Introduction
Bladder cancer is one of the most commonly diagnosed
cancers worldwide (1). The
combination chemotherapy with methotrexate, vinblastine,
doxorubicin, and cisplatin (M-VAC) was reported in 1985 (2) which thereafter became the standard
chemotherapy for patients with bladder cancer (3). Nowadays, the combination chemotherapy
of gemcitabine and cisplatin (GC) has replaced M-VAC therapy as a
first-line treatment for bladder cancer patients, because GC
exhibited similar efficacy to M-VAC with less adverse events
(4,5). Although bladder cancer is relatively
sensitive to chemotherapy, drug-resistance is inevitable and occurs
within a few years in most cases. However, there has been no
clearly established second-line therapy for bladder cancer
resistant to first-line chemotherapy (6).
Androgen receptor (AR), one of the steroid hormone
receptors, is a transcriptional factor whose activity is modulated
by androgens. AR plays an important role in the survival of
prostate cancer cells (7). In
addition, AR has been implicated in other types of cancer including
bladder cancer as well as liver, kidney, breast, and lung cancer
(8–11). A study demonstrated that
downregulation of AR expression inhibited bladder cancer cell
growth in vitro and in vivo (12). It has been also shown that
expression of AR was higher in doxorubicin-resistant bladder cancer
cells than sensitive cells and that androgen/AR signaling
correlated with the effectiveness of doxorubicin (13).
In this study, in an attempt to identify novel
therapeutic targets for gemcitabine-resistance, we developed
gemcitabine-resistant bladder cancer cells from sensitive cells and
performed microarray analysis. We found that AR expression was
upregulated in gemcitabine-resistant cancer cells compared with
parental cells. We then investigated the effects on growth of
gemcitabine-resistant cancer cells of AR knockdown as well as
culture in charcoal-stripped serum (CSS) and treatment with
enzalutamide, a second-generation non-steroidal AR inhibitor used
for castration-resistant prostate cancer (CRPC) patients. We also
studied the effect of enzalutamide on gemcitabineresistance.
Finally, we examined if enzalutamide could inhibit growth of
AR-expressing bladder cancer cells with intrinsic
gemcitabine-resistance. Our results suggested that blockade of AR
signaling by enzalutamide might be effective for patients with
advanced gemcitabine-resistant bladder cancer with increased AR
expression.
Materials and methods
Reagents and antibodies
Gemcitabine and enzalutamide were from Sigma-Aldrich
(St. Louis, MO, USA) and MedChem Express LLC (Princeton, NJ, USA),
respectively. Dihydrotestosterone (DHT) was purchased from Tokyo
Chemical Industry (Tokyo, Japan). Anti-β-actin (#3700), anti-AR
(#5153), anti-cyclin D1 (#2978), anti-cyclin B1 (#12231),
anti-caspase-3 (#9662) and anti-cleaved caspase-3 (#9664)
antibodies were from Cell signaling Technology (Danvers, MA,
USA).
Cell culture and establishment of
gemcitabine-resistant cells
T24 human bladder cancer cell line was obtained from
RIKEN Cell Bank (Tsukuba, Japan). Cells were cultured in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS). T24 cells
were initially cultured in the medium containing 5 nM gemcitabine
and the dose was gradually increased over 3 months. Finally, we
obtained gemcitabine-resistant T24 cells (T24GR) viable in the
presence of 50 nM gemcitabine. In specified experiments, cells were
cultured in CSS from Thermo Fisher Scientific (Waltham, MA, USA).
HTB5 human bladder cancer cell line was obtained from American Type
Culture Collection (Manassas, VA, USA) and cultured in RPMI-1640
medium supplemented with 10% FBS.
Cell growth and viability assay
The number of living cells was counted manually or
automatically by using Countess Automated Cell Counter (Invitrogen,
Carlsbad, CA, USA) after staining with trypan blue. The sensitivity
to gemcitabine and enzalutamide was determined by WST-1 assay using
the kit from Roche Diagnostics (Mannheim, Germany).
Microarray analysis of mRNA
Total RNA extracted by the miRNeasy Mini kit
(Qiagen, Hilden, Germany) was used for the labeling and
hybridization to the Agilent SurePrint G3 Human GE Microarray 8×60K
(Agilent Technologies, Santa Clara, CA, USA). After washing, the
microarrays were scanned on the Agilent DNA Microarray scanner
(Agilent Technologies). Raw data were collected using Agilent
Feature Extraction software and analyzed using the GeneSpring GX11
(Agilent Technologies). The signal intensity of each probe was
normalized by a percentile shift, in which each value was divided
by the 75th percentile of all values in its array. Only the probes
that had expression flags present under at least one condition were
considered. Data were compared using t-tests, with a significance
level set at P<0.05. The remaining probes were selected using
the criterion of at least a 3-fold change. The microarray data have
been deposited in the NCBI Gene Expression Omnibus (GEO) and are
accessible through the GEO Series accession no. GSE77883
(http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE77883).
Quantitative RT-PCR
Total RNA extracted using the miRNeasy Mini kit with
on-column DNase I treatment was reverse transcribed using the
ReverTra Ace qPCR RT Master Mix (Toyobo, Osaka, Japan).
Quantitative RT-PCR was performed on the StepOnePlus Real-Time PCR
system (Applied Biosystems, Foster City, CA, USA) using the
Thunderbird SYBR qPCR Mix (Toyobo). PCR conditions include a
denaturation at 95°C 1 min, followed by 40 cycles of 15 sec at 95°C
and 1 min at 60°C. Primer sequences were as follows: AR forward,
5′-TGTACACGTGGTCAAGTGGG-3′; reverse, 5′-GTGCATGCGGTACTCATTGAA-3′.
18S ribosomal RNA forward, 5′-GTAACCCGTTGAACCCCATT-3′; reverse,
5′-CCATCCAATCGGTAGTAGCG-3′. The relative expression was expressed
as a ratio of AR to 18S rRNA.
Western blot analysis
Whole cell lysates were prepared in lysis buffer [1%
Nonidet P-40, 0.1% sodium deoxycholate, 0.1% sodium dodecyl
sulfate, 1 mM EDTA, 150 mM NaCl, and 10 mM Tris-HCl (pH 7.4)]
containing protease inhibitor cocktail (Sigma-Aldrich). Cell
lysates were subjected to electrophoresis on 4–15%
SDS-polyacrylamide gels (Bio-Rad, Hercules, CA, USA) and
transferred to polyvinylidene difluoride membranes. After blocking
in non-fat milk, membranes were incubated overnight at 4°C with
primary antibodies followed by incubation for 60 min at room
temperature with a horseradish peroxidase-linked secondary
antibodies. Immunoreactive bands were detected by using the
enhanced chemiluminescence (ECL) detection system (GE
Healthcare).
Luciferase reporter assay
Twenty-four hours after seeding cells
(2.5×105) into a 12-well plate, the pGL4 firefly
luciferase reporter gene (Promega, Madison, WI, USA) containing 4
copies of the androgen response element (ARE;
5′-GCTAGCACTTGCTGTTCTGCA-3′) (14)
upstream of the minimal promoter were co-transfected for 6 h with
the Renilla expressing plasmid (phRL-TK, Promega) using
Lipofectamine 2000 (Thermo Fisher Scientific). Firefly and
Renilla luciferase activities were measured using the
Pikkagene Dual Luciferase assay systems (Toyo B-Net, Tokyo, Japan).
The AR transcriptional activity was expressed as a ratio of firefly
to Renilla luciferase activity.
AR copy number assay
Genomic DNA was extracted using the NucleoSpin
Tissue (Takara, Kusatsu, Japan). The copy number variation of AR
gene was determined by using the TaqMan Copy Number assay (assay
id: Hs00034522_cn, Applied Biosystems). The TaqMan Copy Number
Reference assay (RNaseP, assay id: 4403326, Applied Biosystems) was
used as an internal control. The assay was performed on the
StepOnePlus PCR system using the TaqMan Genotyping Master Mix
(Applied Biosystems) and 20 ng of genomic DNA. PCR conditions were
95°C for 10 min for initial denaturation and enzyme activation,
followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C. The copy
number of the AR gene was estimated from the Ct values by using the
CopyCaller software (Applied Biosystems).
Small interfering RNA transfection
Two small interfering RNAs (siRNAs) for human AR
gene were from Dharmacon GE Healthcare (Lafayette, CO, USA). Target
sequences of the AR siRNAs were as follows: siRNA1,
5′-GGAACUCGAUCGUAUCAUU-3′, siRNA2, 5′-UCAAGGAACUCGAUCGUAU-3′. As a
negative control siRNA, siGENOME non-targeting siRNA control gene
was used (Thermo Fisher Scientific). Each siRNA was transfected
into cells for 6 h at 10 nM using Lipofectamine 2000.
Evaluation of drug interaction
Drug interaction was assessed based on the method
described by Chou and Talalay (15) and the combination index (CI) was
determined using the CompuSyn software.
Statistical analysis
Student's t-test was used for comparison between two
groups. Statistical significance was defined as a P-value of
<0.05.
Results
Establishment of gemcitabine-resistant
T24 bladder cancer cells
We developed gemcitabine-resistant T24 cells from
parental T24 cells by treating cells with increasing concentrations
of gemcitabine over 3 months. We performed WST-1 assay to examine
the sensitivity of T24 and T24GR cells to gemcitabine. As shown in
Fig. 1, T24GR cells were resistant
to gemcitabine compared to parental T24 cells. The IC50
values of T24 cells and T24GR were 5.99 nM and 3.46 µM,
respectively (577.4-fold).
Increased expression of AR in T24GR
cells
We investigated mRNA expression profiles of T24 and
T24GR cells. Microarray analysis of mRNA expression showed that 596
genes were upregulated >3-fold in T24GR cells compared with T24
cells (data not shown). AR expression was 32-fold higher in T24GR
cells than in T24 cells (Table I).
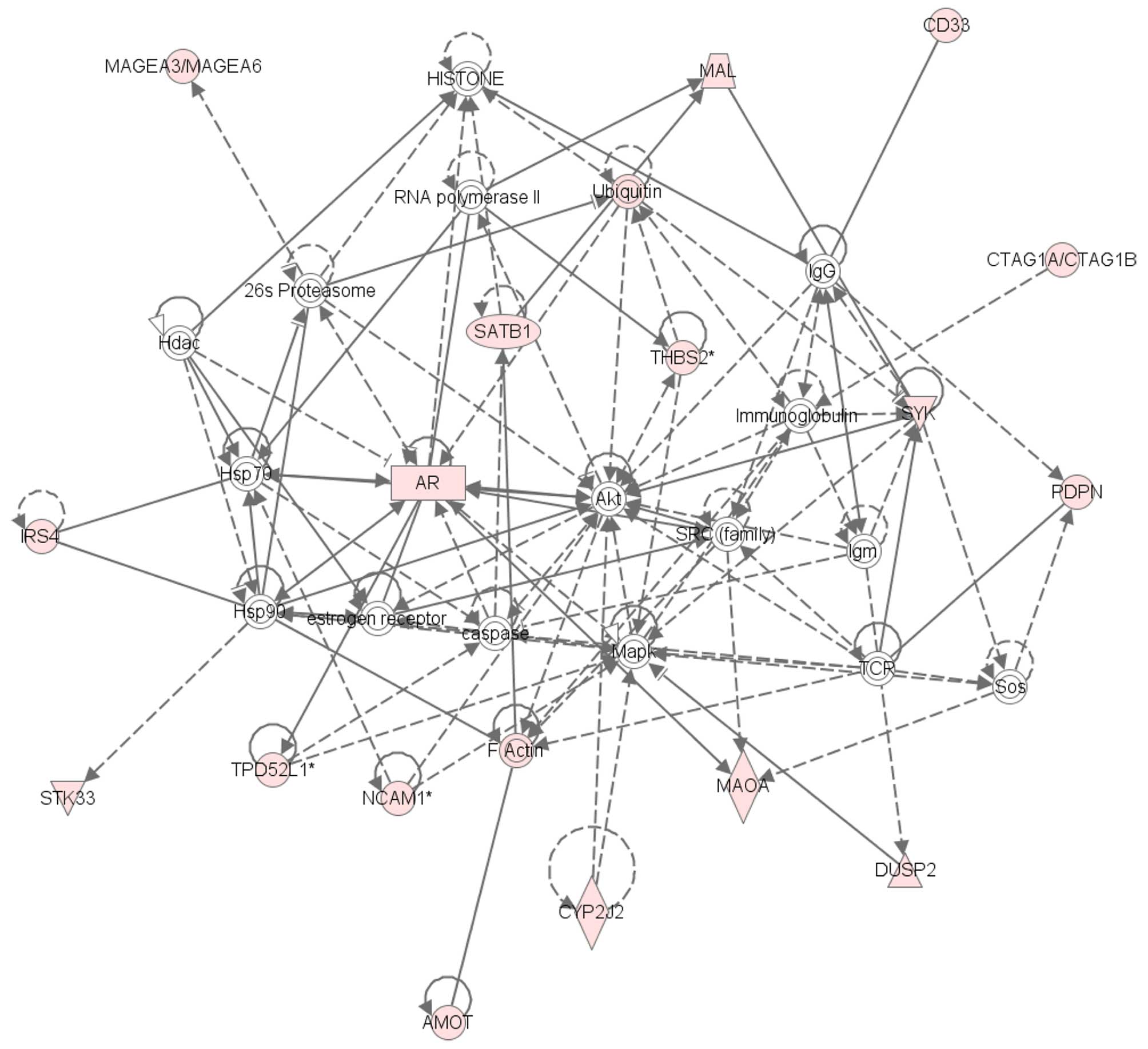
Network analysis of 118 genes whose expression was elevated by
>10-fold in T24GR cells than in T24 cells identified 5 gene
networks, one of which was AR-related gene network (Fig. 2). As shown in Fig. 3A, quantitative RT-PCR confirmed
that AR mRNA level in T24GR cells was significantly higher than
that in T24 cells (40.5-fold). In accordance with the mRNA change,
AR protein expression was markedly increased in T24GR cells
compared with T24 cells (Fig. 3B).
We also investigated the AR transcriptional activity in T24 and
T24GR cells by luciferase reporter assays. Consistent with the
increased expression of AR mRNA and protein, the AR transcriptional
activity in T24 GR cells was about 6 times higher than that in T24
cells in normal medium (Fig. 3C).
These results indicated that AR expression was increased in
gemcitabine-resistant T24GR cells compared with
gemcitabine-sensitive T24 cells, which was also shown to be
functional. Since AR signaling has been implicated in bladder
cancer (16), we selected and
focused on AR in subsequent studies.
 | Table IThe genes with expression showing a
>20-fold increase in T24GR cells compared with T24 cells. |
Table I
The genes with expression showing a
>20-fold increase in T24GR cells compared with T24 cells.
| Gene symbol | Description | Fold change |
|---|
| LRRC38 | Homo sapiens
leucine rich repeat containing 38 (LRRC38) | 5,918.9 |
| PASD1 | Homo sapiens
PAS domain containing 1 (PASD1) | 849.3 |
| FGF13 | Homo sapiens
fibroblast growth factor 13 (FGF13), transcript variant 1 | 747.7 |
| PDPN | Homo sapiens
podoplanin (PDPN), transcript variant 2 | 499.8 |
| ACTG2 | Homo sapiens
actin, γ2, smooth muscle, enteric (ACTG2), transcript variant
1 | 428.6 |
| HS6ST2 | Homo sapiens
heparan sulfate 6-O-sulfotransferase 2 (HS6ST2), transcript variant
L | 259.3 |
| GOLT1A | Homo sapiens
golgi transport 1A (GOLT1A) | 106.2 |
| CXorf57 | Homo sapiens
chromosome X open reading frame 57 (CXorf57), transcript variant
1 | 104.5 |
| CYP2J2 | Homo sapiens
cytochrome P450, family 2, subfamily J, polypeptide 2 (CYP2J2) | 86.3 |
| TSPAN18 | Homo sapiens
tetraspanin 18 (TSPAN18) | 82.9 |
| NELL2 | Homo sapiens
NEL-like 2 (chicken) (NELL2), transcript variant 2 | 82.1 |
| AMOT | Homo sapiens
angiomotin (AMOT), transcript variant 2 | 74.8 |
| KISS1 | Homo sapiens
KiSS-1 metastasis-suppressor (KISS1) | 71.9 |
| FAM155B | Homo sapiens
family with sequence similarity 155, member B (FAM155B) | 69.5 |
| XK | Homo sapiens
X-linked Kx blood group (McLeod syndrome) (XK) | 68.7 |
| SYK | Homo sapiens
spleen tyrosine kinase (SYK), transcript variant 1 | 61.7 |
| XLOC_003505 | BROAD institute
lincRNA (XLOC_003505) | 61.0 |
| BAGE | Homo sapiens
B melanoma antigen (BAGE) | 59.2 |
| STXBP6 | Homo sapiens
syntaxin binding protein 6 (amisyn) (STXBP6) | 55.0 |
| PGM5 | Homo sapiens
phosphoglucomutase 5 (PGM5) | 52.9 |
| MAGEB2 | Homo sapiens
melanoma antigen family B, 2 (MAGEB2) | 51.9 |
| NCAM1 | Homo sapiens
neural cell adhesion molecule 1 (NCAM1), transcript variant 5 | 51.7 |
| XLOC_010251 | BROAD Institute
lincRNA (XLOC_010251) | 49.7 |
| UBD | Homo sapiens
ubiquitin D (UBD) | 49.2 |
| JPH1 | Homo sapiens
junctophilin 1 (JPH1) | 49.1 |
| FAM230C | PREDICTED: Homo
sapiens family with sequence similarity 230, member C
(FAM230C), transcript variant X1 | 48.9 |
| LOC100134317 | Homo sapiens
uncharacterized LOC100134317 (LOC100134317) | 44.0 |
| ITM2A | Homo sapiens
integral membrane protein 2A (ITM2A), transcript variant 1 | 40.3 |
| XAGE2B | Homo sapiens
X antigen family, member 2B (XAGE2B) | 40.1 |
| GDA | Homo sapiens
guanine deaminase (GDA), transcript variant 2 | 39.3 |
| GPC4 | Homo sapiens
glypican 4 (GPC4) | 38.9 |
| SLITRK4 | Homo sapiens
SLIT and NTRK-like family, member 4 (SLITRK4), transcript variant
2 | 37.5 |
| FAM110C | Homo sapiens
family with sequence similarity 110, member C (FAM110C) | 35.6 |
| IRS4 | PREDICTED: Homo
sapiens insulin receptor substrate 4 (IRS4), transcript variant
X1 | 35.4 |
| FOXR2 | Homo sapiens
forkhead box R2 (FOXR2) | 34.6 |
| IGFBP5 | Homo sapiens
insulin-like growth factor binding protein 5 (IGFBP5) | 34.1 |
| AR | Homo sapiens
androgen receptor (AR), transcript variant 1 | 32.2 |
| CNN1 | Homo sapiens
calponin 1, basic, smooth muscle (CNN1) | 32.0 |
| BMP2 | Homo sapiens
bone morphogenetic protein 2 (BMP2) | 31.4 |
| PCSK6 | Homo sapiens
proprotein convertase subtilisin/kexin type 6 (PCsK6), transcript
variant 3 | 31.1 |
| WNT5A | Homo sapiens
wingless-type MMTV integration site family, member 5A (WNT5A),
transcript variant 1 | 30.1 |
| CECR7 | Homo sapiens
cat eye syndrome chromosome region, candidate 7 (non-protein
coding) (CECR7) | 29.3 |
| PLCB4 | Homo sapiens
phospholipase C, β4 (PLCB4), transcript variant 2 | 29.1 |
| IQCF1 | Homo sapiens
IQ motif containing F1 (IQCF1) | 29.1 |
| AWAT2 | Homo sapiens
acyl-CoA wax alcohol acyltransferase 2 (AWAT2) | 29.1 |
| XLOC_014512 | BROAD Institute
lincRNA (XLOC_014512) | 26.9 |
| FTCDNL1 | Homo sapiens
formiminotransferase cyclodeaminase N-terminal like (FTCDNL1) | 24.5 |
| XLOC_005442 | BROAD Institute
lincRNA (XLOC_005442) | 24.5 |
| PLAC4 | Homo sapiens
placenta-specific 4 (PLAC4) | 24.1 |
| POTEB | Homo sapiens
POTE ankyrin domain family, member B (POTEB) | 22.9 |
| GAL | Homo sapiens
galanin/GMAP prepropeptide (GAL) | 22.4 |
| MAL | Homo sapiens
mal, T-cell differentiation protein (MAL), transcript variant
a | 21.7 |
| GABRG2 | Homo sapiens
γ-aminobutyric acid (GABA) A receptor, γ2 (GABRG2), transcript
variant 1 | 20.7 |
| LOC728485 | PREDICTED: Homo
sapiens uncharacterized LOC728485 (LOC728485) | 20.7 |
| FAM89A | Homo sapiens
family with sequence similarity 89, member A (FAM89A) | 20.5 |
| CTAG1A | Homo sapiens
cancer/testis antigen 1A (CTAG1A) | 20.4 |
| ANGPTL2 | Homo sapiens
angiopoietin-like 2 (ANGPTL2) | 20.4 |
| HBE1 | Homo sapiens
hemoglobin, ε1 (HBE1) | 20.4 |
| ITGA11 | Homo sapiens
integrin, α11 (ITGA11) | 20.3 |
AR gene copy number change contributed to
increased AR expression in T24GR cells
In an attempt to identify the mechanism underlying
the increased expression of AR in T24GR cells, we examined whether
the AR gene copy number was changed between T24 and T24GR cells.
The results showed that the number of AR gene copy in T24GR cells
was twice as many as that of T24 cells (Fig. 3D), which was supposed to be one of
the reasons for upregulation of AR in T24GR cells.
AR knockdown inhibited proliferation of
T24GR cells
It has been reported that AR plays an important role
in growth of bladder cancer cells (12,13).
Here we inhibited AR expression by siRNA transfection and examined
the effect on proliferation of T24GR cells cultured in the medium
containing FBS. As shown in Fig.
4A, AR protein expression was effectively reduced by both AR1
siRNA and AR2 siRNA. The proliferation of AR siRNA-transfected
cells was significantly inhibited compared to control
siRNA-transfected cells (Fig. 4B).
These results suggested that elevated AR expression was involved in
proliferation of T24GR cells.
Culture in CSS and treatment with
enzalutamide inhibited growth of T24GR cells
Since AR knockdown reduced growth of T24GR cells, we
examined the effect of androgen depletion and enzalutamide
treatment. In order to deplete androgens, we cultured T24 and T24GR
cells in the medium containing CSS or FBS and compared the cell
viability. Culture in CSS did not affect the viability of T24
cells, but the number of viable T24GR cells cultured in CSS was
lower than that cultured in FBS (Fig.
5A). However, the addition of DHT to the medium containing CSS
did not promote growth of T24GR cells (data not shown). Next, we
investigated the effects of enzalutamide, an AR inhibitor, on
growth of both cell types. Enzalutamide inhibited the viability of
T24GR cells significantly, whereas it also reduced that of T24
cells expressing little or no AR, although to a lesser extent
(Fig. 5B). These results suggested
that proliferation of T24GR cells depended on AR signaling and was
inhibited by culture in CSS and treatment with enzalutamide. The
absence of DHT effect on proliferation in CSS also suggested that
AR might be activated other than androgens, which were present in
FBS but not in CSS.
Growth inhibition of T24GR cells by
culture in CSS and treatment with enzalutamide was mediated by cell
cycle arrest
In order to investigate whether the inhibitory
effects of culture in CSS and treatment with enzalutamide on the
growth of T24GR cells are mediated through cell cycle arrest or
apoptosis, we performed western blot analysis for cyclins and
caspase-3. As shown in Fig. 6,
expression of cyclin D1 was significantly lower in T24GR cells
cultured in CSS and in those treated with enzalutamide. In T24
cells treated with enzalutamide, cyclin D1 expression was slightly
decreased compared with those treated with vehicle, which was
consistent with the enzalutamide effect on growth of T24 cells
(Fig. 5B). Expression of cyclin B1
was not affected by culture in CSS and treatment with enzalutamide
in either T24 or T24GR cells. On the other hand, cleaved caspase-3
was not detected in either T24 or T24GR cells under any
experimental conditions tested, although caspase-3 was expressed at
the same level (data not shown). These results suggested that
culture in CSS and treatment with enzalutamide might inhibit growth
of T24GR cells via induction of cell cycle arrest but not of
apoptosis.
Inhibition of AR transcriptional activity
by enzalutamide treatment in T24GR cells
Enzalutamide exerts an antitumor activity through
preventing androgen binding to the ligand-binding domain of AR, and
also through inhibiting AR nuclear translocation, DNA binding, and
coactivator recruitment (17,18).
Here we measured the AR transcriptional activity in T24GR cells
treated with enzalutamide. The AR transcriptional activity was
decreased in T24GR cells treated with enzalutamide compared with
those treated with vehicle (Fig.
7).
Effects of enzalutamide on
gemcitabine-resistance in T24GR cells
In order to study the combined effects, we treated
cells with enzalutamide and gemcitabine. Treatment with
enzalutamide reduced viability of T24GR cells treated with
gemcitabine (Fig. 8A). The
calculated CI was 0.847 at Fa of 0.5, showing synergy between these
two drugs. Western blot analysis demonstrated that gemcitabine,
enzalutamide and both decreased cyclin D1 expression. However, the
level of cleaved caspase-3 was decreased by the co-presence of
enzalutamide (Fig. 8B), indicating
that enzalutamide did not enhance but rather diminished cleavage of
caspase-3 mediated by gemcitabine. These results suggested that
enzalutamide could attenuate gemcitabine-resistance of T24GR
cells.
Growth inhibition of HTB5 cells by
enzalutamide
Lastly, we investigated the effects of enzalutamide
in HTB5 bladder cancer cell line that was shown to express AR and
possess intrinsic resistance to gemcitabine (19,20).
Treatment with enzalutamide inhibited cell proliferation (Fig. 9A), which was accompanied by a
decrease in cyclin D1 expression (Fig.
9B). These results suggest that enzalutamide inhibits growth of
AR-expressing bladder cancer cells with intrinsic (HTB5) as well as
acquired (T24GR) resistance to gemcitabine.
Discussion
Gemcitabine, difluorodeoxycytidine, is widely used
for treatment of many types of cancer including lung, pancreatic,
biliary tract, breast, and urothelial cancer, but most patients
treated with gemcitabine eventually acquire resistance. Although
genes involved in drug transport and those involved in drug
metabolism have been proposed to be involved in gemcitabine
resistance (21–25) the underlying mechanisms remain
still unclear. In an attempt to identify novel therapeutic targets
for gemcitabine-resistance, we developed gemcitabine-resistant
bladder cancer cells from sensitive cells and performed microarray
analysis. We found that expression of functional AR was elevated in
T24GR cells compared with T24 cells, which was shown to be caused
in part due to increased AR gene copy number.
AR plays an important role in the survival of
prostate cancer cells (7).
Androgen deprivation therapy therefore has been used as a primary
treatment for patients with prostate cancer, but eventually becomes
ineffective, the condition of which is designated as CRPC. AR
mutation and amplification as well as deregulation of AR
co-regulator/activator have been proposed to be responsible for
castration-resistance (26,27).
It has been also demonstrated that AR splice variants influence
sensitivity to taxane, which is used for the treatment of CRPC
(28). On the other hand, AR has
been implicated in a variety of cancers including bladder cancer
(8–11) and also in drug-resistance cancers
such as doxorubicin-resistant bladder cancer (13) as well as cisplatin-resistant
endometrial cancer and taxol-resistant ovarian cancer (29,30).
As far as we know, our study is the first report that associated AR
with gemcitabine-resistance of cancer cells.
Since AR signaling has been implicated in cancer, it
would be possible that the high level of AR might favor growth and
survival of T24GR cells. Indeed, AR knockdown reduced growth of
gemcitabine-resistant T24GR cells. In addition, culture in the
medium containing CSS inhibited growth of T24GR cells
overexpressing AR, which was found to be mediated in part by
induction of cell cycle arrest. However, DHT did not promote
proliferation of T24GR cells cultured in the medium containing CSS.
CSS is depleted not only of steroid hormones but also of some
growth factors, cytokines and nutrients. The lack of proliferation
by DHT might be because AR signaling was activated by other factors
than androgens, which were present in FBS but absent in CSS. Growth
inhibition in the medium containing CSS may be ascribed to the
depletion of some important factors for the survival of T24GR
cells.
These results led us to hypothesize that
anti-androgen drugs could be a new treatment for chemoresistant
bladder cancer. Enzalutamide is a second-generation non-steroidal
AR inhibitor used for CRPC patients, which has a greater affinity
for AR than conventional anti-androgen agents such as bicalutamide
(18). It has been also shown that
the underlying mechanisms of enzalutamide action are distinct from
other anti-androgen drugs in that enzalutamide inhibits not only
androgen binding to the ligand-binding domain of AR but also AR
nuclear translocation, DNA binding and coactivator recruitment
(17). We demonstrated that
enzalutamide inhibited growth of T24GR cells more potently than T24
cells through inducing cell cycle arrest. This is consistent with
the finding that AR expression was much higher in T24GR cells than
in T24 cells. However, enzalutamide also inhibited growth of T24
cells expressing little or no AR, although to a lesser extent,
which was also associated with cell cycle arrest. These results
suggest the presence of AR-independent mechanisms of enzalutamide
to inhibit cell growth. Alternatively, a residual amount of AR in
T24 cells might take part in the inhibition. The underlying
mechanism remains to be clarified by future studies. Lastly, we
showed that the AR transcriptional activity was reduced by
enzalutamide treatment in T24GR cells cultured in normal medium.
Taken together, it was suggested that enzalutamide inhibits growth
of AR-positive T24GR cells through suppressing AR signaling which
is presumably activated by non-androgen ligands and also of
AR-negative T24 cells through AR-independent mechanism.
A recent report revealed that a risk for bladder
cancer recurrence was significantly lower in patients having
received androgen deprivation therapy for prostate cancer (31), suggesting a role of AR signaling
for cancer initiation, promotion and progression. Mashhadi et
al evaluated 120 bladder cancer patients and examined relations
between AR expression and tumor stage or grade (32). The patients with AR-positive tumor
showed worse prognosis than those with AR-negative tumor. However,
there are some conflicting reports showing that loss of AR was
associated with high-grade and high-stage bladder cancer (33). Thus, the significance of AR in
bladder cancer appears to be controversial in pathological and
clinical studies. It remains to be studied whether AR is
upregulated in patients with gemcitabine-resistant bladder cancer
and whether AR expression correlates with gemcitabine-efficacy.
In this study, we showed that enzalutamide inhibits
growth of AR-expressing bladder cancer cells with both acquired and
intrinsic gemcitabine-resistance through cell cycle arrest. We also
found that enzalutamide could attenuate gemcitabineresistance of
T24GR cells. However, enzalutamide that causes a cytostatic
response did not enhance but rather diminished an apoptotic
response mediated by gemcitabine. The precise mechanisms underlying
the combined effects remain to be studied.
It has been very recently reported that AR
expression is elevated in cisplatin-resistant bladder cancer cells
and is associated with resistance to cisplatin-based neoadjuvant
chemotherapy (34). Furthermore,
AR inactivation sensitized bladder cancer cells to cisplatin
treatment. The authors also showed that enzalutamide inhibits the
growth and AR expression/transcriptional activity in bladder cancer
cells (35). Taken together, our
study and others provided evidence that AR could be a therapeutic
target for bladder cancer patients who exhibited resistance to GC
therapy.
In conclusion, we demonstrated that enzalutamide
inhibits proliferation of AR-expressing bladder cancer cell lines
with acquired and intrinsic gemcitabine-resistance, suggesting that
enzalutamide might be effective for patients with advanced
gemcitabine-resistant bladder cancer with increased AR expression.
The efficacy of enzalutamide treatment in patients with advanced
bladder cancer needs to be established by clinical studies.
Abbreviations:
|
AR
|
androgen receptor
|
|
T24GR
|
gemcitabine-resistant T24 cells
|
|
CSS
|
charcoal-stripped serum
|
|
CRPC
|
castration-resistant prostate
cancer
|
|
FBS
|
fetal bovine serum
|
|
siRNA
|
small interfering RNA
|
|
DHT
|
dihydrotestosterone
|
|
CI
|
combination index
|
Acknowledgments
This study was supported in part by the Grant-in-Aid
from The Ministry of Education, Culture, Sports, Science, and
Technology of Japan (C-25861416 to T.K.).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sternberg CN, Yagoda A, Scher HI, Watson
RC, Ahmed T, Weiselberg LR, Geller N, Hollander PS, Herr HW, Sogani
PC, et al: Preliminary results of M-VAC (methotrexate, vinblastine,
doxorubicin and cisplatin) for transitional cell carcinoma of the
urothelium. J Urol. 133:403–407. 1985.PubMed/NCBI
|
|
3
|
Loehrer PJ Sr, Einhorn LH, Elson PJ,
Crawford ED, Kuebler P, Tannock I, Raghavan D, Stuart-Harris R,
Sarosdy MF, Lowe BA, et al: A randomized comparison of cisplatin
alone or in combination with methotrexate, vinblastine, and
doxorubicin in patients with metastatic urothelial carcinoma: A
cooperative group study. J Clin Oncol. 10:1066–1073.
1992.PubMed/NCBI
|
|
4
|
von der Maase H, Hansen SW, Roberts JT,
Dogliotti L, Oliver T, Moore MJ, Bodrogi I, Albers P, Knuth A,
Lippert CM, et al: Gemcitabine and cisplatin versus methotrexate,
vinblastine, doxorubicin, and cisplatin in advanced or metastatic
bladder cancer: Results of a large, randomized, multinational,
multicenter, phase III study. J Clin Oncol. 18:3068–3077.
2000.PubMed/NCBI
|
|
5
|
von der Maase H, Sengelov L, Roberts JT,
Ricci S, Dogliotti L, Oliver T, Moore MJ, Zimmermann A and Arning
M: Long-term survival results of a randomized trial comparing
gemcitabine plus cisplatin, with methotrexate, vinblastine,
doxorubicin, plus cisplatin in patients with bladder cancer. J Clin
Oncol. 23:4602–4608. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yafi FA, North S and Kassouf W: First- and
second-line therapy for metastatic urothelial carcinoma of the
bladder. Curr Oncol. 18:e25–e34. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Feldman BJ and Feldman D: The development
of androgenindependent prostate cancer. Nat Rev Cancer. 1:34–45.
2001. View
Article : Google Scholar
|
|
8
|
Chia K, O'Brien M, Brown M and Lim E:
Targeting the androgen receptor in breast cancer. Curr Oncol Rep.
17:42015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ha YS, Lee GT, Modi P, Kwon YS, Ahn H, Kim
WJ and Kim IY: Increased expression of androgen receptor mRNA in
human renal cell carcinoma cells is associated with poor prognosis
in patients with localized renal cell carcinoma. J Urol.
194:1441–1448. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma WL, Lai HC, Yeh S, Cai X and Chang C:
Androgen receptor roles in hepatocellular carcinoma, fatty liver,
cirrhosis and hepatitis. Endocr Relat Cancer. 21:R165–R182. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mikkonen L, Pihlajamaa P, Sahu B, Zhang FP
and Jänne OA: Androgen receptor and androgen-dependent gene
expression in lung. Mol Cell Endocrinol. 317:14–24. 2010.
View Article : Google Scholar
|
|
12
|
Wu JT, Han BM, Yu SQ, Wang HP and Xia SJ:
Androgen receptor is a potential therapeutic target for bladder
cancer. Urology. 75:820–827. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shiota M, Takeuchi A, Yokomizo A,
Kashiwagi E, Tatsugami K, Kuroiwa K and Naito S: Androgen receptor
signaling regulates cell growth and vulnerability to doxorubicin in
bladder cancer. J Urol. 188:276–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Siddique HR, Mishra SK, Karnes RJ and
Saleem M: Lupeol, a novel androgen receptor inhibitor: Implications
in prostate cancer therapy. Clin Cancer Res. 17:5379–5391. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li Y, Izumi K and Miyamoto H: The role of
the androgen receptor in the development and progression of bladder
cancer. Jpn J Clin Oncol. 42:569–577. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen Y, Clegg NJ and Scher HI:
Anti-androgens and androgen-depleting therapies in prostate cancer:
New agents for an established target. Lancet Oncol. 10:981–991.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tran C, Ouk S, Clegg NJ, Chen Y, Watson
PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, et al:
Development of a second-generation antiandrogen for treatment of
advanced prostate cancer. Science. 324:787–790. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jeon HG, Yoon CY, Yu JH, Park MJ, Lee JE,
Jeong SJ, Hong SK, Byun SS and Lee SE: Induction of caspase
mediated apoptosis and down-regulation of nuclear factor-κB and Akt
signaling are involved in the synergistic antitumor effect of
gemcitabine and the histone deacetylase inhibitor trichostatin A in
human bladder cancer cells. J Urol. 186:2084–2093. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miyamoto H, Yang Z, Chen YT, Ishiguro H,
Uemura H, Kubota Y, Nagashima Y, Chang YJ, Hu YC, Tsai MY, et al:
Promotion of bladder cancer development and progression by androgen
receptor signals. J Natl Cancer Inst. 99:558–568. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Duxbury MS, Ito H, Zinner MJ, Ashley SW
and Whang EE: RNA interference targeting the M2 subunit of
ribonucleotide reductase enhances pancreatic adenocarcinoma
chemosensitivity to gemcitabine. Oncogene. 23:1539–1548. 2004.
View Article : Google Scholar
|
|
22
|
Giovannetti E, Del Tacca M, Mey V, Funel
N, Nannizzi S, Ricci S, Orlandini C, Boggi U, Campani D, Del Chiaro
M, et al: Transcription analysis of human equilibrative nucleoside
transporter-1 predicts survival in pancreas cancer patients treated
with gemcitabine. Cancer Res. 66:3928–3935. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakahira S, Nakamori S, Tsujie M,
Takahashi Y, Okami J, Yoshioka S, Yamasaki M, Marubashi S, Takemasa
I, Miyamoto A, et al: Involvement of ribonucleotide reductase M1
subunit overexpression in gemcitabine resistance of human
pancreatic cancer. Int J Cancer. 120:1355–1363. 2007. View Article : Google Scholar
|
|
24
|
Ohhashi S, Ohuchida K, Mizumoto K, Fujita
H, Egami T, Yu J, Toma H, Sadatomi S, Nagai E and Tanaka M:
Down-regulation of deoxycytidine kinase enhances acquired
resistance to gemcitabine in pancreatic cancer. Anticancer Res.
28B:2205–2212. 2008.
|
|
25
|
Skrypek N, Duchêne B, Hebbar M, Leteurtre
E, van Seuningen I and Jonckheere N: The MUC4 mucin mediates
gemcitabine resistance of human pancreatic cancer cells via the
concentrative nucleoside transporter family. Oncogene.
32:1714–1723. 2013. View Article : Google Scholar
|
|
26
|
Harris WP, Mostaghel EA, Nelson PS and
Montgomery B: Androgen deprivation therapy: Progress in
understanding mechanisms of resistance and optimizing androgen
depletion. Nat Clin Pract Urol. 6:76–85. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tombal B: What is the pathophysiology of a
hormone-resistant prostate tumour? Eur J Cancer. 47(Suppl 3):
S179–S188. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Thadani-Mulero M, Portella L, Sun S, Sung
M, Matov A, Vessella RL, Corey E, Nanus DM, Plymate SR and
Giannakakou P: Androgen receptor splice variants determine taxane
sensitivity in prostate cancer. Cancer Res. 74:2270–2282. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen L, Chang WC, Hung YC, Chang YY, Bao
BY, Huang HC, Chung WM, Shyr CR and Ma WL: Androgen receptor
increases CD133 expression and progenitor-like population that
associate with cisplatin resistance in endometrial cancer cell
line. Reprod Sci. 21:386–394. 2014. View Article : Google Scholar :
|
|
30
|
Sun NK, Huang SL, Lu HP, Chang TC and Chao
CC: Integrative transcriptomics-based identification of cryptic
drivers of taxol-resistance genes in ovarian carcinoma cells:
Analysis of the androgen receptor. Oncotarget. 6:27065–27082. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Izumi K, Taguri M, Miyamoto H, Hara Y,
Kishida T, Chiba K, Murai T, Hirai K, Suzuki K, Fujinami K, et al:
Androgen deprivation therapy prevents bladder cancer recurrence.
Oncotarget. 5:12665–12674. 2014. View Article : Google Scholar
|
|
32
|
Mashhadi R, Pourmand G, Kosari F, Mehrsai
A, Salem S, Pourmand MR, Alatab S, Khonsari M, Heydari F, Beladi L,
et al: Role of steroid hormone receptors in formation and
progression of bladder carcinoma: A case-control study. Urol J.
11:1968–1973. 2014.PubMed/NCBI
|
|
33
|
Miyamoto H, Yao JL, Chaux A, Zheng Y, Hsu
I, Izumi K, Chang C, Messing EM, Netto GJ and Yeh S: Expression of
androgen and oestrogen receptors and its prognostic significance in
urothelial neoplasm of the urinary bladder. BJU Int. 109:1716–1726.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kashiwagi E, Ide H, Inoue S, Kawahara T,
Zheng Y, Reis LO, Baras AS and Miyamoto H: Androgen receptor
activity modulates responses to cisplatin treatment in bladder
cancer. Oncotarget. Jun 14–2016.Epub ahead of print. View Article : Google Scholar
|
|
35
|
Kawahara T, Ide H, Kashiwagi E,
El-Shishtawy KA, Li Y, Reis LO, Zheng Y and Miyamoto H:
Enzalutamide inhibits androgen receptor-positive bladder cancer
cell growth. Urol Oncol. 34:432.e15–23. 2016. View Article : Google Scholar
|