Introduction
Colon cancer is one of the most common malignacies
and the fourth most frequent cause of cancer death (1–4).
Metastasis is a major cause of death in colon cancer patients.
Although the 5-year survival rate for colon cancer patients in
early stages without metastasis is high, this rate drops
significantly in patients diagnosed with regional lymph node
metastases and further decreases to less than 10% in patients with
distant metastases (5–7). Major efforts in the recent years to
improve the therapy for metastatic colon cancer patients include
the development of drugs that target antiangiogenesis and inhibit
EGFR signaling with noticeable success (8–10).
Despite such progress, major challenges still remain in clinical
treatment of colon cancer with distant metastasis. Understanding of
the biological properties of the metastastic cancer cells and the
underlying mechanisms would provide an important basis for the
development of more effective agents and therapeutic
strategies.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is
a key enzyme that catalyzes the redox reaction in the glycolytic
pathway by converting glyceraldehyde-3-phosphate to
1,3-bisphosphoglycerate with a reduction of NAD+ to
NADH. Although GAPDH has long been considered as a house-keeping
enzyme and thus, commonly used as an internal reference in western
blotting and RT-PCR analyses (11,12),
accumulating evidence suggest that GAPDH may also play
non-enzymatic roles with diverse functions and distinct subcellular
distributions including cytoplasma, cell membranes and nucleus
(13–16). Its upregulation seems related to
cancer development, evident by its higher expression in Dunning
R-3327 rat prostatic adenocarcinoma cells compared to normal rat
ventral prostate tissue (17).
Furthermore, our previous study showed that GAPDH expression was
elevated in colon cancer tissue and further increased in metastasis
liver tissue (18), which suggests
that GAPDH may contribute to colon cancer metastasis. These results
indicate an important role of GAPDH in tumor growth and metastasis,
and therefore may be a potential candidate as a molecular target
for cancer therapy. However, the mechanism by which GAPDH promotes
cancer growth and metastasis remains unclear.
The purpose of the present study was to use both
in vitro and in vivo experimental systems to evaluate
the role of GAPDH in colon cancer metastasis and to investigate the
underlying mechanism. Both molecular and biochemical approaches
were employed to test the biological consequences of GAPDH
silencing and the relevant regulatory events. Since
epithelial-mesenchymal transition (EMT) is tightly associated with
the key events during cancer cell detachment from primary tumor
site and seems to endow cell mobility and invasiveness (19), we evaluated the potential link
between GAPDH and EMT in this study. Our results showed that GAPDH
could physically interact with the transcriptional factor Sp1 and
promote the expression of SNAIL, leading to EMT and an increased
cell mobility and cancer metastasis.
Materials and methods
Cell culture and transfection
Human colon cell lines HCT116 and LoVo cells were
purchased from the ATCC (Manassas, VA, USA). HT29 cell line was
obtained from Shanghai Cell Bank (Shanghai, China). The
GAPDH-knockdown HCT116 and LoVo cells (designated as HCT116 sh-GAP
and LoVo sh-GAP, respectively) were established by stable
transfection with short hairpin RNA (shRNA) against GAPDH as
described below. The control cells (designated as HCT116-NC and
LoVo-NC) were transfected with scrambled shRNA. HCT116 sh-GAP and
HCT116-NC cells were cultured in McCoy's 5A medium supplemented
with 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO,
USA). LoVo sh-GAP and LoVo-NC were cultured in F12K medium
supplemented with 10% FBS (Sigma-Aldrich). Cells were incubated at
37°C in a humidified atmosphere with 5% CO2 and were
routinely sub-cultured using 0.25% (w/v) trypsin-EDTA solution to
detach cells from the culture surface.
The short hairpin RNA (shRNA) expression plasmids
(PGLV-H1-GFP/Puro) and lentiviral vectors containing GAPDH-shRNA
(5′-GTATGACAACAGCCTCAAG-3′) or scrambled shRNA
(5′-TTCTCCGAACGTGTCACG-3′) were obtained from Shanghai GenePharma,
Co., Ltd. (Shanghai, China). HCT116 and LoVo cells were infected
with lentiviral particles using the protocol recommended by the
manufacturer, and the transfected cells were selected using 2.5
µg/ml puromycin. Silencing to GAPDH expression was confirmed
by qRT-PCR and western blot analysis.
Antibodies and reagents
The following antibodies were used for
immunoblotting analyses using standard western blotting procedures:
E-cadherin, vimentin, β-catenin, SNAIL, α-tubulin (all from Cell
Signaling Technology, Beverly, CA, USA), GAPDH (Abcam, Cambridge,
UK), NOTCH3 (Cell Signaling Technology). Anti-mouse and anti-rabbit
secondary antibodies conjugated with HRP were purchased from Santa
Cruz Biotechnology (Santa Cruz, CA, USA). Matrigel and Transwell
chambers were purchased from BD Biosciences (San Jose, CA, USA).
TRIzol reagent and puromycin were purchased from Invitrogen
(Carlsbad, CA, USA).
Assays of cellular metabolism
Lactate and glucose concentrations in the cell
culture medium were measured using a SBA-40D analyzer (Institute of
Biology, Shandong Academy of Sciences (Jinan, China). Each
experiment was repeated at least three times independently.
Cellular oxygen consumption rate (OCR) and extracellular
acidification rate (ECAR) were measured by XF24 analyzer (Seahorse
Bioscience, Inc., North Billerica, MA, USA). Briefly, cells were
seeded in McCoy'5A medium at 40,000 cells/well in XF24 cell plate
24 h before the assay. On the day of the assay, the medium was
changed to DMEM (without serum, no glucose, no bicarbonate,
supplemented with 2 mM glutamine), and incubated at 37°C in an
incubator without CO2 for 1 h before measurement of OCR
and ECAR. Glucose (10 mM), oligomycin (3 mM) and 2-DG (0.1 M) were
prepared in the Dulbecco's modified Eagle's medium (DMEM) and
loaded onto ports A, B and C, respectively. Metabolic analysis was
performed using the glycolytic stress test protocol recommended by
the manufacturer. Glycolytic rate was estimated from the ECAR after
the injection of glucose. Maximum glycolytic capacity was
calculated by the rate of increase in ECAR after the injection of
oligomycin following glucose. The glycolytic reserve was estimated
using the difference between maximum glycolytic capacity and
glycolytic rate. Glycolytic responds was determined using the ratio
of maximum glycolytic capacity to glycolysis rate. Each assay was
repeated at least 3 times.
Assay of GAPDH enzyme activities
GAPDH enzyme activity was measured using an assay
kit (AM1639) purchased from Thermo Fisher Scientific. Cells were
plated in 96-well plate (5,000 cells/well), 200 µl KDalert
lysis buffer was added to each well, and the samples were then
incubated at 4°C for 15 min. The cell lysate was homogenized by
pipetting up and down 4–5 times, and 10 µl of each lysate
was transfered to a clean 96-well dark plate. Next, 90 µl of
KDalert Master Mix was added to each well. The increase in
fluorescence (the excitation wavelength at 560 nm and the emission
wavelength at 590 nm) was measured every minute for up to 8 min at
room temperature, using a Fluoroskan spectrometer.
Cell proliferation and colony formation
assays
Cell proliferation was assessed using MTT assay.
After the cells in 96-well plates were incubated with the MTT
reagent (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide) at 37°C for 3 h, the culture medium was removed, and then
200 µl dimethyl sulfoxide (DMSO) was added to dissolve the
formazan. The OD values were recorded at 569 nm using a microplate
reader (Bio-Rad Laboratories, Hercules, CA, USA). For colony
formation assay, cells were seeded in a 6-well plate (500
cells/well) and were cultured in McCoy'5A medium (HCT116 cells) or
F12 K medium (LoVo cells) supplemented with 10% FBS for 14 days.
The cells were washed twice with phosphate-buffered saline (PBS),
fixed with methanol for 15 min, and the colonies were stained with
crystal violet for 15 min at room temperature. After the samples
were washed with water and air-dried, colonies of >50 cells were
counted.
RNA isolation and qRT-PCR
Total RNA was extracted from cultured cells with
TRIzol and RNA concentration was calculated by measuring the OD
value at 260 nm. RNA (0.5–1 µg) was reverse-transcribed
using an M-MLV Reverse Transcriptase kit according to the
manufacturer's protocol. The resulting cDNA (20 ng) was mixed with
SYBR-Green Master Mix, amplified by PCR with the respective
primers, and analyzed using the Universal Probe Library Center
software (Bio-Rad Laboratories). The PCR primers used in the
present study are listed in Table
I.
 | Table IThe primers used for qRT-PCR in the
present study. |
Table I
The primers used for qRT-PCR in the
present study.
| Genes | Forward primer | Reverse primer |
|---|
| GAPDH |
5′-AATGGGCAGCCGTTAGGAAA-3′ |
5′-GCCCAATACGACCAAATCAGAG-3′ |
| Actin |
5′-AACTCCATCATGAAGTGTGACG-3′ |
5′-GATCCACATCTGCTGGAAGG-3′ |
| ABCG2 |
5′-AGGCAGATGCCTTCTTCGTT-3′ |
5′-TGAGATTGACCAACAGACCATCA-3′ |
| ALDH1 |
5′-GGCCCTCAGATTGACAAGGA-3′ |
5′-AACACTGTGGGCTGGACAAA-3′ |
| NOTCH1 |
5′-GACAGCCTCAACGGGTACAA-3′ |
5′-CACACGTAGCCACTGGTCAT-3′ |
| NOTCH3 |
5′-GCCAAGCGGCTAAAGGTAGA-3′ |
5′-ATTAGCGGGGTGAAGCCATC-3′ |
| NANOG |
5′-CAATGGTGTGACGCAGGGAT-3′ |
5′-TGCACCAGGTCTGAGTGTTC-3′ |
| SOX2 |
5′-GGGGAAAGTAGTTTGCTGCC-3′ |
5′-CGCCGCCGATGATTGTTATT-3′ |
| BMI1 |
5′-CTGGTTGCCCATTGACAGCG-3′ |
5′-AAAAATCCCGGAAAGAGCAGCC-3′ |
| SNAIL |
5′-CGAGTGGTTCTTCTGCGCTA-3′ |
5′-CTGCTGGAAGGTAAACTCTGGA-3′ |
| α-Tublin |
5′-GTCTCGCGTTGTTCTCTGGG-3′ |
5′-GCACTCACGCATGTTTTCCC-3′ |
Western blot analysis and
co-immunoprecipitation (IP)
For preparation of cell extracts, cells were washed
twice with cold PBS, and lysed in RIPA lysis buffer containing
protease inhibitors and phosphatase inhibitors on ice for 20 min.
Proteins (30 µg) were separated by electrophoresis using 10%
SDS-PAGE. After electrophoresis, proteins were transferred to
polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA),
and incubated with the following primary antibodies at the
dilutions recommended by the manufacturers: E-cadherin (1:1,000),
vimentin (1:1,000), β-catenin (1:1,000), SNAIL (1:1,000), GAPDH
(1:10,000) or NOTCH3 (1:1,000). The samples were incubated
overnight at 4°C on a rocking-platform, and then incubated with
horseradish peroxidase-conjugated secondary antibody (dilution,
1:10,000) for 1 h at room temperature. Protein bands were
visualized using enhanced chemiluminescence reagent ECL (Nanjing
KeyGen Biotech, Co., Ltd., Nanjing, China). α-Tubulin was also
probed as a loading control. Co-immunoprecipitation assays were
performed using the Pierce Co-immunoprecipitation kits (Thermo
Fisher Scientific, Rockford, IL, USA). Cell lysates were
solubilized using Nonidet P-40 at a final concentration of 0.5%.
Soluble fractions (200–600 µg) from cell lysates were
incubated overnight at 4°C with antibody-immobilized beads. Beads
were then centrifuged at 1000 × g for 1 min, and washed three times
with PBS before elution. The eluted proteins were heated at 90°C
for 5 min and analyzed by western blotting.
Chromatin immunoprecipitation (ChIP)
The kit was used for ChIP assays according to the
protocol recommended by the manufacturer (Active Motif, Carlsbad,
CA, USA). Cells (5×106) were cross-linked with 1%
formaldehyde, and DNA was isolated and sheared to an approximate
length of 200–1000 bp by sonication using an SLPe sonicator
(Branson Ultrasonics Corp., Danbury, CT, USA). Sheared DNA was
incubated with 1.5 µg anti-GAPDH antibody or control IgG
overnight at 4°C, followed by immunoprecipitation with 20 µl
of protein A beads. Enriched DNA was extracted from the
DNA/antibody/protein A/beads complexes by proteinase K digestion
and purified using spin columns. Promoter sequence was analyzed by
PCR. SNAIL minimal promoter primers were as follows: forward
primer, ATTCGTGGGTGCTCAAGAGG (nucleotide positions −103 to −84);
reverse primer, GCCCAGTCCTGGTGAATCTC (nucleotide positions +31 to
+50).
Cell migration and invasion assays
For 'wound healing' assay, cells were seeded in a
6-well culture plate, grown to 80% confluence, and then the plate
was scratched across the surface of the cell monolayer with a
sterile pipette tip. The cell debris was removed by washing with
fresh medium. At the indicated time points (24–48 h), the cells
that migrated into the wounded area or protruded from the border of
the scratched area were visualized and photographed under an
inverted microscope.
Cell migration ability was further assessed using
6.5-mm Transwell chambers. Cell invasion was assessed using the
Matrigel invasion chambers (24-well DI kit from BD Biosciences).
The assays were performed according to the manufacturer's
instructions. Briefly, 5.0×105 cells were suspended in
serum-free medium and seeded into the upper chamber. The lower
chamber was filled with medium containing 10% FBS. After 24 h of
incubation, the migrated/invaded cells that passed the filter were
fixed with 4% paraformaldehyde, stained with crystal violet
solution and counted under a microscope. The cell counts in five
random fields were averaged for each filter from triplicate
experiments.
Phalloidine staining of F-actin
Cells were fixed with absolute ethyl alcohol for 20
min and permeabilized with 0.25% of Triton X-100 in PBS at room
temperature for 10 min. Samples were blocked with 1% of bovine
serum albumin (BSA; Sigma-Aldrich) and incubated with fluorescent
phalloidine for 30 min in the dark environment at room temperature,
then washed with PBS for 3 times. The samples were also stained
with DAPI for 3 min in the dark environment at room temperature and
washed with PBS for 3 times to reveal the nuclei. Cells were
observed under a scanning confocal microscope (Olympus FV10i;
Olympus).
Xenograft model of tumor growth and
metastasis
To evaluate the impact of GAPDH on tumor growth
in vivo, 1.5×106 cells (NC control or sh-GAP)
were subcutaneously innoculated into the left or right axilla of
BALB/C nude mice (5 mice/group). Tumor length (L) and width (W)
were measured twice a week using a caliper, and tumor volume was
calculated using the formula: V = (W2 × l)/2. After 3
weeks, the mice were sacrificed and the tumors were dissected for
comparison. To analyze metastasis, 1.5×106 HCT116-NC or
HCT116 sh-GAP cells were injected into BALB/C nude mice (12
mice/group) through tail vein. After one and a half months, mice
were sacrificed and the lungs were isolated and inspected for
metastasis. Both primary and metastatic tumors were isolated for
imaging and pathological/histological analyses. The animal
experiments were performed in complaince with the protocol and
procedures approved by the Institutional Animal Care and Use
Committee of Sun Yat-sen University Cancer Center.
Tumor specimens, H&E staining and
immunohistochemistry
Tumor specimens embedded in paraffin slides were
subjected to H&E staining and immunohistochemistry as
previously described (18). For
immunohistochemistry, paraffin embedded specimens were first
treated with 3% H2O2 for 30 min to quench the
endogenous peroxidase activity. The slides were then immersed in
citrate buffer, heated for 5 min at 100°C. After cooling, blocking
with 10% goat serum. Rabbit monoclonal antibody against human
GAPDH, biotinylated goat anti-rabbit antibody, and
streptavidin-peroxidase conjugate were added sequentially. After
the incubation and washing, the tissue slides were stained with the
DAB substrate kit, and then stained with hematoxylin.
Statistical analysis
Statistical analysis was performed using the
GraphPad Prism software package (v.4.02; GraphPad Software, Inc.,
La Jolla, CA, USA). The Student's t-test was used to evaluate the
statistical significance of the two mean values between two groups.
A P<0.05 was considered statistically significant.
Results
Suppression of GAPDH by shRNA and its
effect on glucose metabolism
To investigate the impact of GAPDH on cancer cell
behavior, we first constructed GAPDH knockdown cell lines by stable
transfection of HCT116 and LoVo cells with lentivirus vectors
containing either shRNA against GAPDH (sh-GAP) or scramble shRNA as
a negative control (NC). Stable transfection with sh-GAP
significantly reduced the expression of GAPDH in both HCT116 and
LoVo cells, as revealed by RT-PCR and western blot analysis
(Fig. 1A and B). GAPDH knockdown
by shRNA consistently reduced GAPDH activity in both HCT116 and
LoVo cells (Fig. 1C). As GAPDH is
an important glycolytic enzyme (20), we evaluated glycolytic activity
after silencing of GAPDH expression by shRNA, and showed that the
sh-GAP cells consumed less glucose and produced less lactate than
the NC cells in both HCT116 and LoVo cell pairs (Fig. 1D). These metabolic changes were
futher confirmed using a Seahorse XF analyzer. As shown in Fig. 1E, the basal glycolytic activity
(basal extracellular acidification rate, ECAR) was lower in HCT116
sh-GAP cells than in HCT116-NC cells, and there was significantly
less increase in ECAR in HCT116 sh-GAP cells than in HCT116-NC
cells after glucose was injected to the culture medium, indicating
that GAPDH knockdown substantially impaired glycolytic capacity.
Furthermore, we examined the maximum glycolytic capacity in these
two cell lines by using oligomycin to inhibit oxidative
phosphorylation. Our results showed that the maximum glycolytic
capacity decreased substantially after GAPDH silencing, and that
glycolytic reserve was also significantly reduced after GAPDH
knockdown in HCT116 cells (Fig.
1E–G). Oxygen consumption rate (OCR) also decreased in HCT116
sh-GAP cells compared with HCT116-NC cells (data not shown).
Notably, unlike the control cells that showed an increase in OCR
when exogenous glucose was added, injection of glucose into sh-GAP
cells did not increase OCR, suggesting that silencing of GAPDH
compromised the ability of cells to use glucose as a substrate for
oxidative metabolism in the mitochondria.
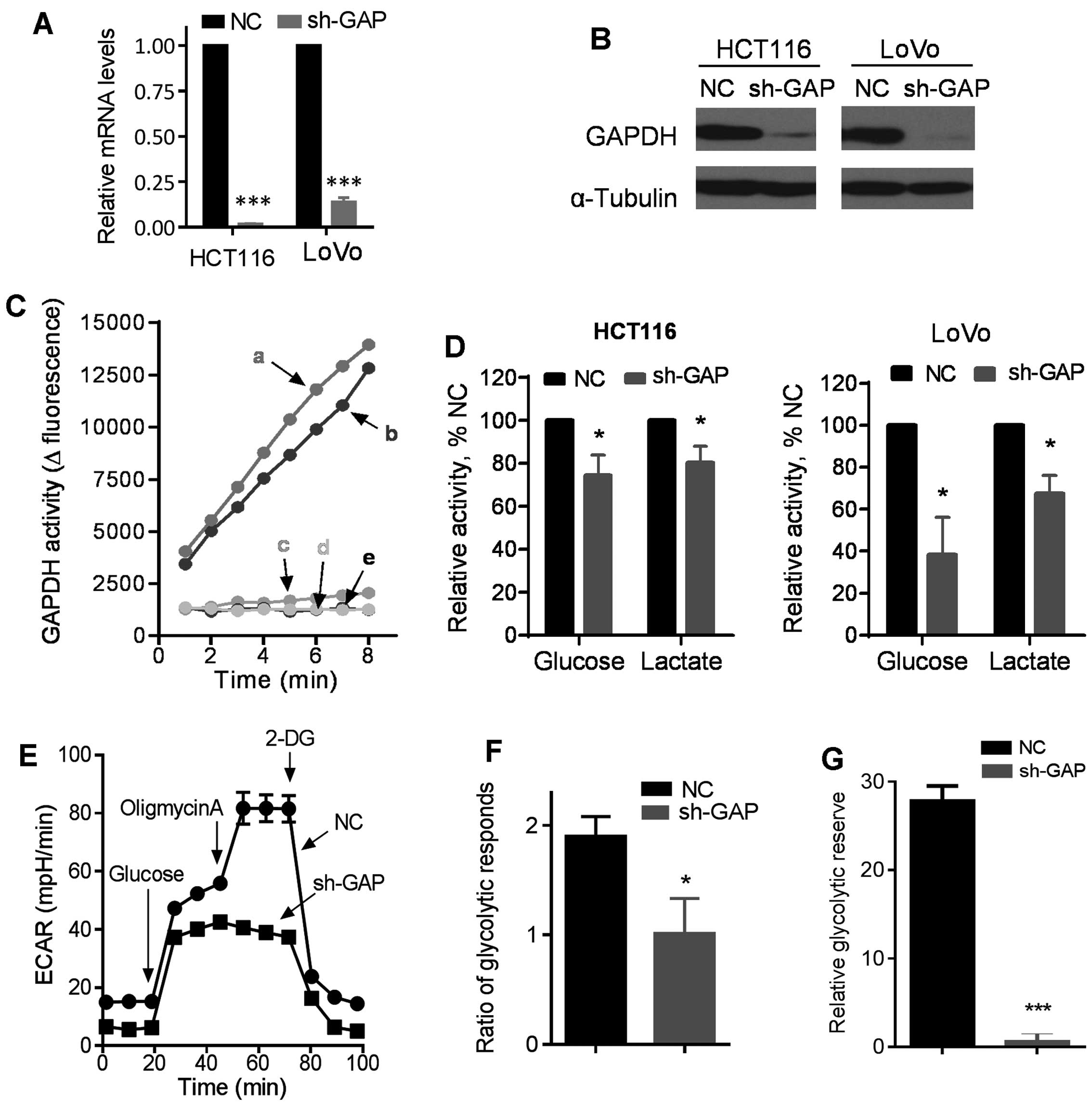 | Figure 1Impact of GAPDH silencing by shRNA on
glucose metabolism in colon cancer cells. (A) Expression of GAPDH
mRNA in HCT116 and LoVo cells infected with lentiviral vector
containing shRNA (sh-GAP) or negative control shRNA (NC). (B)
Western blot analysis of GAPDH levels in sh-GAP and NC cells.
α-Tubulin was blotted as the protein loading control. (C) GAPDH
enzyme activity was measured in sh-GAP and NC cell lines. a, b, c,
d and e represent LoVo-NC, HCT116-NC, HCT116 sh-GAP, LoVo sh-GAP
and black control, respectively. (D) Comparison of glucose
consumption and lactate production in the medium of sh-GAP and NC
cells in both HCT116 and LoVo cell pairs after cells were cultured
in fresh medium for 12 h. (E) Glycolysis was measured by an XF24
metabolic analyzer using the glycolytic test kit provided by the
manufacturer. A representative ECAR outputs in response to glucose,
oligomycin, and 2-DG in HCT116 sh-GAP cells in comparison with
HCT116-NC cells are shown. (F) Glycoytic respond ratios in HCT116
sh-GAP and HCT116NC were calculated from three separate
experiments. (G) Relative glycoytic reserve calculated from three
separate experiments. Data are shown as mean ± SD. n=3, *P<0.05,
***P<0.001. |
Effect of GAPDH on cancer cell
proliferation and migration/invasion
Uncontrolled proliferation is a characteristic
behavior of cancer cells. MTT and colony formation assays were
performed to test the effect of GAPDH silencing on cell
proliferation in HCT116 and LoVo cells. As shown in Fig. 2A, cell proliferation was markedly
retarded in sh-GAP cells compared to the NC cells in both HCT116
and LoVo cell pairs. Furthermore, GAPDH silencing markedly reduced
colony formation ability in both HCT116 and LoVo cell lines, as
evidenced by the small size and low number of colonies in the
sh-GAP cells compared to NC cells (Fig. 2B and C).
As GAPDH depletion inhibits cell proliferation and
colony formation in vitro, we further evaluated the impact
of GAPDH on tumor formation in vivo. HCT116 sh-GAP and LoVo
sh-GAP cells were inoculated subcutaneously into athymic nude mice
to initiate tumor formation, and the same numbers of the respective
NC cell lines were also inoculated in the same fashion as control
groups. Tumor growth was monitored for three weeks. As shown in
Fig. 2D, tumor growth was
significantly retarded in sh-GAP group compared to the NC group. By
the end of week 3, the animals were sacrificed and the tumors were
isolated for comparison (Fig. 2E).
The data confirmed that suppression of GAPDH expression
significantly inhibited tumor growth in vivo.
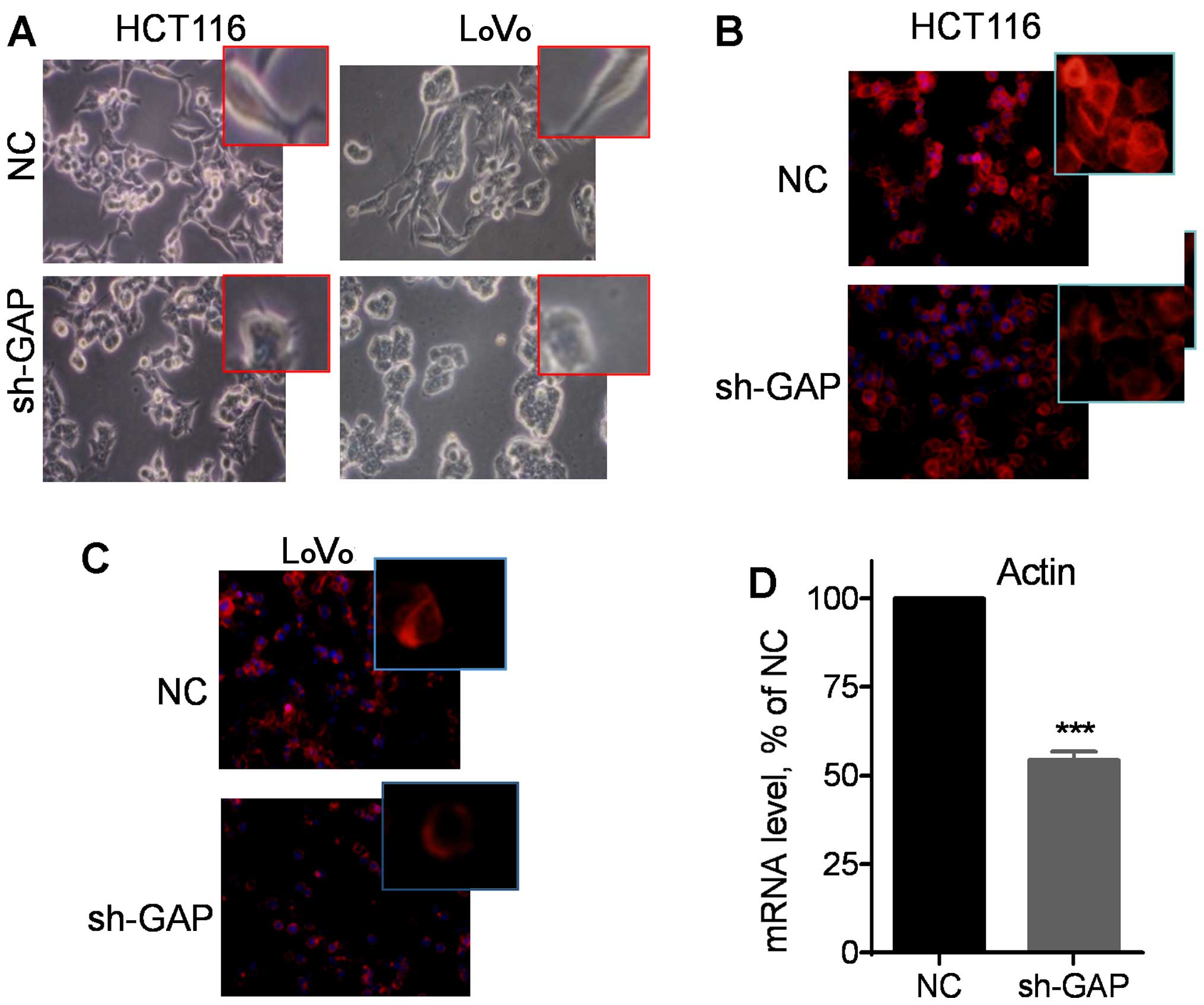
Notably, microscopic examination revealed that
silencing of GAPDH expression led to apparent changes in cell
morphology from the original spindle-like mesenchymal shape to a
more polygonal epithelial appearance (Fig. 3A). Fluorescent staining of F-actin
using phalloidin revealed that suppression of GAPDH substantially
reduced the amount of F-actin in the cytoskeleton filaments, as
evidenced by a significant decrease in fluorescent intensity in
HCT116 sh-GAP cells (Fig. 3B) and
LoVo sh-GAP cells (Fig. 3C).
Consistently, quantitative analysis of gene expression by qRT-PCR
using a pair of primers for both β-actin and γ-actin showed a
significant reduction in actin mRNA expression after silencing of
GAPDH (Fig. 3D). These data
together suggest that GAPDH might play an important role in the
regulation of actin expression and thus, affecting cell
motility.
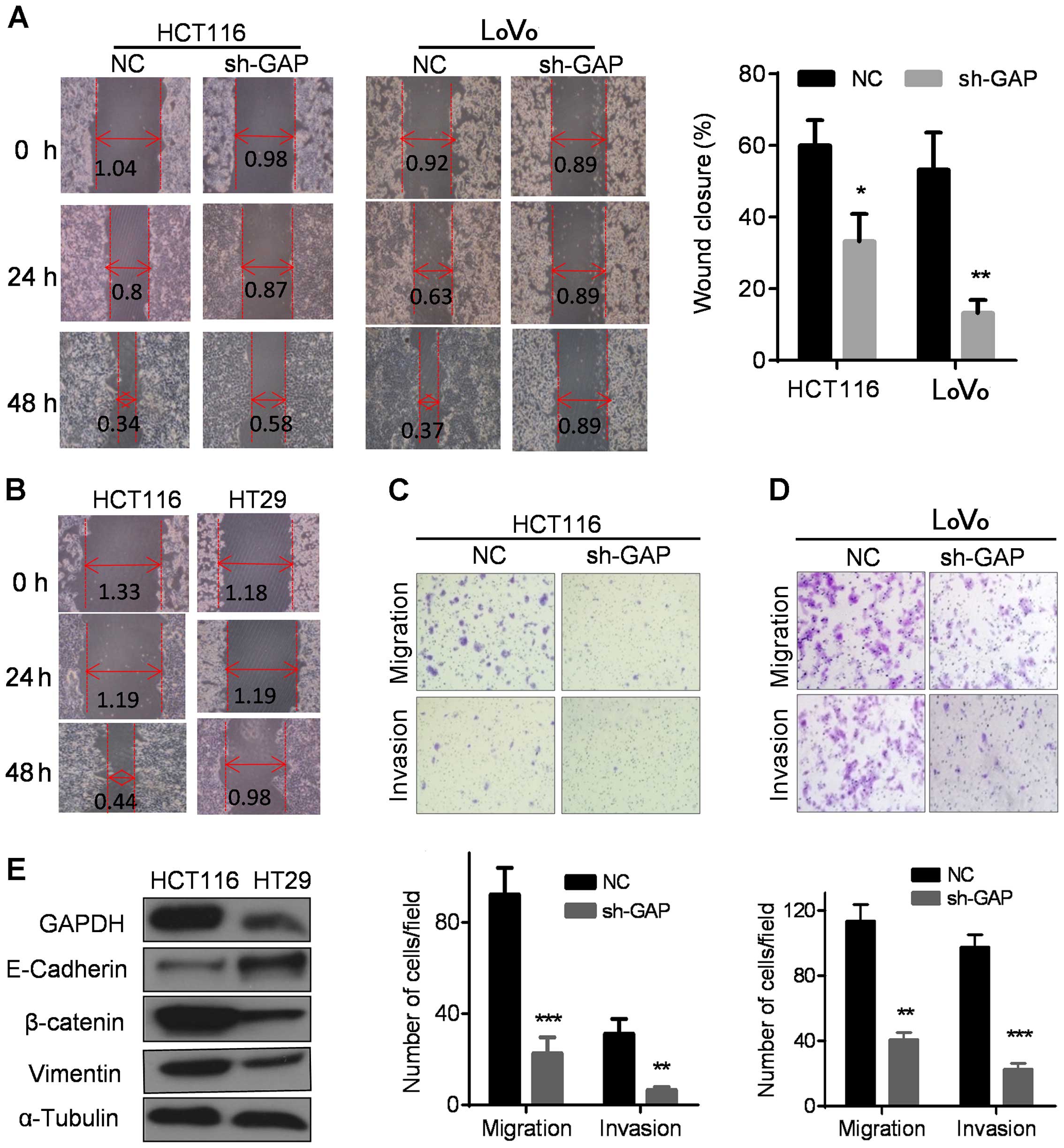
We then used a 'wound healing' assay to evaluate the
impact of GAPDH on cell migration. As shown in Fig. 4A, the control HCT116 and LoVo cells
migrated and covered ~60–70% scratched area after 48 h, wherea
sh-GAP cells only filled approximately 10–30% of the scratched area
during the same period. Theses results indicate that GAPDH
depletion substantially repressed cell migration. Similar
phenomenon was observed in HT29 cells, which expressed much lower
GAPDH and migrated significantly slower than HCT116 cells with
higher GAPDH expression (Fig. 4B).
Furthermore, Transwell and Matrigel invasion assays were performed
to compare the migration and the invasion in the NC and sh-GAP
cells. As shown in Fig. 4C and D,
GAPDH knockdown significantly inhibited cell migration and invasion
in both HCT116 and LoVo cells. Consistently, HT29 with low
expression of GAPDH also exhibited reduction in invasive capacity
compared to HCT116 with high GAPDH expression (Fig. 4E).
Attenuation of EMT and downregulation of
SNAIL by silencing of GAPDH
Previous studies suggest that EMT is essential for
migration and invasion regulated by SNAIL, which suppresses the
expression of E-cadherin and upregulates the vimentin and
β-catenin, leading to enhancement of EMT (21,22).
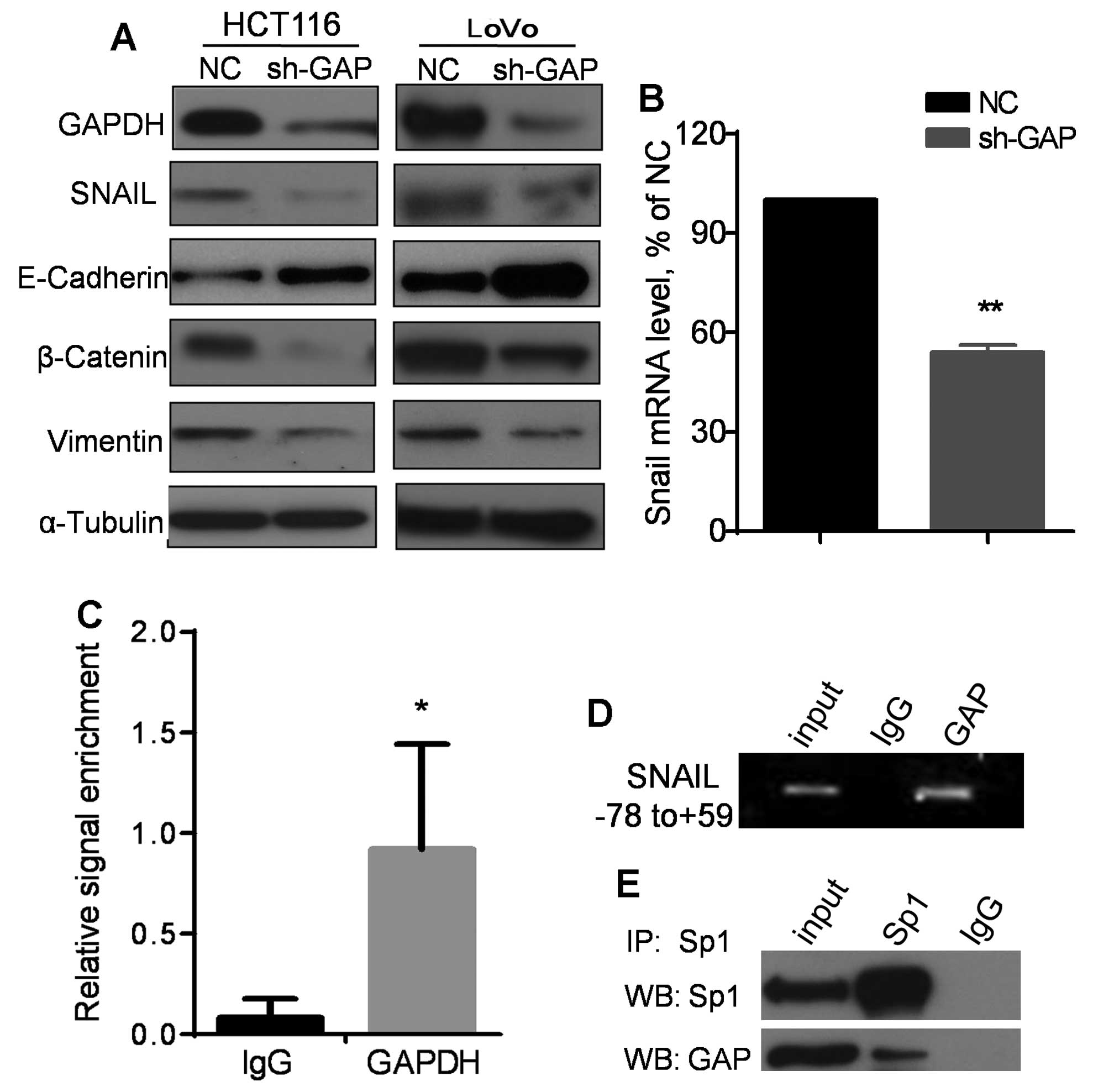
Therefore, we examined the impact of GAPDH knockdown on expression
of EMT-related molecules by western blot analysis. As shown in
Fig. 5A, silencing of GAPDH
resulted in a suppression of SNAIL expression, associated with an
upregulation of E-cadherin and a downregulation β-catenin and
vimentin in both HCT116 and LoVo cells. Consistently, a high
expression of E-cadherin and a low expreesion of β-catenin and
vimentin were also observed in HT29 cells, which expressed a lower
level of GAPDH compared to HCT116 cells (Fig. 4E). Quantitative analysis of mRNA by
qRT-PCR revealed that SNAIL mRNA expression level was significantly
decreased when GAPDH was knocked down by shRNA (Fig. 5B), suggesting that the decrease in
SNAIL protein in sh-GAP cells might likely be due to a reduction at
the transcriptional level. Since GAPDH protein has been observed to
translocate to the nucleus and affect gene expression (16), we speculated that GAPDH might
regulate SNAIL transcription by interacting with its promoter. To
test this possibility, chromatin immunoprecipitation (ChIP) assay
was performed using an antibody against GAPDH to pull down the DNA
fragments associated with GAPDH, followed by PCR using a pair of
primers specific for SNAIL minimal promoter region (nt
positions −78 to +59). Non-specific IgG was used as a control for
the pulldown. As shown in Fig. 5C and
D, pulldown of GAPDH resulted in a sinificant enrichment of PCR
signal for the SNAIL minimal promoter. Since it is known
that the transcriptional factor Sp1 directly binds to SNAIL
minimal promoter (−78/+59) and enhance its expression to promote
EMT (23), we tested if Sp1 and
GAPDH were physically associated with each other by
co-immunoprecipitation assay. As shown in Fig. 5E, Sp1 and GAPDH were
co-precipitated by the Sp1 antibody, while immunoprecipitation
using control IgG yield negative signal, suggesting a direct
physical interaction between GAPDH and Sp1.
Regulation of stem-like cell markers by
GAPDH
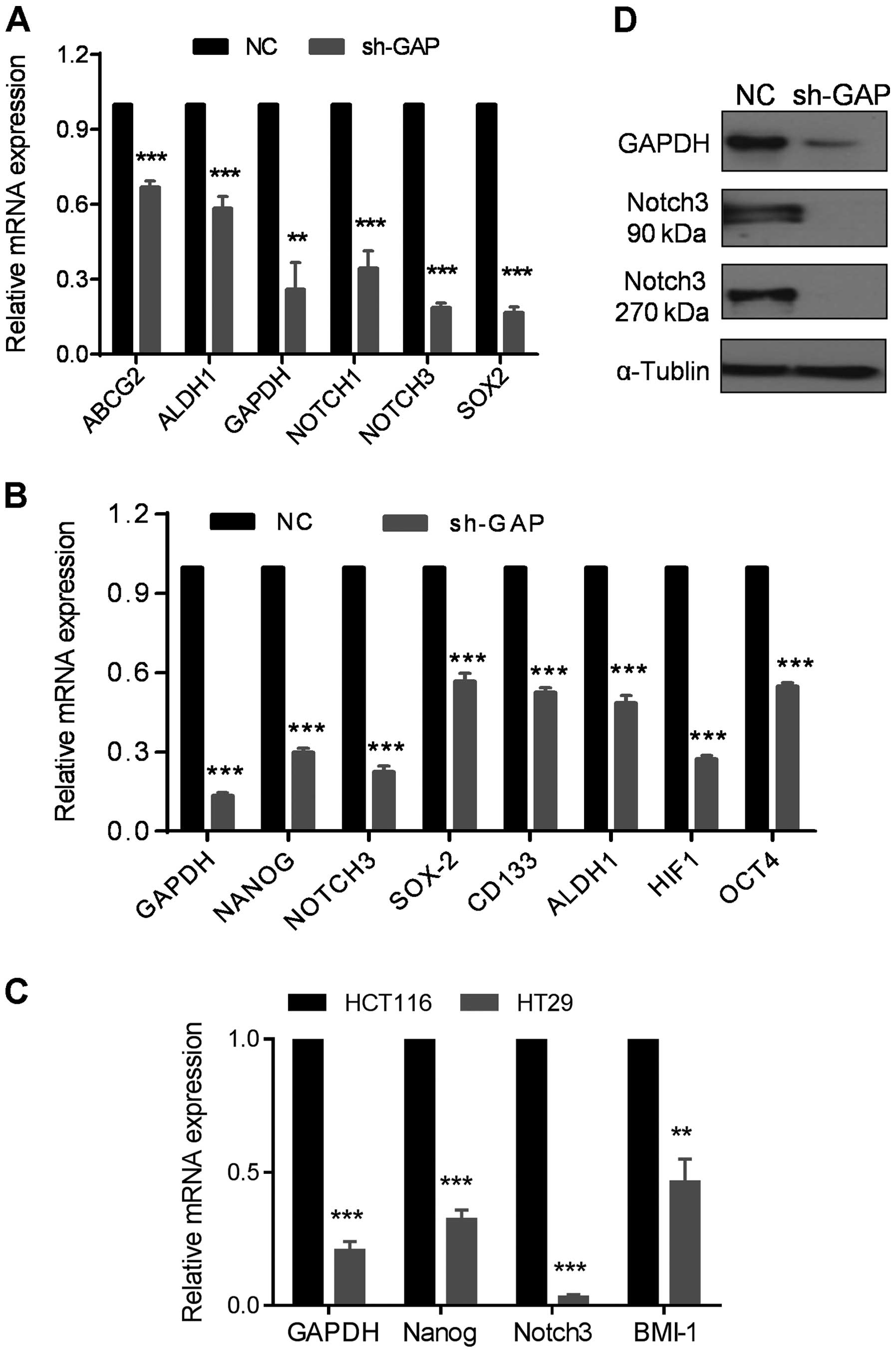
Since EMT is closely associated with stem-like cell
properties, we examined the expression of stem-like cell markers in
sh-GAP and NC cells. Strikingly, most of the stem cell-related
markers including ABCG2, ALDH1, SOX2,
OCT4, CD133, NOTCH1, NOTCH3 and
NANOG were substantially decreased after GAPDH silencing, as
shown by qRT-PCR assay (Fig. 6A and
B). The expressions of NOTCH3, NANOG and
BMI1 were also reduced in HT29 with lower expression of
GAPDH compared to HCT116 (Fig.
6C). Consistently, expression of NOTCH3 protein was abolished
when GAPDH expression was suppressed by shRNA, as evidenced by the
disappearance of both the 90-kDa extracellular domain of NOTCH3 and
the 270-kDa NOTCH3 protein (Fig.
6D). These results indicate that silencing of GAPDH expression
could significantly downregulate the expression of stem
cell-related molecules.
Impact of GAPDH on tumor invasion and
lung metastasis in vivo
Based on the observations that GAPDH depletion
reduced cell migration and invasion in vitro, we conducted
animal experiments to evaluate the impact of GAPDH on tumor
invation and metastasis in vivo. As shown in Fig. 7A and B, two control cancer cell
lines (HCT116-NC and LoVo-NC) with high GAPDH expression exhibited
invasive phenotype in the subcutaneous tumor xenografts with
noticeable clusters of tumor cells invading into the adjacent
tissues, while shRNA silencing of GAPDH expression in HCT116 and
LoVo cells (sh-GAP) suppressed the invasive behavior.
We then compared the abilities of the HCT116 sh-GAP
cells and HCT116-NC cells to form distant metastasis in mouse
models. The same number of sh-GAP and NC cells was injected into
the tail veins of 6-week old nude mice. After 45 days, the mice
were sacrificed and lung metastasis were examined (Fig. 7C), and the expression of GAPDH in
the tumor and adjacent tissue was revealed by immunohistochemistry
staining (Fig. 7D). The results
showed that HCT116-NC cells with high GAPDH expression developed
large tumors in the lung, whereas the ability of sh-GAP cells to
form tumors in the lung was significantly reduced (Fig. 7E). These data further confirm that
GAPDH plays an important role in colon cancer invasion and
metastasis in vivo.
Discussion
Under physiological conditions, normal cells rely on
the more energy-efficient oxidative phosphorylation to generate
ATP. In contrast, many cancer cells actively use the glycolytic
pathway for ATP generation even in the presence of adequate oxygen,
a phenomenon known as the Warburg effect (24). GAPDH is an important glycolytic
enzyme and its high expression in cancer cells seems to be
associated with aggressive malignant behavior of the cancer cells
and is also associated with poor prognosis of lung cancer patients
(25–27). Our previous study showed that high
GAPDH expression was associated with cancer metastasis (18), but the underlying mechanism remains
unclear. The main goals of the study were to use biochemical and
genetic strategies to establish defined cell models to evaluate the
impact of GAPDH on cancer cell behavior in vitro and in
vivo relevant to metastasis, and to investigate the possible
regulatory mechanisms.
Cancer metastasis is a complex process involving
multiple steps from detachment of cancer cells from the
original/primary tumor to establishment of new cancer colonies at
the distant tissue sites. This process requires the concerted
action of various proteins that affect cell adhesion, migration,
invasion and survival at the target tissues (28). EMT is associated with certain key
steps in this process involving cell migration and invasion, and is
also considered as a stem-like cell phenotype (19,29).
In the present study, we demonstrated that silencing of GAPDH
significantly abrogated the EMT phenotype of colon cancer cells in
both HCT116 and LoVo cells lines, and inhibited cell migration and
invasion in vitro and tumor metastasis in vivo. These
data strongly suggest that GAPDH may play an important role in
promoting metastasis, at least in colon cancer. At the molecular
level, GAPDH seems to physically intereact with Sp1, a key
transcriptional factor known to bind to the promoter of SNAIL and
enhance its expression (23,30).
It seems possible that GAPDH forms a protein complex with Sp1 and
enhance to expression of SNAIL, which is a transcriptional inducer
of EMT (31). Indeed, we showed
that shRNA silencing of GAPDH expression led to a significant
decrease of SNAIL expression in both HCT116 and LoVo cells,
accompanied by an upregulation of E-cadherin and a downregulation
of β-catenin and vimentin (Fig.
5A). Consistent with these observations, there was a
significant decrease in F-actin formation and an apparent reversion
of essenchymal cell morphology to epithelial appearance after
knockdown of GAPDH expression. These data together support a
possibility that GAPDH may promote EMT and cancer metastasis by
enhancing Sp1-mediated expression of SNAIL.
GAPDH is a molecule with enzymatic activity in
glycolysis in the cytoplasma and non-enzymatic functions in the
nucleus (13–16,20).
Although we showed that silencing of GAPDH significantly attenuate
glycolysis and other relevant metabolism, it is unclear if such
metabolic changes play any significant role in affecting
metastasis. One possibility is that GAPDH might affect cell
mobility and metastasis indirectly by affecting energy (ATP)
generation through its metabolic function in glycolysis. We indeed
observed that a knockdown of GAPDH expression led to significant
decrease in glycolysis and an inhibition of cell migration and
invasion. However, we could not exclude the possibility that the
reduced glycolysis and decreased cell migration were two parallel
events without causal relationship. Another possibility is that
GAPDH affects cell migration and cancer metastasis mainly through
its non-metabolic function. A previous study showed that GAPDH
would translocate to nucleus when cells encountered certain stress
conditions such as oxidative stress in cancer cells. Oxidative
stress-induced S-nitrosylation of GAPDH could promote translocation
of GAPDH to nucleus (15), where
it could intereact with Sp1 under oxidative stress conditions, and
activate SNAIL transcription. Thus, it is possible that
GAPDH may promote EMT and metastasis through its non-enzymatic
function in the nucleus. Further research is required to clearly
define the relative contribution of metabolic and transcriptional
function of GAPDH in promoting cancer metastasis. Generation of
GAPDH mutants with change in either enzyme activity or
transcriptional activity would provide important tools for such
study.
It is interesting to note that silencing of GAPDH
expression led to downregulation of SNAIL and decreased expression
of stem cell markers including ABCG2, ALDH1, SOX2, OCT4, CD133,
NOTCH1, NOTCH3, NANOG and BMI1. Although the exact relationship
between EMT and cancer stem cells still remains unclear and
somewhat controversial, emerging evidence suggest a close
association between EMT and cancer stem cell phenotype. For
instance, a recent study suggests that SNAIL could promote EMT and
increase the expression of stem cell markers in colon cancer cells
leading to an enhancement of cancer cell invasion and metastasis
(31). Another study showed that
silencing stem cell regulator SOX2 could induced a reversion of EMT
known as mesenchymal-epithelial transition (32). Consistent with these observations,
our findings that a knockdown of GAPDH resulted in a downregulation
of SNAIL and stem-related molecules and led to morphological
changes of colon cancer cells from the spindle-like mesenchymal
shape to a polygonal epithelial appearance support the notion that
EMT and stemness of cancer cells are linked.
In summary, the present study suggests that GAPDH
may play an important role in promoting cancer metastasis through
upregulation of Sp1-mediated expression of SNAIL, leading to
epithelial-mesenchymal transition and enhancement of cellular
migration and invasion. As such, it may be possible to prevent or
inhibit colon cancer metastasis by silencing GAPDH through genetic
manipulation or chemical inhibtion. Future studies are needed to
test GAPDH and its downstream molecules as therapeutic targets in
metastatic colon cancer.
Acknowledgments
The present study was supported in part by grants
from the National Natural Science Foundation of China (nos.
81430060 and 81502573), the Guangzhou Innovation Research Program
(no. LCY201317), the Guangzhou Medicare Collaborative Innovation
Program (no. 201508020250) and the 2014A030310421 from the Natural
Science Foundation of Guangdong Province.
Abbreviations:
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
|
EMT
|
epithelial-mesenchymal transition
|
|
shRNA
|
short hairpin RNA
|
|
OCR
|
oxygen consumption rate
|
|
ECAR
|
extracellular acidification rate
|
|
NC
|
negative control
|
References
|
1
|
Martins SF, Garcia EA, Luz MA, Pardal F,
Rodrigues M and Filho AL: Clinicopathological correlation and
prognostic significance of VEGF-A, VEGF-C, VEGFR-2 and VEGFR-3
expression in colorectal cancer. Cancer Genomics Proteomics.
10:55–67. 2013.PubMed/NCBI
|
|
2
|
Weitz J, Koch M, Debus J, Höhler T, Galle
PR and Büchler MW: Colorectal cancer. Lancet. 365:153–165. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Des Guetz G, Uzzan B, Nicolas P, Cucherat
M, Morere JF, Benamouzig R, Breau JL and Perret GY: Microvessel
density and VEGF expression are prognostic factors in colorectal
cancer. Meta-analysis of the literature. Br J Cancer. 94:1823–1832.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Martins SF, Reis RM, Rodrigues AM,
Baltazar F and Filho AL: Role of endoglin and VEGF family
expression in colorectal cancer prognosis and anti-angiogenic
therapies. World J Clin Oncol. 2:272–280. 2011.PubMed/NCBI
|
|
5
|
Barderas R, Mendes M, Torres S, Bartolomé
RA, López-Lucendo M, Villar-Vázquez R, Peláez-García A, Fuente E,
Bonilla F and Casal JI: In-depth characterization of the secretome
of colorectal cancer metastatic cells identifies key proteins in
cell adhesion, migration, and invasion. Mol Cell Proteomics.
12:1602–1620. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arlt F and Stein U: Colon cancer
metastasis: MACC1 and Met as metastatic pacemakers. Int J Biochem
Cell Biol. 41:2356–2359. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
O'Connell JB, Maggard MA and Ko CY: Colon
cancer survival rates with the new American Joint Committee on
Cancer sixth edition staging. J Natl Cancer Inst. 96:1420–1425.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Murata K and Moriyama M: Isoleucine, an
essential amino acid, prevents liver metastases of colon cancer by
antiangiogenesis. Cancer Res. 67:3263–3268. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Iqbal S and Lenz HJ: Angiogenesis
inhibitors in the treatment of colorectal cancer. Semin Oncol.
31(Suppl 17): 10–16. 2004. View Article : Google Scholar
|
|
10
|
El Zouhairi M, Charabaty A and Pishvaian
MJ: Molecularly targeted therapy for metastatic colon cancer:
Proven treatments and promising new agents. Gastrointest Cancer
Res. 4:15–21. 2011.PubMed/NCBI
|
|
11
|
Mori R, Wang Q, Danenberg KD, Pinski JK
and Danenberg PV: Both beta-actin and GAPDH are useful reference
genes for normalization of quantitative RT-PCR in human FFPE tissue
samples of prostate cancer. Prostate. 68:1555–1560. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Murthi P, Fitzpatrick E, Borg AJ, Donath
S, Brennecke SP and Kalionis B: GAPDH, 18S rRNA and YWHAZ are
suitable endogenous reference genes for relative gene expression
studies in placental tissues from human idiopathic fetal growth
restriction. Placenta. 29:798–801. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harada N, Yasunaga R, Higashimura Y,
Yamaji R, Fujimoto K, Moss J, Inui H and Nakano Y:
Glyceraldehyde-3-phosphate dehydrogenase enhances transcriptional
activity of androgen receptor in prostate cancer cells. J Biol
Chem. 282:22651–22661. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tisdale EJ: Glyceraldehyde-3-phosphate
dehydrogenase is required for vesicular transport in the early
secretory pathway. J Biol Chem. 276:2480–2486. 2001. View Article : Google Scholar
|
|
15
|
Sirover MA: On the functional diversity of
glyceraldehyde-3-phosphate dehydrogenase: Biochemical mechanisms
and regulatory control. Biochim Biophys Acta. 1810:741–751. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zheng L, Roeder RG and Luo Y: S phase
activation of the histone H2B promoter by OCA-S, a coactivator
complex that contains GAPDH as a key component. Cell. 114:255–266.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Epner DE, Partin AW, Schalken JA, Isaacs
JT and Coffey DS: Association of glyceraldehyde-3-phosphate
dehydrogenase expression with cell motility and metastatic
potential of rat prostatic adenocarcinoma. Cancer Res.
53:1995–1997. 1993.PubMed/NCBI
|
|
18
|
Tang Z, Yuan S, Hu Y, Zhang H, Wu W, Zeng
Z, Yang J, Yun J, Xu R and Huang P: Over-expression of GAPDH in
human colorectal carcinoma as a preferred target of 3-bromopyruvate
propyl ester. J Bioenerg Biomembr. 44:117–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sirover MA: New nuclear functions of the
glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in
mammalian cells. J Cell Biochem. 95:45–52. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou BP, Deng J, Xia W, Xu J, Li YM,
Gunduz M and Hung MC: Dual regulation of Snail by
GSK-3beta-mediated phosphorylation in control of
epithelial-mesenchymal transition. Nat Cell Biol. 6:931–940. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Peña C, García JM, Silva J, García V,
Rodríguez R, Alonso I, Millán I, Salas C, de Herreros AG, Muñoz A,
et al: E-cadherin and vitamin D receptor regulation by SNAIL and
ZEB1 in colon cancer: Clinicopathological correlations. Hum Mol
Genet. 14:3361–3370. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barberà MJ, Puig I, Domínguez D,
Julien-Grille S, Guaita-Esteruelas S, Peiró S, Baulida J, Francí C,
Dedhar S, Larue L, et al: Regulation of Snail transcription during
epithelial to mesenchymal transition of tumor cells. Oncogene.
23:7345–7354. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tokunaga K, Nakamura Y, Sakata K, Fujimori
K, Ohkubo M, Sawada K and Sakiyama S: Enhanced expression of a
glyceraldehyde-3-phosphate dehydrogenase gene in human lung
cancers. Cancer Res. 47:5616–5619. 1987.PubMed/NCBI
|
|
26
|
Révillion F, Pawlowski V, Hornez L and
Peyrat JP: Glyceraldehyde-3-phosphate dehydrogenase gene expression
in human breast cancer. Eur J Cancer. 36:1038–1042. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Puzone R, Savarino G, Salvi S, Dal Bello
MG, Barletta G, Genova C, Rijavec E, Sini C, Esposito AI, Ratto GB,
et al: Glyceraldehyde-3-phosphate dehydrogenase gene over
expression correlates with poor prognosis in non small cell lung
cancer patients. Mol Cancer. 12:972013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nguyen DX and Massagué J: Genetic
determinants of cancer metastasis. Nat Rev Genet. 8:341–352. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Beerling E, Seinstra D, de Wit E, Kester
L, van der Velden D, Maynard C, Schäfer R, van Diest P, Voest E,
van Oudenaarden A, et al: Plasticity between epithelial and
mesenchymal states unlinks EMT from metastasis-enhancing stem cell
capacity. Cell Rep. 14:2281–2288. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Higashimura Y, Nakajima Y, Yamaji R,
Harada N, Shibasaki F, Nakano Y and Inui H: Up-regulation of
glyceraldehyde-3-phosphate dehydrogenase gene expression by HIF-1
activity depending on Sp1 in hypoxic breast cancer cells. Arch
Biochem Biophys. 509:1–8. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fan F, Samuel S, Evans KW, Lu J, Xia L,
Zhou Y, Sceusi E, Tozzi F, Ye XC, Mani SA, et al: Overexpression of
snail induces epithelial-mesenchymal transition and a cancer stem
cell-like phenotype in human colorectal cancer cells. Cancer Med.
1:5–16. 2012. View
Article : Google Scholar
|
|
32
|
Han X, Fang X, Lou X, Hua D, Ding W, Foltz
G, Hood L, Yuan Y and Lin B: Silencing SOX2 induced
mesenchymal-epithelial transition and its expression predicts liver
and lymph node metastasis of CRC patients. PLoS One. 7:e413352012.
View Article : Google Scholar : PubMed/NCBI
|