a membrane-bound
energy-dependent efflux pump, in neoplastic tissue is responsible
for MDR formation. Correspondingly, it has been reported that
As2O3 inhibits the P-gp expression of
leukemia cells (29). To examine
whether As2O3, DDP and Vp16 affect P-gp
expression in SK-N-SH cells, we initially monitor the expression of
P-gp at various time-points and various concentrations using
western blotting. The concentrations were based on our results of
IC50 of As2O3, DDP and Vp16 (the
concentration gradients based on the results of preliminary
experiments). Each group was set in three parallel wells.
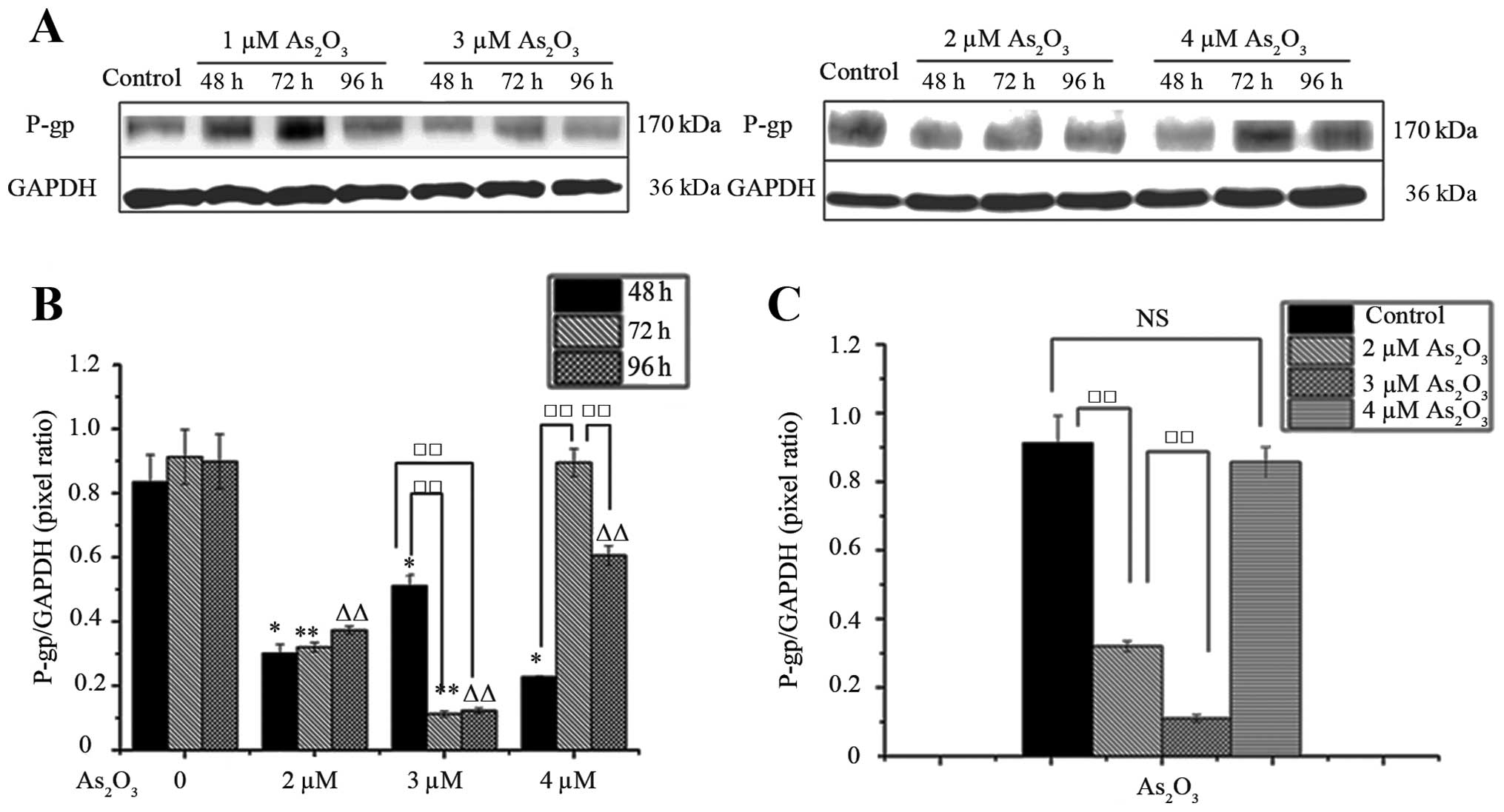
As shown in Fig. 3A and
B, western blotting revealed that As2O3
treatment did not result in a time- and dose-dependent upregulation
of P-gp (P<0.05). To determine the extent of the observed
striking reduction in P-gp expression, we performed western blot
analysis on P-gp expression using the column chart (Fig. 3B). We found that 2 and 3 μM
As2O3 treatment for 48, 72 and 96 h leads to
reduced expression of P-gp (Fig.
3B), and the expression levels of P-gp were downregulated in 3
μM As2O3 treatment for 72 h more notably than
in 2 μM As2O3 for 72 h (Fig. 3C). As2O3 did
not lead to decreasing levels of P-gp expression in a time- and
dose-dependent manner. In particular, P-gp expression was
profoundly decreased under 3 μM (IC50)
As2O3 treatment for 72 h. At 4 μM
As2O3 for 48 and 96 h, the expression of P-gp
was noted to gradually decrease, but no differences were found at
72 h after As2O3 exposure. Taken together,
our data reveal that after As2O3 treatment,
we observed a reproducible reduction in the induced P-gp expression
(Fig. 3B). Upon treatment with
different concentrations of As2O3, expression
of P-gp was substantially decreased (Fig. 3B). These results may imply that
As2O3 may not be a substrate of P-gp and so
it cannot stimulate and sustain the expression of P-gp in SK-N-SH
cells during incubation.
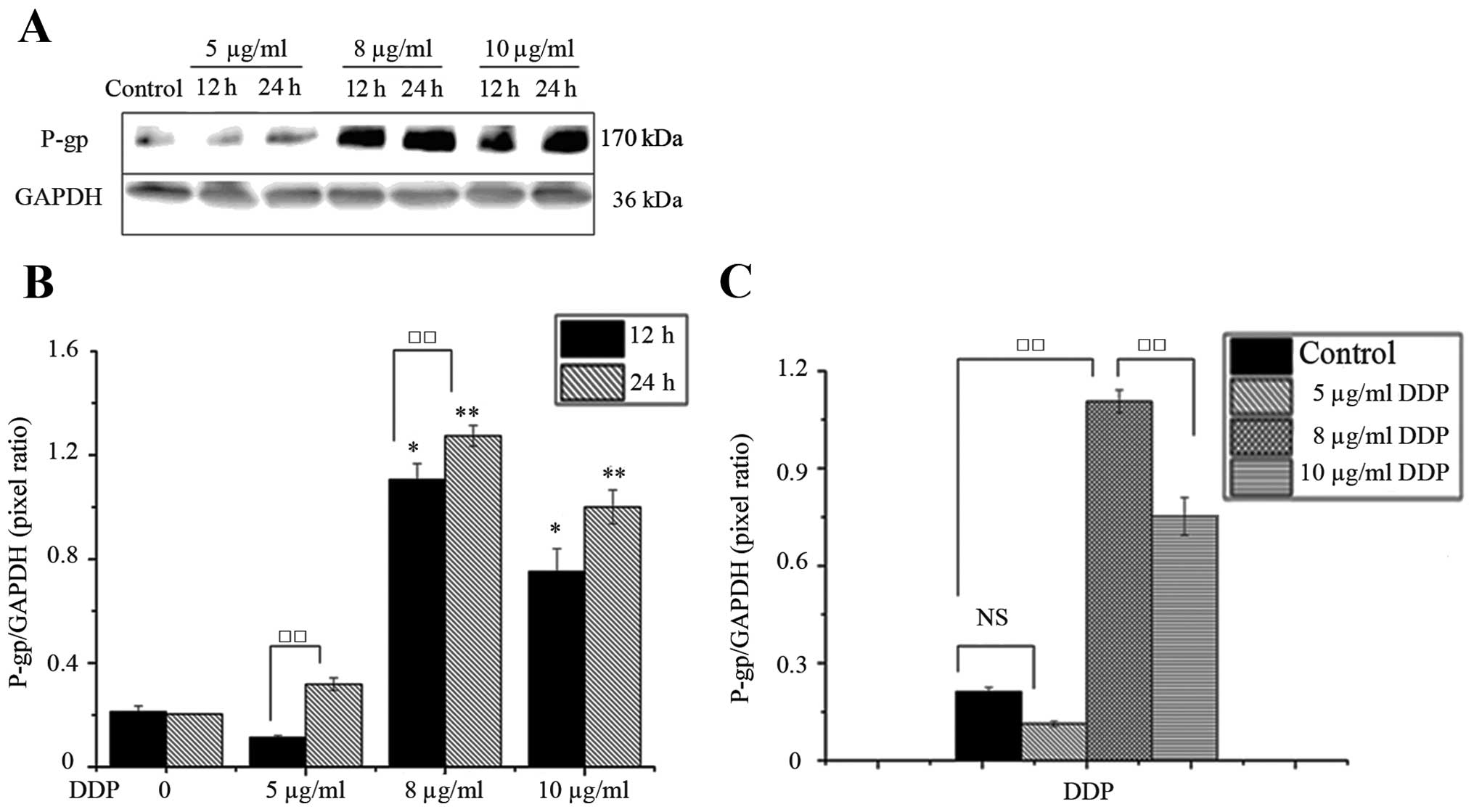
On the contrary, the expression of P-gp in SK-N-SH
cells was significantly retained and upregulated when treating with
DDP and VP16. Western blots further identified that substantial
amounts of P-gp were expressed in treatment with 8 and 10 μg/ml DDP
for 12 and 24 h in comparison to the control (Fig. 4). As shown in Fig. 4B, expression levels of P-gp, were
significantly lower in the same concentration of DDP for 24 h
compared to DDP for 12 h. Furthermore, the highest levels of P-gp
were observed in SK-N-SH cells with IC50 of DDP for 24
h. This difference was statistically significant in 5 μg/ml DDP for
12 and 24 h.
Similarly, the increase in the P-gp expression level
was detected in SK-N-SH cells that were treated with Vp16 in
comparison with the basal level identified in control cells
(Fig. 5). Furthermore, 50 and 100
μg/ml of Vp16 for 24 and 48 h elicited increase over the P-gp
expression in SK-N-SH cells. These histograms illustrate the
pattern of the dose-dependent response to various concentrations of
Vp16 (Fig. 5B and C). As shown in
Fig. 5B, the P-gp expression was
higher in 100 μg/ml Vp16 of 48 h than in 100 μg/ml Vp16 of 24 h. On
the other hand, upon IC50 of Vp16 administration for 48
h, the P-gp expression was the highest. The P-gp was consistently
induced by 150 μg/ml Vp16 (Fig.
5B).
To examine whether P-gp levels correlated
with apoptosis, western blot analyses and flow cytometry were
performed
Our results showed that DDP and Vp16 induced
apoptosis in SK-N-SH cells, and they increased the level of P-gp
expression (Fig. 1C and D). In
contrast, As2O3 in the micromolar range had a
concentration-dependent toxic effect on SK-N-SH cells with
decreased P-gp expression (Fig.
1B). In conclusion, this study demonstrates that
As2O3 is efficacious in reversing
P-gp-mediated MDR by inhibiting transport function and expression
of P-gp.
Discussion
NB is characterised as the most common extracranial
solid tumor with great malignancy that ranges from spontaneous
regression in 10% of all cases to rapid and largely treatment
resistant progression with fatal outcome (30). Despite treatment advances including
chemotherapy, radiotherapy and stem cell transplantation, advanced
NB still remains one of the most challenging problems that
physicians must deal with in pediatric oncology and long-term
survival rate is <40% (6).
However, much of the literature points to the fact that the
prolonged treatment with chemotherapy is often ineffectual due to
the appearance of MDR.
The availability of reducing MDR drugs is a
prerequisite to develop novel effective therapies. Decades ago,
As2O3 was known as a carcinogen, however,
according to research at present it now becomes a hotspot in the
treatment of tumors. The clinical use of
As2O3 as an anticancer agent is based on the
drug’s ability to induce APL cell differentiation and promotes
apoptosis. As2O3 is a poor substrate for
transport by P-glycoprotein and recommended for cancer therapy
especially in reversing the MDR in vitro, such as leukemia
(31), osteosarcoma (32), myeloma (33), gastric (34) and hepatic carcinoma (35). In addition, we found that
As2O3 lacked cross-resistance in both
P-glycoprotein- and MRP-overexpressing cell lines (36).
As2O3 partially reduce MDR to
DOX in K562/A02 cells via reducing function of efflux pumps such as
GST-p and modulate the expression of MDR-related proteins such as
Topo-II and bcl-2 (31). Although
our understanding of As2O3 clinical treatment
of MDR NB has many advantages, less is known about the effect of
As2O3 on the P-gp expression in NB.
To address the possibility that
As2O3 has a role in reducing drug resistance,
the expression levels of P-gp were assayed by western blotting. In
this study we investigated the mechanism involved in the
suppression of MDR NB. We analyzed the effect of
As2O3 on P-gp expression, in order to
investigate the mechanism involved in As2O3
treatment of MDR NB and provide new evidence for
As2O3 clinical treatment NB and thereby
reinforcing that As2O3 has potential in
clinical treatment of NB, contributing to reducing drug resistance.
To our knowledge, this is the first demonstration of
As2O3 directly affecting expression levels of
P-gp in SK-N-SH cell lines. Therefore, our approach, which utilizes
As2O3 and conventional chemotherapy drug
treatment of NB cell lines and detects the P-gp levels, provides a
more comprehensive system in which to investigate the roles of
As2O3 in apoptosis events, ultimately leading
to establishment of the possibility of As2O3
clinical treatment MDR NB. This is the first report characterizing
the P-gp alterations in SK-N-SH cells treated with
As2O3, DDP and VP16.
Herein, we report the effect of
As2O3 on cytotoxicity of SK-N-SH human
neuroblastoma cells in vitro. Following treatment with
As2O3 at different low doses for 48 h,
apoptosis was observed in SK-N-SH cells, and the apoptosis rate has
a positive correlation with acting time; coincident with the
results of Karlsson et al (28). Our data also demonstrate that
As2O3 is efficacious in inhibiting the
proliferation of SK-N-SH cells in a dose-dependent manner, with an
IC50 of ~3 μM following treatment with
As2O3 for 72 h. In this study,
As2O3 inhibited cell viability and/or induced
apoptosis in SK-N-SH cells depending on the drug concentration,
suggesting that by prolonging the time of effective drug and
increased the drug concentration, can significantly improve drug
efficacy.
In a previous report, we first demonstrated that
TrkA, TrkB and TrkC gene expression were upregulated with
increasing concentration of As2O3
administration, more apparent at 48 h than 24 h. Advanced NB tumors
carrying amplified MYCN oncogene which is the most unfavorable
prognostic factor and prominent genetic marker of high-stage
disease often exhibit an aggressive phenotype (37). Yin et al (38) found that MYCN mRNA in LA-N-5 cell
lines was strongly inhibited by treatment with different
concentrations of As2O3, and decline the most
obvious at 3 μM As2O3. This, together with
our study, indicates that As2O3 may exert a
greater anti-carcinogenesis effect on MYCN positive high-risk NB
than other drugs.
To overcome the limitation, one strategy is to
increase doses of chemotherapy drugs; however, it causes serious
toxic effects on heart, liver, kidney and hearing damage, which
patients are not able to tolerate, abandoning the treatment. When
As2O3 was combined with DDP or DOX in ALL, it
showed strong synergy with chemotherapeutic agent in vitro
and reversed the effect of DDP or DOX on multidrug resistance of
tumor cell lines (15,31,39).
In fact, a number of recent publications indicate that
As2O3 combined with DDP or DOX effect on
leukemia cells lead to synergy and reverse tumor resistance. We
speculate that the cytotoxic efficiency of
As2O3 combined with conventional chemotherapy
drugs, is important from a clinical point of view.
In conclusion, our data shows that the
IC50 value for As2O3 in SK-N-SH
cells was ~3 μM, a concentration very close to clinically
achievable concentration for successfully treating patients with
acute promyelocytic leukemia (40). Thus it is suggested that
As2O3 might be suitable in the treatment of
some instances of NB.
Suppression of cell growth by
As2O3 can be explained in part by its
capacity to affect cell cycle distribution. Cell cycle analysis has
revealed that As2O3 significantly induced a
time-dependent accumulation of cells in the G2/M phase more
significantly increased 72 h later. Therefore, to explain the
mechanism that As2O3 induced rapid and
extensive apoptosis concomitant with arrest of cells in G2/M phase
of the cell cycle, due to G2/M block, unlike G1 block, is more
toxic to the cells, and not sreversible. Thus
As2O3-induced G2/M arrest may be a
requirement for the activation of apoptotic pathways.
Park et al (41) showed that
As2O3 arrest cell cycle in G2/M phase in
leukemia U937 cells and trigger apoptosis. Gao et al (42)
showed that As2O3 could arrest NB4 cell lines
in G2/M phase, and G2/M cells which possessed higher ROS levels
were more susceptible to As2O3-induced
apoptosis (42). More strikingly,
Hassani et al (43) showed
that the G2/M arrest plays a critical role in the induction of
apoptosis by As2O3 in NB cells.
Here, we confirmed that that
As2O3 treatment resulted in accumulation of
cells in the G2/M phase in a time-dependent manner. Furthermore,
activation of caspases occurred only in
As2O3-induced mitotic cells, not in
interphase cells (36), suggesting
As2O3 has a higher cytotoxicity against NB
cells blocked in G2/M phase. This finding provides a more
comprehensive understanding of restraining cell cycle at G2/M
closely related to the initiation of apoptosis.
The exact mechanisms underlying
As2O3 that could markedly arrest cell cycle
in G2/M phase are not fully understood. It is possible that
As2O3 affects the expression of the cell
cycle regulatory elements and AKt signal transduction pathway. The
results of Han et al (44)
suggest that, the levels of cdc2 protein which is related to the
progression of G2/M phase were downregulated in cells treated with
As2O3. There are a variety of reports
suggesting that Akt is one of the important survival factors and
plays an essential role stimulating of cellular growth and
chemotherapeutic resistance to apoptosis (45,46).
PTEN as the tumor suppressor gene inactivated in a wide variety of
cancers downregulates active Akt (47) regulates many biological processes
including metabolism, growth, survival, and proliferation. Data
from Zhang et al (48) and
Li et al (49) showed that
As2O3 is associated with upregulated Wee1 and
PTEN levels and downregulated cdc2 protein and Akt signal pathway
resulting in induction of G2/M cell cycle arrest.
In addition, As2O3 treatment
blocked the cell cycle in G1 or at G2/M depending on the cell line.
We have also observed that As2O3 was able to
induce an M phase arrest of the cell cycle in MCF-7, H460, HeLa and
K562 cell lines (36), related
with tubulin aggregation, like paclitaxel (50). Liu et al (51) reported that
As2O3-induced G1 arrest in myeloma cells with
wild-type (wt) p53 but G2/M arrest in myeloma cells with mutated
p53. Thus As2O3 induces cell cycle arrest in
two distinct pathways including G1 or G2/M cell cycle arrest,
depending on varying p53 status.
Here, we confirmed that As2O3
could markedly arrest cell cycle in G2/M phase with a
time-dependent manner. Furthermore, activation of caspases occurred
only in As2O3-induced mitotic cells, not in
interphase cells (36), suggesting
As2O3 has a higher cytotoxicity against NB
cells blocked in G2/M phase. This finding provides a more
comprehensive understanding of As2O3-induced
mitotic arrest closely related to the initiation of apoptotic
pathways. Taken together, our data demonstrate that
As2O3 markedly arrests cell cycle at G2/M
phase, and induces apoptosis. Our study on cell cycle revealed that
As2O3 causes accumulation of SK-N-SH cells in
the G2/M phase, in rational combinatorial strategies involving
As2O3 in combination with vinorelbine (NVB)
that G2/M phase is sensitive to and might clinically intensify
As2O3-induced apoptosis.
Currently, the overexpression of membrane proteins
in cancer cells functioning as drug pumbs results in MDR to
chemotherapeutic agents is a major impediment to the successful
treatment of cancer. The adenosine triphosphate (ATP)-binding
cassette (ABC) membrane transporter family is well known including
ABCB1 (P-glycoprotein, P-gp), ABCC1 (multidrug-resistant protein 1,
MRP1), ABCC2 (MRP2) and ABCG2 (breast cancer resistance protein,
BCRP). As far as is know, multidrug resistance P-glycoprotein, an
ABCB1 member of the ABC transporter family, acts as a
membrane-bound energy-dependent efflux pump lowering intracellular
drug levels to sublethal concentrations and helps cells to escape
from death. P-gp which pumps a broad range of structurally
unrelated cells encoded by the MDR1 gene, overexpression of it in
neoplastic tissue is responsible for the emergence of the MDR
phenotype in cancer cells. P-gp has a basal ATPase activity and can
be activated in the presence of its substrates such as commonly
used chemotherapy drugs of VCR, DOX and Vp16, then expelling
antitumor agents from the cytoplasm by ATP hydrolysis (52). Overexpression of P-gp confers
reduced sensitivity to large group of cytotoxic drugs.
It is generally believed that the overexpression of
P-gp pumping out intracellular commonly used chemotherapy drugs in
NB chemotherapy regiments such as anthracycline, epipodophyllotoxin
and vinca alkaloids, leads to NB developing MDR. Gibalová et
al (53) indicated that P-gp,
independent of its drug efflux activity as DDP is not transportable
by P-gp, altering DDP-induced apoptosis pathway of a decrease in
DDP-induced caspase-3 activation that confers a partial loss of DDP
sensitivity. Moreover, Topo-II (54) and glutathione S-transferase π
(GSTπ) (55) overexpression
contributes to MDR formation. In contrast, acumulating evidence
indicates that P-gp has an antiapoptotic function by pumping some
caspase-dependent apoptotic substance and toxic chemicals (56,57).
Up to now, the regulatory mechanisms underlying the
expression of P-gp have not yet been well clarified. Duraj et
al (58) verified that that
mitogen-activated protein kinase (MAPK) pathway was associated with
regulatory P-gp expression in P-gp-mediated MDR. In addition, Wei
et al (52) demonstrated
that tetrandrine, an inhibitor of MAPK pathway, effectively
reverses the MDR of KBv200 and MCF-7/adr cells presumably via
downregulating P-gp expression of SK-N-SH. In addition, the
hepatocarcinogen 2-acetylaminofluorene 2-AAF could also upregulate
the human MDR1 expression in human hepatoma (59). Zhou et al (60) provided the first direct evidence
that the c-Jun NH 2-terminal kinase (JNK) pathway could indeed
downregulate P-glycoprotein in a dose- and time-dependent manner,
and reverses P-glycoprotein mediated MDR in cancer cells.
Although As2O3 had potent
reversal effect on leukemia cell P-gp-mediated MDR in vitro
(29), little information is
available regarding this compound’s effect on the P-gp expression
in NB cells. Our study aims to verify that. We actually did observe
that As2O3 inhibited P-gp expression in
SK-N-SH cells. In particular, P-gp protein levels were strongly
decreased with 2 and 3 μM As2O3 for 48, 72
and 96 h. The data in this study showed upregulation of P-gp on
SK-N-SH cells following treatment with DDP or VP16, which is
consistent with Gu et al (61) and Yang and Page (62). Collectively, our result showed high
levels of P-gp observed when SK-N-SH cells were treated with DDP
and Vp16, however, P-gp expression is strongly reduced upon
treatment with different concentrations of
As2O3. It is therefore very likely that
appropriately increased As2O3 accumulation
and acting time, P-gp expression would not be upregulated. In
general, the results presented in this study indicate
As2O3 is a poor substrate for transport by
P-glycoprotein, suggesting that As2O3 may be
useful for treatment of human solid tumors, particularly in
patients with MDR. There is evidence that decreasing protein
expression level of P-gp is one of the effective ways to reverse
P-gp-mediated MDR (52).
Therefore, these findings support the notion that
inhibition of P-gp expression may reverse the MDR phenotype through
enhancing intracellular accumulation of anticancer drugs. To
circumvent MDR, much effort has focused on finding P-gp inhibitors
and identification of molecules targeting multiple drug-resistant
mechanisms to effectively reverse P-gp-mediated MDR. However, the
failure of this type of drugs with poor clinical effectiveness has
substantially dampened the initial enthusiasm. One reasonable
explanation is the side-effects of these drugs which now has
affected its wide application in the clinic.
In conclusion, As2O3
inhibited the growth of SK-N-SH cells by inducing a G2/M arrest of
the cell cycle and by triggering apoptosis. These findings
demonstrated that As2O3 is effective and a
potential agent in reversing P-gp-mediated MDR. Our results support
As2O3 as an effective complement to
conventional chemotherapy for high-risk NB patients and possibly
also for potential use for reversing P-gp-mediated MDR when
synergistic with chemotherapy.
Acknowledgements
This study was supported by grant 2014A030313024
from the Guang Dong Natural Science Foundation; grant from the
Guangdong Science and Technology Department (2015B050501004).
References
|
1
|
Li T, Cui ZB, Ke XX, Tan J, Li FF, Li T,
Wang XW and Cui HJ: Essential role for p53 and caspase-9 in DNA
damaging drug-induced apoptosis in neuroblastoma IMR32 cells. DNA
Cell Biol. 30:1045–1050. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wolter J, Angelini P and Irwin M: p53
family: Therapeutic targets in neuroblastoma. Future Oncol.
6:429–444. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cohn SL, Pearson AD, London WB, Monclair
T, Ambros PF, Brodeur GM, Faldum A, Hero B, Iehara T, Machin D, et
al; INRG Task Force. The International Neuroblastoma Risk Group
(INRG) classification system: An INRG Task Force report. J Clin
Oncol. 27:289–297. 2009. View Article : Google Scholar :
|
|
4
|
Sato Y, Kurosawa H, Sakamoto S, Kuwashima
S, Hashimoto T, Okamoto K, Tsuchioka T, Fukushima K and Arisaka O:
Usefulness of 18F-fluorodeoxyglucose positron emission
tomography for follow-up of 13-cis-retinoic acid treatment for
residual neuroblastoma after myeloablative chemotherapy. Medicine
(Baltimore). 94:e12902015. View Article : Google Scholar
|
|
5
|
Yu AL, Gilman AL, Ozkaynak MF, London WB,
Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay
KK, et al; Children’s Oncology Group. Anti-GD2 antibody with
GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J
Med. 363:1324–1334. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lim JY, Kim YS, Kim KM, Min SJ and Kim Y:
B-carotene inhibits neuroblastoma tumorigenesis by regulating cell
differentiation and cancer cell stemness. Biochem Biophys Res
Commun. 450:1475–1480. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pettersson HM, Karlsson J, Pietras A, Øra
I and Påhlman S: Arsenic trioxide and neuroblastoma cytotoxicity. J
Bioenerg Biomembr. 39:35–41. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Keshelava N, Davicioni E, Wan Z, Ji L,
Sposto R, Triche TJ and Reynolds CP: Histone deacetylase 1 gene
expression and sensitization of multidrug-resistant neuroblastoma
cell lines to cytotoxic agents by depsipeptide. J Natl Cancer Inst.
99:1107–1119. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yonehara A, Tanaka Y, Kulkeaw K, Era T,
Nakanishi Y and Sugiyama D: Aloe vera extract suppresses
proliferation of neuroblastoma cells in vitro. Anticancer Res.
35:4479–4485. 2015.PubMed/NCBI
|
|
10
|
Goldsmith KC and Hogarty MD: Targeting
programmed cell death pathways with experimental therapeutics:
Opportunities in high-risk neuroblastoma. Cancer Lett. 228:133–141.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Street CA, Routhier AA, Spencer C, Perkins
AL, Masterjohn K, Hackathorn A, Montalvo J, Dennstedt EA and Bryan
BA: Pharmacological inhibition of Rho-kinase (ROCK) signaling
enhances cisplatin resistance in neuroblastoma cells. Int J Oncol.
37:1297–1305. 2010.PubMed/NCBI
|
|
12
|
Qiu YY, Mirkin BL and Dwivedi RS:
Inhibition of DNA methyltransferase reverses cisplatin induced drug
resistance in murine neuroblastoma cells. Cancer Detect Prev.
29:456–463. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pajic M, Norris MD, Cohn SL and Haber M:
The role of the multidrug resistance-associated protein 1 gene in
neuroblastoma biology and clinical outcome. Cancer Lett.
228:241–246. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang JM, Sheard MA, Ji L, Sposto R and
Keshelava N: Combination of vorinostat and flavopiridol is
selectively cytotoxic to multidrug-resistant neuroblastoma cell
lines with mutant TP53. Mol Cancer Ther. 9:3289–3301. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Iwasaki I, Sugiyama H, Kanazawa S and
Hemmi H: Establishment of cisplatin-resistant variants of human
neuroblastoma cell lines, TGW and GOTO, and their drug
cross-resistance profiles. Cancer Chemother Pharmacol. 49:438–444.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gillet JP, Efferth T and Remacle J:
Chemotherapy-induced resistance by ATP-binding cassette transporter
genes. Biochim Biophys Acta. 1775:237–262. 2007.PubMed/NCBI
|
|
17
|
Gesundheit B, Malach L, Or R and Hahn T:
Neuroblastoma cell death is induced by inorganic arsenic trioxide
(As (2)O (3)) and inhibited by a normal human bone marrow
cell-derived factor. Cancer Microenviron. 1:153–157. 2008.
View Article : Google Scholar
|
|
18
|
Au WY, Li CK, Lee V, Yuen HL, Yau J, Chan
GC, Ha SY and Kwong YL: Oral arsenic trioxide for relapsed acute
promyelocytic leukemia in pediatric patients. Pediatr Blood Cancer.
58:630–632. 2012. View Article : Google Scholar
|
|
19
|
Iland HJ, Bradstock K, Supple SG, Catalano
A, Collins M, Hertzberg M, Browett P, Grigg A, Firkin F, Hugman A,
et al; Australasian Leukaemia and Lymphoma Group. All-transretinoic
acid, idarubicin, and IV arsenic trioxide as initial therapy in
acute promyelocytic leukemia (APML4). Blood. 120:1570–1580; quiz
1752. 2012. View Article : Google Scholar
|
|
20
|
Haga N, Fujita N and Tsuruo T: Involvement
of mitochondrial aggregation in arsenic trioxide
(As2O3)-induced apoptosis in human
glioblastoma cells. Cancer Sci. 96:825–833. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pettersson HM, Pietras A, Munksgaard
Persson M, Karlsson J, Johansson L, Shoshan MC and Påhlman S:
Arsenic trioxide is highly cytotoxic to small cell lung carcinoma
cells. Mol Cancer Ther. 8:160–170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lallemand-Breitenbach V, Zhu J, Chen Z and
de Thé H: Curing APL through PML/RARA degradation by
As2O3. Trends Mol Med. 18:36–42. 2012.
View Article : Google Scholar
|
|
23
|
Woo SY, Lee MY, Jung YJ, Yoo ES, Seoh JY,
Shin HY, Ahn HS and Ryu KH: Arsenic trioxide inhibits cell growth
in SH-SY5Y and SK-N-AS neuroblastoma cell lines by a different
mechanism. Pediatr Hematol Oncol. 23:231–243. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim DW, Ahan SH and Kim TY: Enhancement of
arsenic trioxide (As(2)O(3))-mediated apoptosis using berberine in
human neuroblastoma SH-SY5Y Cells. J Korean Neurosurg Soc.
42:392–399. 2007. View Article : Google Scholar
|
|
25
|
Florea AM, Splettstoesser F and Büsselberg
D: Arsenic trioxide (As2O3) induced calcium
signals and cytotoxicity in two human cell lines: SY-5Y
neuroblastoma and 293 embryonic kidney (HEK). Toxicol Appl
Pharmacol. 220:292–301. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Karlsson J, Edsjö A, Påhlman S and
Pettersson HM: Multidrug-resistant neuroblastoma cells are
responsive to arsenic trioxide at both normoxia and hypoxia. Mol
Cancer Ther. 4:1128–1135. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ryu KH, Woo SY, Lee MY, Jung YJ, Yoo ES,
Seoh JY, Kie JH, Shin HY and Ahn HS: Morphological and biochemical
changes induced by arsenic trioxide in neuroblastoma cell lines.
Pediatr Hematol Oncol. 22:609–621. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Karlsson J, Øra I, Pörn-Ares I and Påhlman
S: Arsenic trioxide-induced death of neuroblastoma cells involves
activation of Bax and does not require p53. Clin Cancer Res.
10:3179–3188. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qian XP, Liu BR, Yin HT, Wang LF, Zou ZY
and Du J: Effect of arsenic trioxide on drug transporting molecules
in acute promyelocytic leukemia cell line. Zhonghua Zhong Liu Za
Zhi. 26:601–605. 2004.(In Chinese).
|
|
30
|
Lang WH and Sandoval JA: Detection of PI3K
inhibition in human neuroblastoma using multiplex luminex bead
immunoassay: A targeted approach for pathway analysis. J Biomol
Screen. 19:1235–1245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao D, Jiang Y, Dong X, Liu Z, Qu B,
Zhang Y, Ma N and Han Q: Arsenic trioxide reduces drug resistance
to adriamycin in leukemic K562/A02 cells via multiple mechanisms.
Biomed Pharmacother. 65:354–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhao H, Guo W, Peng C, Ji T and Lu X:
Arsenic trioxide inhibits the growth of adriamycin resistant
osteosarcoma cells through inducing apoptosis. Mol Biol Rep.
37:2509–2515. 2010. View Article : Google Scholar
|
|
33
|
Abeltino M, Bonomini S, Bolzoni M, Storti
P, Colla S, Todoerti K, Agnelli L, Neri A, Rizzoli V and Giuliani
N: The proapoptotic effect of zoledronic acid is independent of
either the bone microenvironment or the intrinsic resistance to
bortezomib of myeloma cells and is enhanced by the combination with
arsenic trioxide. Exp Hematol. 39:55–65. 2011. View Article : Google Scholar
|
|
34
|
Xue YW, Han JG, Li BX and Yang BF:
Reversal effect and mechanism of arsenic trioxide on multidrug
resistance of gastric carcinoma cells SGC7901. Yao Xue Xue Bao.
42:949–953. 2007.(In Chinese). PubMed/NCBI
|
|
35
|
Chan JY, Siu KP and Fung KP: Effect of
arsenic trioxide on multidrug resistant hepatocellular carcinoma
cells. Cancer Lett. 236:250–258. 2006. View Article : Google Scholar
|
|
36
|
Ling YH, Jiang JD, Holland JF and
Perez-Soler R: Arsenic trioxide produces polymerization of
microtubules and mitotic arrest before apoptosis in human tumor
cell lines. Mol Pharmacol. 62:529–538. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Paffhausen T, Schwab M and Westermann F:
Targeted MYCN expression affects cytotoxic potential of
chemotherapeutic drugs in neuroblastoma cells. Cancer Lett.
250:17–24. 2007. View Article : Google Scholar
|
|
38
|
Yin H, Tang SQ and Zhao QY: Effect of
arsenic trioxide on MYCN mRNA expression in neuroblastoma cells.
Shandong Med J. 37:57–58. 2008.
|
|
39
|
D’Aguanno S, D’Alessandro A, Pieroni L,
Roveri A, Zaccarin M, Marzano V, De Canio M, Bernardini S, Federici
G and Urbani A: New insights into neuroblastoma cisplatin
resistance: A comparative proteomic and meta-mining investigation.
J Proteome Res. 10:416–428. 2011. View Article : Google Scholar
|
|
40
|
Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM,
Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, et al: Use of arsenic
trioxide (As2O3) in the treatment of acute
promyelocytic leukemia (APL): II. Clinical efficacy and
pharmacokinetics in relapsed patients. Blood. 89:3354–3360.
1997.PubMed/NCBI
|
|
41
|
Park JW, Choi YJ, Jang MA, Baek SH, Lim
JH, Passaniti T and Kwon TK: Arsenic trioxide induces G2/M growth
arrest and apoptosis after caspase-3 activation and bcl-2
phosphorylation in promonocytic U937 cells. Biochem Biophys Res
Commun. 286:726–734. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gao F, Yi J, Yuan JQ, Shi GY and Tang XM:
The cell cycle related apoptotic susceptibility to arsenic trioxide
is associated with the level of reactive oxygen species. Cell Res.
14:81–85. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hassani S, Ghaffari SH, Zaker F, Mirzaee
R, Mardani H, Bashash D, Zekri A, Yousefi M, Zaghal A, Alimoghaddam
K, et al: Azidothymidine hinders arsenic trioxide-induced apoptosis
in acute promyelocytic leukemia cells by induction of p21 and
attenuation of G2/M arrest. Ann Hematol. 92:1207–1220. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Han YH, Kim SZ, Kim SH and Park WH:
Arsenic trioxide inhibits the growth of Calu-6 cells via inducing a
G2 arrest of the cell cycle and apoptosis accompanied with the
depletion of GSH. Cancer Lett. 270:40–55. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kandasamy K and Srivastava RK: Role of the
phosphatidylinositol 3′-kinase/PTEN/Akt kinase pathway in tumor
necrosis factor-related apoptosis-inducing ligand-induced apoptosis
in non-small cell lung cancer cells. Cancer Res. 62:4929–4937.
2002.PubMed/NCBI
|
|
46
|
Chen X, Thakkar H, Tyan F, Gim S, Robinson
H, Lee C, Pandey SK, Nwokorie C, Onwudiwe N and Srivastava RK:
Constitutively active Akt is an important regulator of TRAIL
sensitivity in prostate cancer. Oncogene. 20:6073–6083. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chalhoub N and Baker SJ: PTEN and the
PI3-kinase pathway in cancer. Annu Rev Pathol. 4:127–150. 2009.
View Article : Google Scholar :
|
|
48
|
Zhang X, Jia S, Yang S and Yang Y, Yang T
and Yang Y: Arsenic trioxide induces G2/M arrest in hepatocellular
carcinoma cells by increasing the tumor suppressor PTEN expression.
J Cell Biochem. 113:3528–3535. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li Y, Qu X, Qu J, Zhang Y, Liu J, Teng Y,
Hu X, Hou K and Liu Y: Arsenic trioxide induces apoptosis and G2/M
phase arrest by inducing Cbl to inhibit PI3K/Akt signaling and
thereby regulate p53 activation. Cancer Lett. 284:208–215. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li YM and Broome JD: Arsenic targets
tubulins to induce apoptosis in myeloid leukemia cells. Cancer Res.
59:776–780. 1999.PubMed/NCBI
|
|
51
|
Liu Q, Hilsenbeck S and Gazitt Y: Arsenic
trioxide-induced apoptosis in myeloma cells: p53-dependent G1 or
G2/M cell cycle arrest, activation of caspase-8 or caspase-9, and
synergy with APO2/TRAIL. Blood. 101:4078–4087. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wei N, Sun H, Wang F and Liu G: H1, a
novel derivative of tetrandrine reverse P-glycoprotein-mediated
multidrug resistance by inhibiting transport function and
expression of P-glycoprotein. Cancer Chemother Pharmacol.
67:1017–1025. 2011. View Article : Google Scholar
|
|
53
|
Gibalová L, Sereš M, Rusnák A, Ditte P,
Labudová M, Uhrík B, Pastorek J, Sedlák J, Breier A and Sulová Z:
P-glycoprotein depresses cisplatin sensitivity in L1210 cells by
inhibiting cisplatin-induced caspase-3 activation. Toxicol In
Vitro. 26:435–444. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Pommier Y, Leteurtre F, Fesen MR, Fujimori
A, Bertrand R, Solary E, Kohlhagen G and Kohn KW: Cellular
determinants of sensitivity and resistance to DNA topoisomerase
inhibitors. Cancer Invest. 12:530–542. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Burg D, Riepsaame J, Pont C, Mulder G and
van de Water B: Peptide-bond modified glutathione conjugate analogs
modulate GSTpi function in GSH-conjugation, drug sensitivity and
JNK signaling. Biochem Pharmacol. 71:268–277. 2006. View Article : Google Scholar
|
|
56
|
Lee WK, Torchalski B, Kohistani N and
Thévenod F: ABCB1 protects kidney proximal tubule cells against
cadmium-induced apoptosis: Roles of cadmium and ceramide transport.
Toxicol Sci. 121:343–356. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhang X, Wu X, Li J, Sun Y, Gao P, Zhang
C, Zhang H and Zhou G: MDR1 (multidrug resistence 1) can regulate
GCS (glucosylceramide synthase) in breast cancer cells. J Surg
Oncol. 104:466–471. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Duraj J, Zazrivcova K, Bodo J, Sulikova M
and Sedlak J: Flavonoid quercetin, but not apigenin or luteolin,
induced apoptosis in human myeloid leukemia cells and their
resistant variants. Neoplasma. 52:273–279. 2005.PubMed/NCBI
|
|
59
|
Kuo MT, Liu Z, Wei Y, Lin-Lee YC, Tatebe
S, Mills GB and Unate H: Induction of human MDR1 gene expression by
2-acetylaminofluorene is mediated by effectors of the
phosphoinositide 3-kinase pathway that activate NF-kappaB
signaling. Oncogene. 21:1945–1954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhou J, Liu M, Aneja R, Chandra R, Lage H
and Joshi HC: Reversal of P-glycoprotein-mediated multidrug
resistance in cancer cells by the c-Jun NH2-terminal kinase. Cancer
Res. 66:445–452. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Gu J, Tang Y, Liu Y, Guo H, Wang Y, Cai L,
Li Y and Wang B: Murine double minute 2 siRNA and wild-type p53
gene therapy enhances sensitivity of the SKOV3/DDP ovarian cancer
cell line to cisplatin chemotherapy in vitro and in vivo. Cancer
Lett. 343:200–209. 2014. View Article : Google Scholar
|
|
62
|
Yang X and Pagé M: P-glycoprotein
expression in ovarian cancer cell line following treatment with
cisplatin. Oncol Res. 7:619–624. 1995.PubMed/NCBI
|