Introduction
Esophageal squamous cell carcinoma (ESCC), a
heterogeneous tumor containing various genetic and epigenetic
changes, is a leading cause of cancer death worldwide. Unlike
esophageal adenoma carcinoma (EAC), which is common in some
developed countries, ESCC is most prevalent in underdeveloped
regions (1). Despite recent
therapeutic advances, ESCC outcomes are unsatisfactory; only 15–25%
of ESCC patients survive 5 years (2). To investigate the driving mutation
behind ESCC and find a potential therapeutic target, recent
genomics studies using high-throughput sequencing have been done by
our group and others. However, these studies yielded few mutated
genes with frequencies as high as already known mutated genes such
as TP53, NOTCH1 and NFE2L2 (3–7).
RNA editing is an epigenetic process of
post-transcriptional modification. The most common human RNA
editing is the conversion of adenosine (A) to inosine (I), which is
catalyzed by adenosine deaminases acting on the RNA protein family
(ADARs). Three members of the ADAR gene family exist, ADAR1, ADAR2
(or ADARB1), and ADAR3. ADAR1 and ADAR2 are ubiquitously expressed,
whereas ADAR3 is localized to the brain (8). Because I is recognized as guanosine
(G) by translation machinery, ADARs may recode the transcriptome.
If the editing event occurs in the coding region, the protein
sequence, structure, and function can be altered.
Since the first report of altered RNA editing in
brain cancer in 2001 (9), more
studies have supported the idea that misregulation of ADARs are key
to tumorigenesis. ADAR1 and ADAR2 can edit many similar sites, but
other sites are clearly specific for one or the other (10), so dysregulation of either can
uniquely affect tumorigenesis. In fact, ADAR1 appears to act as an
oncogene in cancers (11–14), including ESCC (15), whereas ADAR2 was reported to be a
suppressor of brain tumors (16)
and hepatocellular carcinoma (14). Our previous genomic study indicated
that a gene amplification region containing microRNA, miR-4707-5p,
could aberrantly suppress ADAR2 in ESCC (3), but the function of ADAR2 in ESCC has
historically been poorly understood.
IGFBP7, or IGFBP-related protein 1 (IGFBP-rP1), has
been shown to bind to (and interfere with the activation of) IGF1R,
block downstream Akt signaling, and induce apoptosis (17). Although IGFBP7 is reported to be an
editing target (18,19), the relationship between ADAR2 and
IGFBP7 is not clear in ESCC. Thus, we report that IGFBP7 is a novel
target gene of ADAR2 in ESCCs, as evidenced by whole transcriptome
data and direct sequencing. We also describe the antitumor role and
mechanism of ADAR2 in ESCC in vitro and in vivo.
Materials and methods
Cell lines
An ESCC EC109 cell line was obtained from the Cell
Bank of the Chinese Academy of Sciences (Shanghai, China). The
Japanese ESCC KYSE30 cell line was generously provided by Dr Guan
Xinyuan from Sun Yat-sen University Cancer Center (SYSUCC). These
two cell lines were maintained in RPMI-1640. The HEK293T cell line
(used for lentivirus packaging) was obtained from SYSUCC and
maintained in DMEM. We added 10% fetal bovine serum and 1 mM
penicillin/streptomycin to the media.
Vector construction and transfection
assay
For siRNA transfection, Lipofectamine RNAiMAX
reagent (Invitrogen) was used according to kit directions. siRNAs
were purchased from RiboBio (Guangzhou, China). For gene transient
transfection, coding sequences were cloned into pcDNA3.1, and the
plasmids were transfected with Lipofectamine 2000 (Invitrogen). For
gene stable transfection, pLVX-IRES-Puro lentiviral expression
vector (Clontech) was used according to the manufacturer's
instructions.
Cell growth and clone formation
Cell growth was measured by MTT assay as previously
reported (20), using
2×103 cells seeded into each well of a 96-well plate. A
clone formation assay was carried out as previously reported
(15), using 1×103
cells seeded into each well of a 24-well plate.
Apoptosis assay
Apoptosis was measured using an Annexin V-FITC and
PI staining kit (Bestbio, Shanghai, China) according to the
manufacturer's protocol. Briefly, cells were collected, washed and
resuspended in 400 µl binding buffer containing 5 µl
Annexin V-FITC and 10 µl PI. After incubation, samples were
assessed with flow cytometry. We summed percents of early apoptotic
cells (Annexin V-positive, PI-negative) and late apoptotic or
necrotic cells (Annexin V/PI-double-positive) to obtain percent
apoptotic cells.
Animal tumorigenicity experiments
Animal experiments were approved by the
Institutional Animal Care and Use Committee at SYSUCC. Five-week
old female BALB/C-nu/nu nude mice were purchased from Experimental
Animal Center of Guangdong Province and used for the tumorigenicity
assay. Briefly, 1×106 cells resuspended in RPMI-1640
were injected into each mouse scapula. After 4 weeks, mice were
sacrificed and xenograft tumors were removed, photographed, and
weighed.
RNA sequencing
High-quality total RNA was extracted with TRIzol
(Invitrogen), and polyA+ mRNA was selected with
oligo(dT) magnetic beads and chemically fragmented. RNA fragments
were reverse transcribed to cDNA and the ~200 bp cDNA fraction was
selected to construct a sequencing library. RNA-Seq was carried out
on a HiSeq 2000 platform according to Illumina's protocol. After
filtration, clean reads were aligned to the human reference genome
(hg19) with Tophat2 (21). Single
nucleotide variants were assessed with GATK2 (22). SnpSift was used for variant
functional annotation (23).
Analysis of RNA editing with
direct-sequencing
Site-specific RNA editing was measured with RT-PCR
and direct sequencing (24).
Editing was calculated with ImageJ software (http://rsb.info.nih.gov/ij/). Primers for PCR include
FLNA-forward (CTTTGTTCCCGCTGAGATGG), FLNA-reverse
(TTCCAGACCTGCTCCGTAAG), FLNB-forward (GAAGAACTCACACTGCGTCC),
FLNB-reverse (CCTGCTCGGGTGGTGTTAAT), COPA-forward
(GCGAGGCATTGACTTCCATA), COPA-reverse (GGTCTCTGCGGAAAGTCTGA),
IGFBP7-forward (CTGCCCCTCTCCTCTTCCT), IGFBP7-reverse
(GGGATTCCGATGACCTCACA).
Western blotting
Western blotting was carried out according to
published standard methods. Antibodies used included ADAR2 (Sigma,
1:1,000), IGFBP7 (Abcam, 1:1,000), Matriptase (GeneTex, 1:1000),
pAkt (Ser473) (CST, 1:1,000), total Akt (CST, 1:1,000), pBAD (SAB,
1:1,000), BAD (CST, 1:1,000), GAPDH (CST, 1:1,000), His-tag (CST,
1:1000), c-Myc-tag (MBL, 1:1,000).
Clinical samples
Tissue microarray (TMA) blocks with 116 ESCC
samples, and three pairs of frozen ESCC primary tumor tissues and
adjacent normal esophageal tissues were obtained from the Biobank
of SYSUCC. Tissue samples were surgically removed between 2011 and
2014. Each participant signed an informed consent form and the
study was approved by the Human Ethics Committee.
Immunohistochemical staining and
assessment
Immunohistochemical staining for pAkt (antibody was
purchased from CST) and pBAD (antibody was purchased from SAB) was
conducted as previously reported (25). Two investigators scored
immunoreactivity independently, and staining was scored as follows:
i) percent positively staining cells: zero (0–10%), 1 (11–25%), 2
(26–50%), 3 (51–75%), and 4 (76–100%); and ii) staining intensity:
zero (no signal), 1 (weak), 2 (moderate), and 3 (strong). Staining
scores were multiplied by percent scores for a final score (range
0–12).
Statistical analysis
Unless otherwise specified, data are means ± SD of
three experiments. SPSS (version 16.0) software was used for data
analysis. P<0.05 was considered statistically significant.
Results
ADAR2 functions as a tumor suppressor
during ESCC progression
Previously, we reported that ADAR2 expression was
downregulated in ESCC compared with adjacent normal tissues, and
patients with less tumor ADAR2 expression had worse prognoses
(3). Furthermore, we found that
several known editing targets of ADAR2 were edited in normal
esophageal tissue, but they were almost not edited in the seven
ESCC cell lines (data not shown), which suggested that ADAR2 lost
its function in ESCC. To investigate the ADAR2 role during ESCC
progression, we used ADAR2 lentivirus to overexpress ADAR2 in ESCC
KYSE30 and EC109 cell lines. Overexpressing lines were KYSE30-AR2
and EC109-AR2, and the control lentiviral-infected cells were
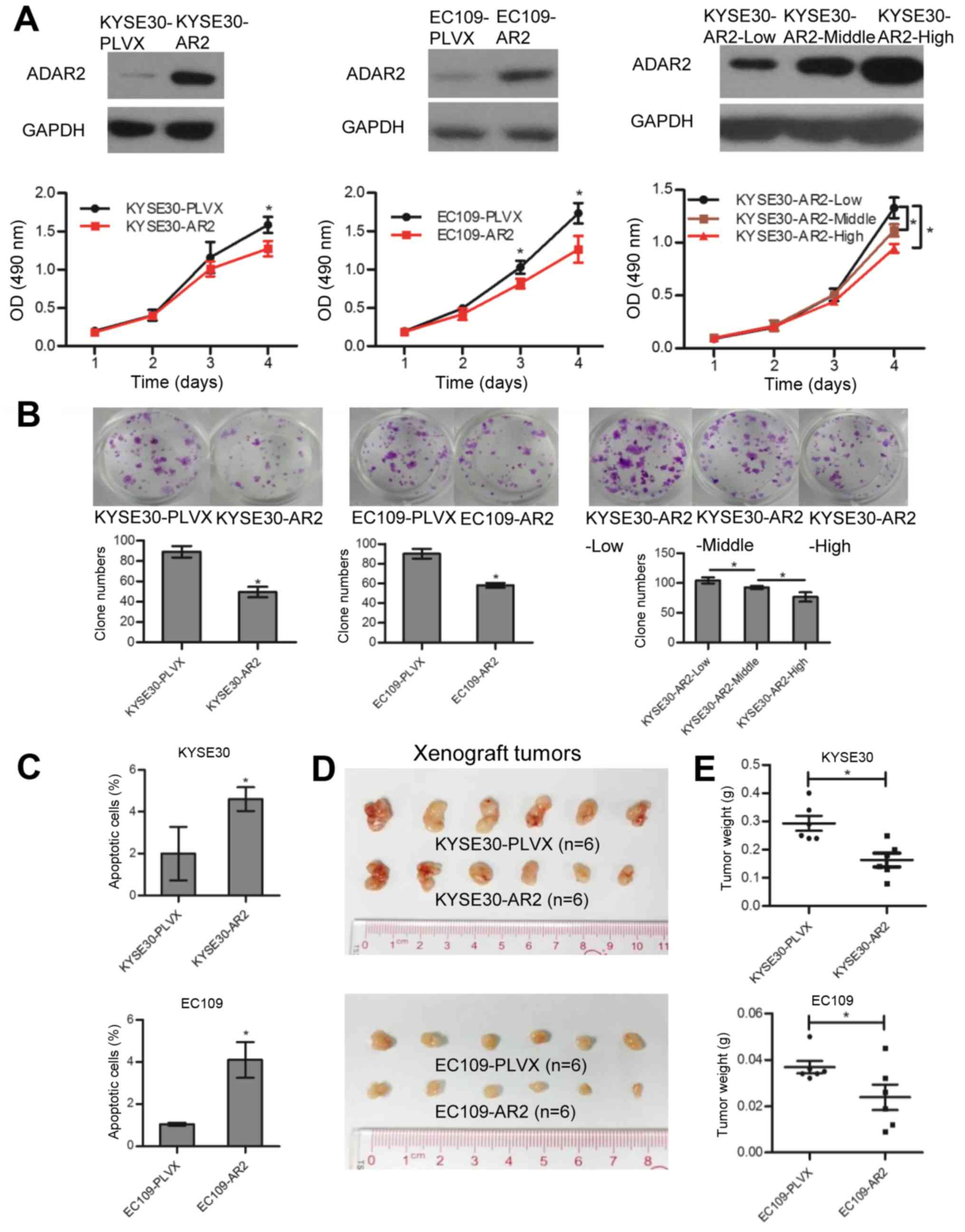
KYSE30-PLVX and EC109-PLVX. Upregulation of ADAR2 was confirmed
with western blotting (Fig. 1A).
In vitro studies confirmed that cells transfected with ADAR2
had less growth (Fig. 1A) and
colony formation (Fig. 1B)
compared to controls. Annexin V and PI staining revealed that
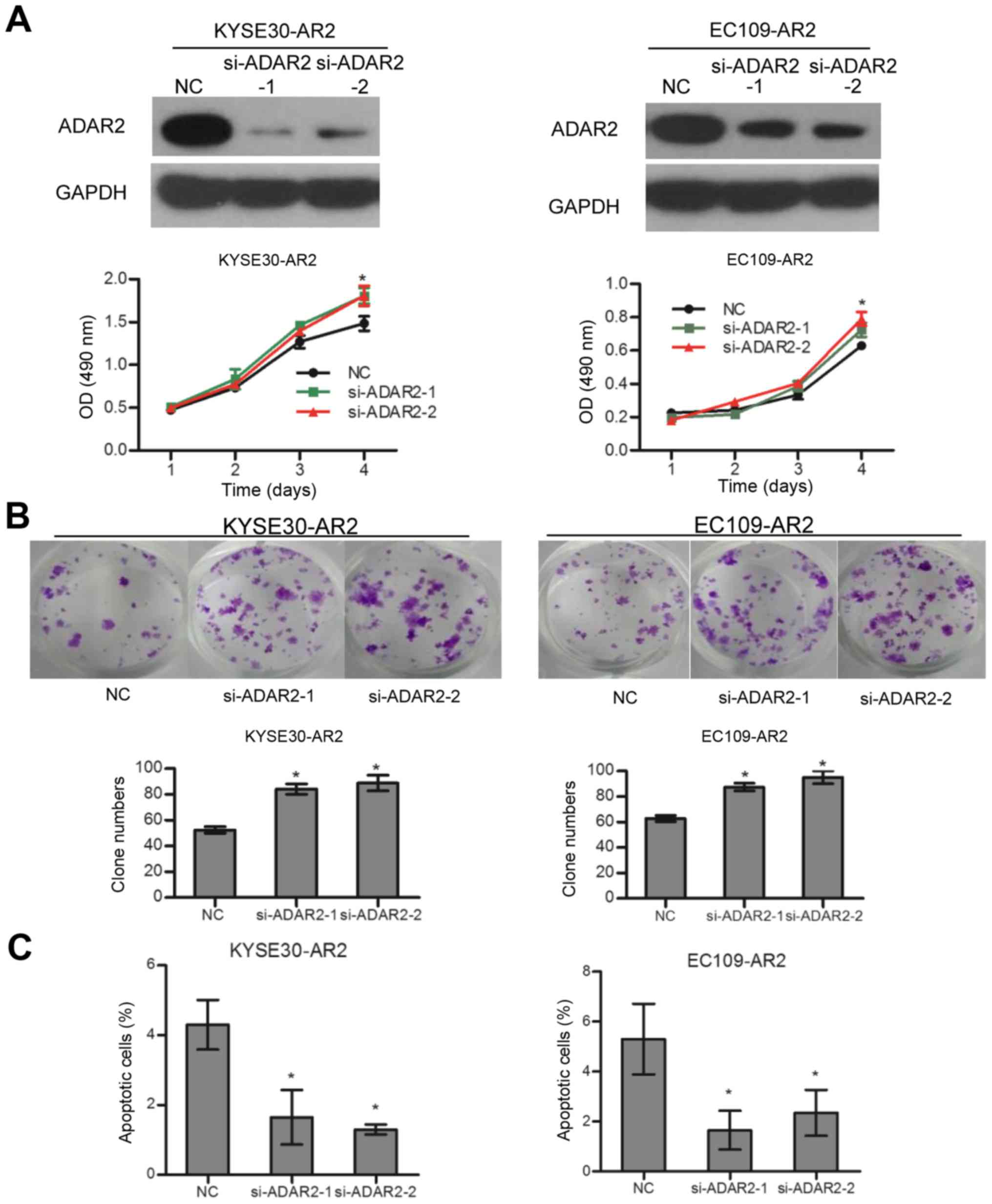
overexpression of ADAR2 induced apoptosis (Fig. 1C). In contrast, knockdown of ADAR2
in KYSE30-AR2 and EC109-AR2 reversed cell growth and apoptosis
effects to some extent (Fig. 2).
Moreover, ADAR2 overexpression decreased tumor growth in xenograft
tumor models (Fig. 1D and E).
Exogenous overexpression of ADAR2 can
edit the IGFBP7 transcript
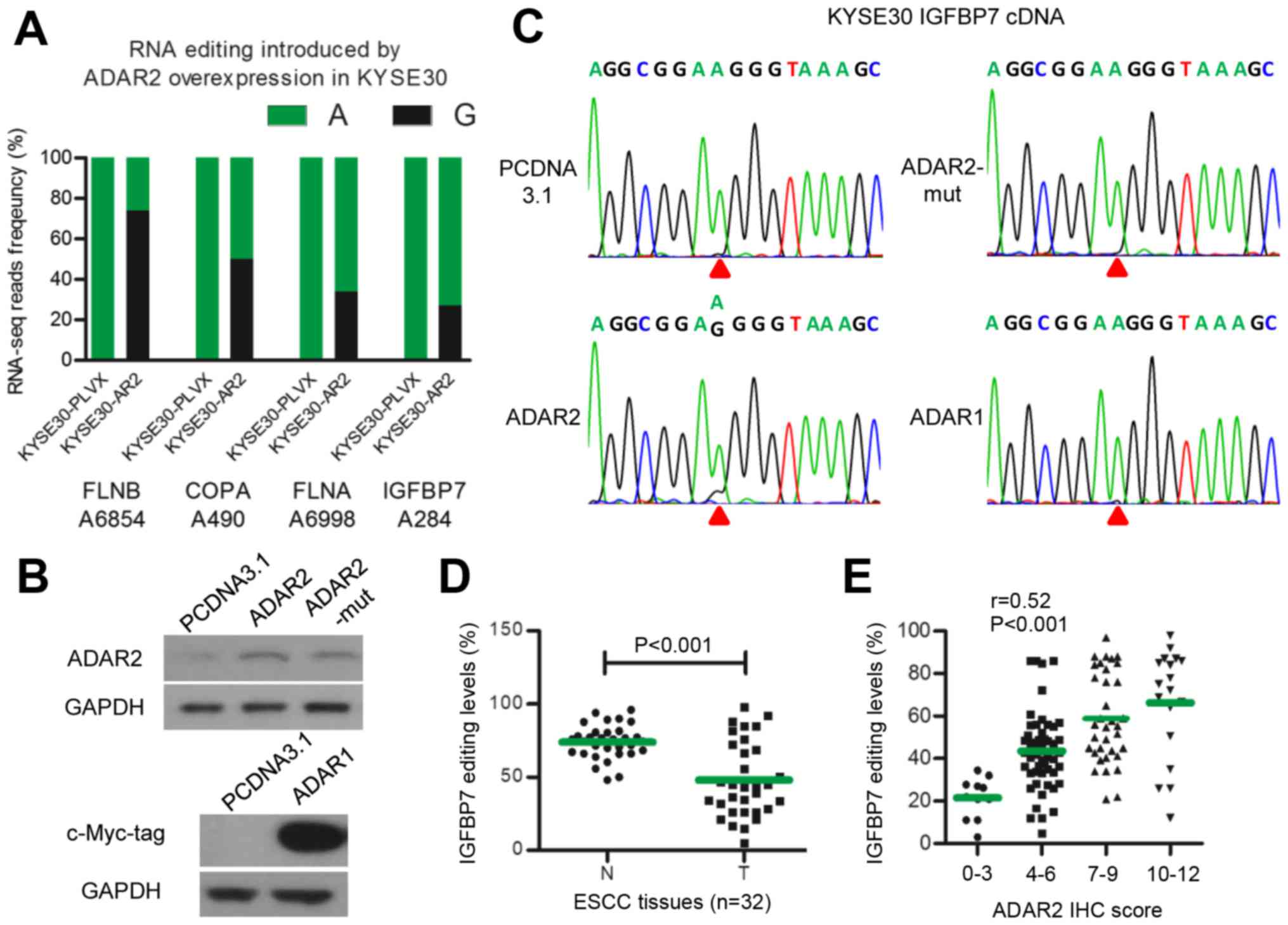
To explore target genes edited by ADAR2 in ESCC, we
used RNA-seq assay in KYSE30-PLVX and KYSE30-AR2 cells. We noted 19
A-to-G single nucleotide variation sites, G fractions of which were
≥10% higher in KYSE30-AR2 than in KYSE30-PLVX cells. These were
selected as potential A-to-I editing sites catalyzed by ADAR2. We
validated these sites by assaying corresponding sequences of DNA
and mRNA in KYSE30-AR2 and KYSE30-PLVX cells with direct
sequencing. Finally, transcripts of four genes were identified as
substrates of ADAR2 in ESCCs, including IGFBP7, FLNB, COPA, and
FLNA (Fig. 3A). IGFBP7 was the
only one reported to be associated with apoptosis (26–29),
so we focused on IGFBP7. The editing site in IGFBP7 identified with
RNA-seq was located at position 284 of the coding sequence,
changing codon 95 from AAG (lysine) to AIG (arginine), henceforth
termed K95R. K95R was validated in EC109-AR2 successfully (data not
shown). We constructed and transfected three plasmids expressing wt
ADAR1 and ADAR2, and RNA editing function loss type ADAR2
(ADAR2-mut) caused by deaminase domain mutation (30), respectively (Fig. 3B). Direct sequencing showed that
only wt ADAR2 could edit IGFBP7 K95R, and neither ADAR1 nor
ADAR2-mut could (Fig. 3C). We then
measured IGFBP7 editing in 32 pairs of ESCC tumors and adjacent
normal tissues. The data show that, similar to ADAR2 protein,
IGFBP7 K95R editing decreased in tumors (Fig. 3D). Moreover, IGFBP7 K95R editing
was moderately correlated with ADAR2 expression in 116 ESCC tumors
(r=0.52, P<0.001) (Fig. 3E).
Thus, these results suggested that IGFBP7 K95R is an ADAR2-specific
editing target in ESCCs.
ADAR2 promotes ESCC apoptosis depending
on IGFBP7 editing
IGFBP7 was previously reported to be an apoptotic
promoter in several other cancers (26–29).
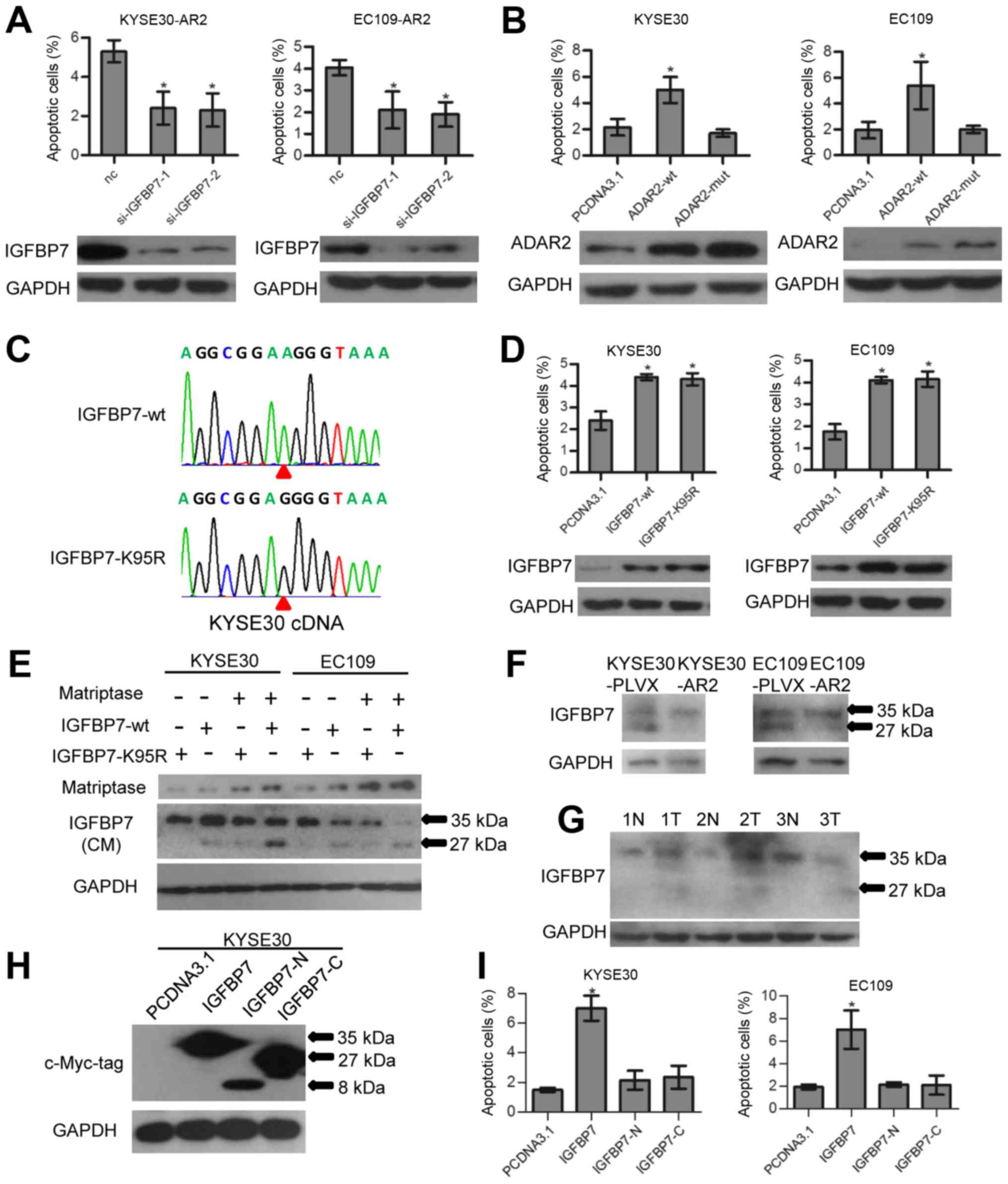
So, we investigated the role of IGFBP7 in ADAR2-mediated apoptosis
in ESCCs. At 48 h after transfection of IGFBP7 siRNAs and the siRNA
negative control (nc) in KYSE30-AR2 and EC109-AR2, cells were
harvested and apoptosis was assayed. Data show that knockdown of
IGFBP7 abolished ADAR2-induced apoptosis (Fig. 4A). Transfection of ADAR2-mut, the
RNA editing function loss type of ADAR2, did not induce apoptosis
as wt did (Fig. 4B). Thus, the
pro-apoptotic ability of ADAR2 might depend on IGFBP7 editing.
However, overexpression of wt IGFBP7 (IGFBP7-wt) could promote ESCC
apoptosis as well as the edited type (IGFBP7-K95R) (Fig. 4C and D). We conjectured that RNA
editing could fine tune IGFBP7 function by affecting its RNA or
protein stability, however, this fine-tuning was trivial when cells
expressed exogenous IGFBP7 abundantly.
RNA editing of K95R protects IGFBP7 from
matriptase proteolysis
According to previous reports, IGFBP7 is a secreted
protein and IGFBP7 K95R editing is located within a matriptase
protease recognition site (PRS) and inhibited matriptase mediated
proteolysis in test tubes (31).
In our study, we validated this phenomenon in culture medium of
ESCC cells. Matriptase and IGFBP7 (IGFBP7-wt and IGFBP7-K95R)
expression vectors were co-transfected into KYSE30 and EC109 as
described in Fig. 4E. Then, 48 h
after transfection, media were collected and assayed with western
blotting. Data show that IGFBP7-wt could be partially cleaved at
the 27 kDa C-terminus and the 8 kDa N-terminus (only the intact and
27 kDa C-terminus was measured with western blotting) by
overexpression of matriptase, whereas little IGFBP7-K95R was
cleaved (Fig. 4E). Moreover,
western blotting showed that ADAR2 overexpressing xenograft tumor
had less cleaved endogenous IGFBP7 (Fig. 4F). Thus, RNA editing of K95R can
protect IGFBP7 against matriptase proteolysis in ESCC culture and
xenografts. Measuring IGFBP7 expression in 3 pairs of surgically
removed ESCC tumors and adjacent normal tissues revealed that more
cleaved IGFBP7 was apparent in tumors (Fig. 4G), indicating a positive
correlation between IGFBP7 editing and tissue stability.
Intact rather than cleaved IGFBP7
promotes cell apoptosis
Full-length IGFBP7, and not the truncated
C-terminus, was reported to bind to IGF1R and inhibit downstream
Akt signaling in mouse embryonic fibroblasts (MEFs) (17). We investigated the function of
intact and cleaved IGFBP7 in ESCC cells with regard to apoptosis.
The intact form, N- and C-terminus of IGFBP7 expression constructs
(each contained a BM40 signal peptide sequence in the vector) were
transfected into ESCC cells separately. Only the intact form
induced apoptosis 48 h after transfection (Fig. 4H and I). Thus, protein integrity is
essential for IGFBP7 pro-apoptotic ability.
ADAR2 inhibits Akt signaling depending on
IGFBP7
IGFBP7 was reported to mediate apoptosis via
regulating Akt signaling (17), so
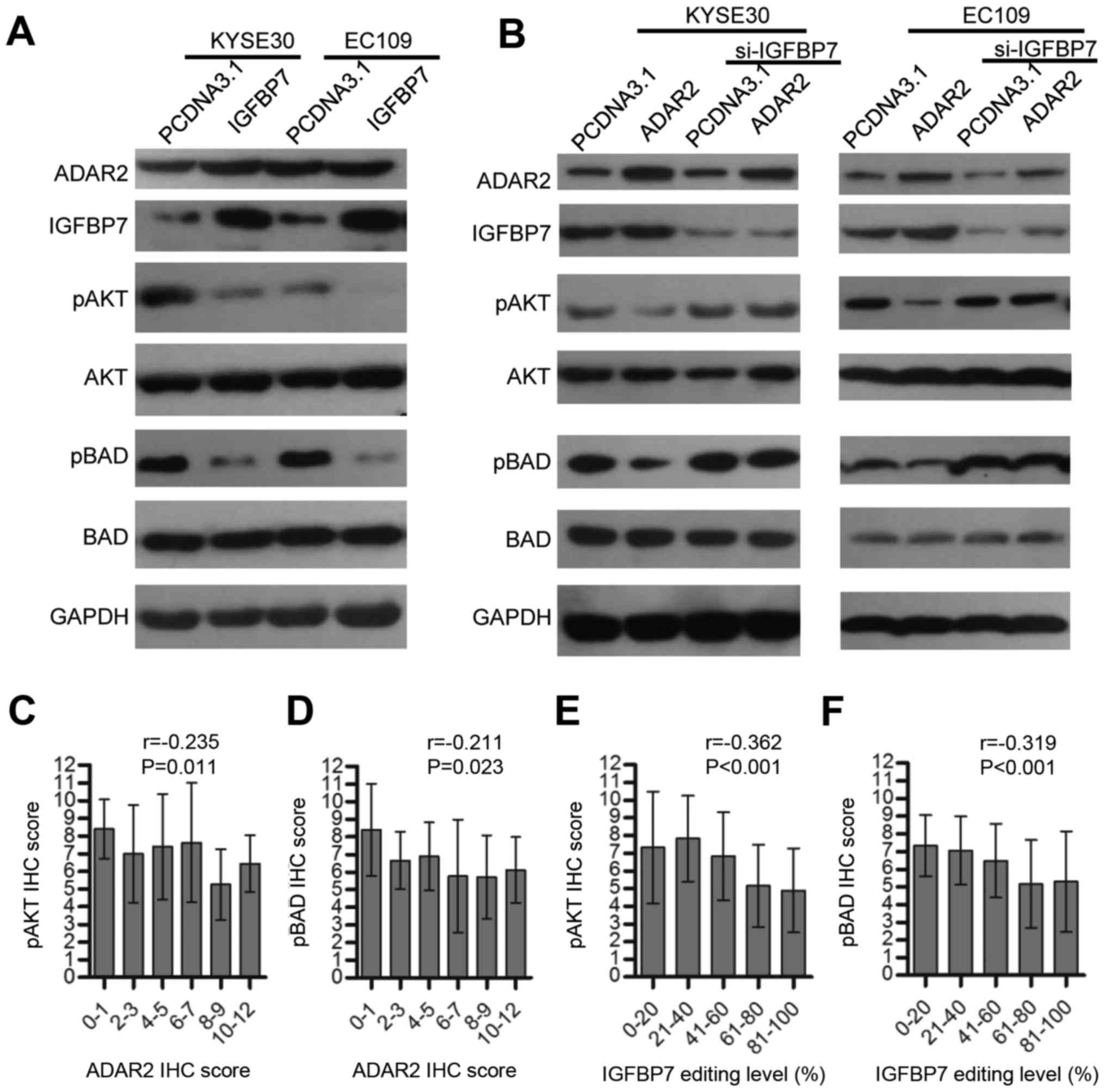
we validated this phenomenon in ESCC (Fig. 5A), and investigated whether ADAR2
could influence Akt signaling. As expected, ADAR2 overexpression in
KYSE30 and EC109 cells inhibited phosphorylation of Akt and a
downstream molecular BAD (BCL2 associated agonist of cell death)
(Fig. 5B). However, when IGFBP7
was knocked down, this effect was abolished (Fig. 5B). Tissue arrays containing 116
ESCC tumors were immunohistochemically assayed for pAkt and pBAD,
and data show that both were negatively correlated with ADAR2
(Fig. 5C and D) or IGFBP7 K95R
editing (Fig. 5E and F) to a
certain degree. Therefore, ADAR2 might protect BAD from
phosphorylation and induce apoptosis by promoting IGFBP7-mediated
Akt signaling inhibition.
Discussion
We report that ADAR2 promotes apoptosis by editing
and stabilizing IGFBP7 in ESCC. This is in contrast to ADAR1, which
acts as an oncogene in ESCC, and has a different editing profile
(15). On the other hand, editing
profiles of ADAR2 vary among different contexts. For example, in
glial cells, ADAR2 can edit GluR-B, an AMPA receptor subunit, and
recode Gln607 to Arg (Q/R). Decreased activity of ADAR2 leads to
reduced editing at GluR-B Q/R and promotes cell malignant
transformation (9,32,33).
However, in ESCC, GluR-B expression was below the detection
threshold for RT-PCR in our study (data not shown). Our findings
strongly suggest that, in esophageal epithelia, the
IGFBP7-IGF1R-Akt-BAD axis is a major ADAR2 target, which, when
inhibited, leads to abnormal cell survival. These observations are
consistent with previous findings that ESCC patients with tumors
strongly expressing both IGF-II and IGF1R had worse survival rates
(34), indicating that IGF1R might
be crucial for ESCC therapy.
Until now, only a few editing sites of ADAR2 have
been reported in neuronal tissues and beyond (14,35,36).
More detailed editing information for ADAR2 must be gathered,
especially for extraneuronal tissues. Here, we focused on editing
targets of ADAR2 in ESCC. As we previously found, ADAR2 expression
decreased in ESCC tumors compared to normal tissues, and the
preliminary experiment showed that ADAR2 activity in ESCC cell
lines were disrupted. Therefore, we overexpressed ADAR2 in KYSE30
cell line and RNAseq data revealed several editing targets of
ADAR2. All four genes were previously reported as A-to-I RNA
editing targets, and among them, FLNB, COPA, and FLNA were
confirmed ADAR2 substrates. None of the four ADAR2 target genes has
been correlated to ADAR2 function in any tissue type. One novelty
of our data is that we were the first to report that ADAR2 was
responsible for editing of IGFBP7 in ESCC and that ADAR2 regulates
IGFBP7.
As previously reported, overexpression of IGFBP7 can
promote apoptosis in cancers of the prostate (26), colorectum (27), thyroid (28), and breast (29). IGFBP7 could be silenced by promoter
hyper-methylation in multiple neoplastic tissues (37–39).
In this study, we report that ADAR2-mediated editing can protect
IGFBP7 from cleavage in vitro and in vivo, and the
integrity is essential for IGFBP7 to induce apoptosis in ESCCs.
This suggests a novel regulatory mechanism for IGFBP7 in ESCCs.
However, RNA editing may be a mild regulation mechanism of IGFBP7.
We found that overexpression of wt IGFBP7 could promote ESCC
apoptosis as did the edited type (Fig.
4C and D). Perhaps the abundance of exogenous IGFBP7 was beyond
the regulating range of RNA editing.
BAD is a key regulator of programmed cell death, and
its pro-apoptotic activity can be inhibited by phosphorylation
regulated by Akt. Our results suggest that ADAR2 can inhibit Akt
signaling, which, in turn, inhibits phosphorylation and releases
pro-apoptotic activity of BAD. However, cancer cells might escape
pro-apoptotic effects of ADAR2 by decreased transcription of
IGFBP7, constitutive activation of AKT signaling due to RAS or PTEN
mutations, EGFR or HER2 amplification, or INSR activation.
We did not study the effects of ADAR2 on endogenous
IGFBP7 in a culture medium because it was below the threshold of
detection by western blotting. However, we measured endogenous
IGFBP7 in xenograft tumors and noted significantly less cleaved
IGFBP7 in ADAR2 overexpressing xenograft tumors compared to
control. More sensitive methods will be required in future
studies.
Several recent whole-genome sequencing studies
indicate that recurrent, somatic single-nucleotide variations are
rare in ESCC (4–7). A-to-I editing reduction of IGFBP7
K95R to cause single nucleotide changes, however, offers consistent
alteration across ESCC patients. In this regard, this
post-transcriptional change might be more essential for cell
transformation. On the other hand, distinct to genomic variations,
RNA editing is a variation that can be spatiotemporally controlled
by ADARs, permitting the design of target therapies to eliminate
these variations. Our previous study indicated that miR-4707-5p
aberrant overexpression in ESCC reduced ADAR2 protein, and patients
with low ADAR2-expressing tumors had worse prognoses. Therefore,
the miR-4707/ADAR2/IGFBP7 axis may be a useful target for ESCC
treatment. Overall, we offer evidence that ADAR2 promotes apoptosis
in ESCC by editing and protecting IGFBP7 from cleavage, and these
data suggest that ADAR2 may have substantial efficacy for treating
ESCC with minimal, if any, effects on normal tissues.
Acknowledgments
We thank Professor Xin-Yuan Guan of Sun Yat-sen
University Cancer Center for the ESCC cell line. We also thank the
Bank of Tumor Resources, Sun Yat-sen University Cancer Center for
providing ESCC samples for this study. This study was supported by
the grants from the National Natural Science Foundation of China
(grant no. 81502056), the National Science Fund for Distinguished
Young Scholars of China (grant no. 81325018) and the Key Project
for International Cooperation and Exchange of the National Natural
Science Foundation of China (grant no. 81220108022).
References
|
1
|
Pickens A and Orringer MB: Geographical
distribution and racial disparity in esophageal cancer. Ann Thorac
Surg. 76:S1367–S1369. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Enzinger PC and Mayer RJ: Esophageal
cancer. N Engl J Med. 349:2241–2252. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qin HD, Liao XY, Chen YB, Huang SY, Xue
WQ, Li FF, Ge XS, Liu DQ, Cai Q, Long J, et al: Genomic
characterization of esophageal squamous cell carcinoma reveals
critical genes underlying tumorigenesis and poor prognosis. Am J
Hum Genet. 98:709–727. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Song Y, Li L, Ou Y, Gao Z, Li E, Li X,
Zhang W, Wang J, Xu L, Zhou Y, et al: Identification of genomic
alterations in oesophageal squamous cell cancer. Nature. 509:91–95.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun
ZM, Zhang F, Zhao ZR, Li ZT, Liu ZY, et al: Genetic landscape of
esophageal squamous cell carcinoma. Nat Genet. 46:1097–1102. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang L, Zhou Y, Cheng C, Cui H, Cheng L,
Kong P, Wang J, Li Y, Chen W, Song B, et al: Genomic analyses
reveal mutational signatures and frequently altered genes in
esophageal squamous cell carcinoma. Am J Hum Genet. 96:597–611.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin DC, Hao JJ, Nagata Y, Xu L, Shang L,
Meng X, Sato Y, Okuno Y, Varela AM, Ding LW, et al: Genomic and
molecular characterization of esophageal squamous cell carcinoma.
Nat Genet. 46:467–473. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nishikura K: Functions and regulation of
RNA editing by ADAR deaminases. Annu Rev Biochem. 79:321–349. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Maas S, Patt S, Schrey M and Rich A:
Underediting of glutamate receptor GluR-B mRNA in malignant
gliomas. Proc Natl Acad Sci USA. 98:14687–14692. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hogg M, Paro S, Keegan LP and O'Connell
MA: RNA editing by mammalian ADARs. Adv Genet. 73:87–120.
2011.PubMed/NCBI
|
|
11
|
Shah SP, Morin RD, Khattra J, Prentice L,
Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, et al:
Mutational evolution in a lobular breast tumour profiled at single
nucleotide resolution. Nature. 461:809–813. 2009. View Article : Google Scholar
|
|
12
|
Chen L, Li Y, Lin CH, Chan TH, Chow RK,
Song Y, Liu M, Yuan YF, Fu L, Kong KL, et al: Recoding RNA editing
of AZIN1 predisposes to hepatocellular carcinoma. Nat Med.
19:209–216. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang Q, Crews LA, Barrett CL, Chun HJ,
Court AC, Isquith JM, Zipeto MA, Goff DJ, Minden M, Sadarangani A,
et al: ADAR1 promotes malignant progenitor reprogramming in chronic
myeloid leukemia. Proc Natl Acad Sci USA. 110:1041–1046. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chan TH, Lin CH, Qi L, Fei J, Li Y, Yong
KJ, Liu M, Song Y, Chow RK, Ng VH, et al: A disrupted RNA editing
balance mediated by ADARs (Adenosine DeAminases that act on RNA) in
human hepatocellular carcinoma. Gut. 63:832–843. 2014. View Article : Google Scholar :
|
|
15
|
Qin YR, Qiao JJ, Chan TH, Zhu YH, Li FF,
Liu H, Fei J, Li Y, Guan XY and Chen L: Adenosine-to-inosine RNA
editing mediated by ADARs in esophageal squamous cell carcinoma.
Cancer Res. 74:840–851. 2014. View Article : Google Scholar
|
|
16
|
Galeano F, Rossetti C, Tomaselli S,
Cifaldi L, Lezzerini M, Pezzullo M, Boldrini R, Massimi L, Di Rocco
CM, Locatelli F, et al: ADAR2-editing activity inhibits
glioblastoma growth through the modulation of the
CDC14B/Skp2/p21/p27 axis. Oncogene. 32:998–1009. 2013. View Article : Google Scholar :
|
|
17
|
Evdokimova V, Tognon CE, Benatar T, Yang
W, Krutikov K, Pollak M, Sorensen PH and Seth A: IGFBP7 binds to
the IGF-1 receptor and blocks its activation by insulin-like growth
factors. Sci Signal. 5:ra922012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gommans WM, Tatalias NE, Sie CP, Dupuis D,
Vendetti N, Smith L, Kaushal R and Maas S: Screening of human SNP
database identifies recoding sites of A-to-I RNA editing. RNA.
14:2074–2085. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Levanon EY, Hallegger M, Kinar Y, Shemesh
R, Djinovic-Carugo K, Rechavi G, Jantsch MF and Eisenberg E:
Evolutionarily conserved human targets of adenosine to inosine RNA
editing. Nucleic Acids Res. 33:1162–1168. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qi F, Cai P, Liu X, Peng M and Si G:
Adenovirus-mediated P311 inhibits TGF-β1-induced
epithelial-mesenchymal transition in NRK-52E cells via
TGF-β1-Smad-ILK pathway. Biosci Trends. 9:299–306. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim D, Pertea G, Trapnell C, Pimentel H,
Kelley R and Salzberg SL: TopHat2: Accurate alignment of
transcriptomes in the presence of insertions, deletions and gene
fusions. Genome Biol. 14:R362013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cingolani P, Patel VM, Coon M, Nguyen T,
Land SJ, Ruden DM and Lu X: Using Drosophila melanogaster as a
model for genotoxic chemical mutational studies with a new program,
SnpSift. Front Genet. 3:352012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li JB, Levanon EY, Yoon JK, Aach J, Xie B,
Leproust E, Zhang K, Gao Y and Church GM: Genome-wide
identification of human RNA editing sites by parallel DNA capturing
and sequencing. Science. 324:1210–1213. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu DQ, Li FF, Zhang JB, Zhou TJ, Xue WQ,
Zheng XH, Chen YB, Liao XY, Zhang L, Zhang SD, et al: Increased
RIPK4 expression is associated with progression and poor prognosis
in cervical squamous cell carcinoma patients. Sci Rep. 5:119552015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mutaguchi K, Yasumoto H, Mita K, Matsubara
A, Shiina H, Igawa M, Dahiya R and Usui T: Restoration of
insulin-like growth factor binding protein-related protein 1 has a
tumor-suppressive activity through induction of apoptosis in human
prostate cancer. Cancer Res. 63:7717–7723. 2003.PubMed/NCBI
|
|
27
|
Ruan W, Xu E, Xu F, Ma Y, Deng H, Huang Q,
Lv B, Hu H, Lin J, Cui J, et al: IGFBP7 plays a potential tumor
suppressor role in colorectal carcinogenesis. Cancer Biol Ther.
6:354–359. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vizioli MG, Sensi M, Miranda C, Cleris L,
Formelli F, Anania MC, Pierotti MA and Greco A: IGFBP7: An
oncosuppressor gene in thyroid carcinogenesis. Oncogene.
29:3835–3844. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Benatar T, Yang W, Amemiya Y, Evdokimova
V, Kahn H, Holloway C and Seth A: IGFBP7 reduces breast tumor
growth by induction of senescence and apoptosis pathways. Breast
Cancer Res Treat. 133:563–573. 2012. View Article : Google Scholar
|
|
30
|
Cenci C, Barzotti R, Galeano F, Corbelli
S, Rota R, Massimi L, Di Rocco C, O'Connell MA and Gallo A:
Down-regulation of RNA editing in pediatric astrocytomas: ADAR2
editing activity inhibits cell migration and proliferation. J Biol
Chem. 283:7251–7260. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Godfried Sie C, Hesler S, Maas S and
Kuchka M: IGFBP7's susceptibility to proteolysis is altered by
A-to-I RNA editing of its transcript. FEBS Lett. 586:2313–2317.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada
N, Hagimura N, Okado H, Miwa A, Kurihara H, Nakazato Y, Tamura M,
et al: Blockage of Ca(2+)-permeable AMPA receptors suppresses
migration and induces apoptosis in human glioblastoma cells. Nat
Med. 8:971–978. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ishiuchi S, Yoshida Y, Sugawara K, Aihara
M, Ohtani T, Watanabe T, Saito N, Tsuzuki K, Okado H, Miwa A, et
al: Ca2+-permeable AMPA receptors regulate growth of
human glioblastoma via Akt activation. J Neurosci. 27:7987–8001.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Imsumran A, Adachi Y, Yamamoto H, Li R,
Wang Y, Min Y, Piao W, Nosho K, Arimura Y, Shinomura Y, et al:
Insulin-like growth factor-I receptor as a marker for prognosis and
a therapeutic target in human esophageal squamous cell carcinoma.
Carcinogenesis. 28:947–956. 2007. View Article : Google Scholar
|
|
35
|
Riedmann EM, Schopoff S, Hartner JC and
Jantsch MF: Specificity of ADAR-mediated RNA editing in newly
identified targets. RNA. 14:1110–1118. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Stulić M and Jantsch MF: Spatio-temporal
profiling of Filamin A RNA-editing reveals ADAR preferences and
high editing levels outside neuronal tissues. RNA Biol.
10:1611–1617. 2013. View Article : Google Scholar
|
|
37
|
Kanemitsu N, Kato MV, Miki T, Komatsu S,
Okazaki Y, Hayashizaki Y and Sakai T: Characterization of the
promoter of the murine mac25 gene. Biochem Biophys Res Commun.
279:251–257. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ahmed S, Yamamoto K, Sato Y, Ogawa T,
Herrmann A, Higashi S and Miyazaki K: Proteolytic processing of
IGFBP-related protein-1 (TAF/angiomodulin/mac25) modulates its
biological activity. Biochem Biophys Res Commun. 310:612–618. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin J, Lai M, Huang Q, Ma Y, Cui J and
Ruan W: Methylation patterns of IGFBP7 in colon cancer cell lines
are associated with levels of gene expression. J Pathol. 212:83–90.
2007. View Article : Google Scholar : PubMed/NCBI
|