Introduction
Classical Hodgkin lymphoma (cHL) predominantly is a
B-cell malignancy with unique features. Only ~1–5% of the whole
tumor mass is represented by Hodgkin and Reed Sternberg (HRS)
cells, the malignant component of cHL. The major part of the tumor
mass is composed of an inflammatory infiltrate that provides a
protective and supportive microenvironment for the tumor cells
(1). Despite the 5 year overall
survival of 81%, cured patients often suffer from treatment-related
complications like pulmonary fibrosis, sterility, and
cardiovascular disorders (2).
Moreover, they may develop secondary tumors such as acute leukemia
and myelodysplastic syndrome (MDS) (2). Thus, a substantial percentage of
treatment failures and the high frequency of adverse effects of the
chemotherapy warrants the search for novel therapeutic
approaches.
Although the HRS cells are derived from germinal or
post germinal center B-cells, they have lost most of their B-cell
phenotype and express surface markers of various other lineages of
the immune system (3). We and
others have shown that epigenetic silencing contributes to the
extinguishing of the B-cell program in cHL (4–6). In
particular, promoter CpG island hyper-methylation was identified as
a major cause of silencing of PU.1/SPI1, BOB.1/OBF.1/POU2AF1 and
other important regulators of the B-cell program (6). Of note, PU.1 has been shown to act as
a tumor suppressor in cHL (7).
Additionally, known tumor suppressor genes like p16 and p18INK4c
are also epigenetically silenced in cHL (5,7–10).
Epigenetic processes are known to contribute to the
oncogenic program in virtually all tumor types by activation of
proto-oncogenes or by silencing of tumor suppressor genes (11,12).
Unlike the genomic aberrations, epigenetic modifications including
CpG dinucleotide methylation and histone deacetylation are
reversible by inhibitors of DNA methylation and of histone
deacetylases (HDAC), respectively. Currently, the constantly
growing list of epigenetic modifiers makes epigenetic therapy one
of the most quickly emerging branches of targeted cancer therapy.
For some tumors epigenetic therapy is already established as an
efficient and less harmful than classical chemotherapy approach to
cancer treatment (13). HDAC
inhibitors were successfully studied for treatment of cHL.
Combination of the pan-HDAC inhibitor vorinostat with mammalian
target of rapamycin (mTOR) inhibitor sirolimus induced complete
regression of primary refractory cHL (14). Another pan-HDAC inhibitor
panobinostat reduced tumor mass in 74% of patients with relapsed
cHL including 23% partial and 4% complete remission (15). HDAC inhibitor entinostat (SNDX-275)
has been shown to induce apoptosis in cHL cell lines and entered
clinical trials (16).
The DNA methyltransferase (DNMT) inhibitors, in
particular decitabine (5-aza-2′-deoxycytidine/5-Aza-dC), is used
for treatment of acute myeloid leukemia (AML) (17) and is already approved for treatment
of patients with MDS (18). To our
knowledge DNMT inhibitors are not specifically tested for use in
patients with cHL. However, complete regression of relapsed
metastatic cHL was described as a fortunate side-effect in a MDS
patient treated with the 5-Aza-dC related demethylating agent
5-azacytidine (19). Both
inhibitors have similar function and demonstrated similar efficacy
against MDS and AML (20).
5-Aza-dC has often been used by us and others to demonstrate
epigenetic mechanisms of gene silencing in cHL cell lines, but
assessment of its cytotoxic effects was neglected (4,6,21).
In this study we focused on the possible mechanisms
of the antitumor effect of demethylating agents in cHL. Using GEP
we could show that along with the reactivation of tumor suppressor
genes 5-Aza-dC also induced several oncogenic pathways such as
MEK/ERK, JAK-STAT and NF-κB. Here we demonstrate that
pharmacological inactivation of some of these pro-survival pathways
significantly increases the anti-tumor effect of 5-Aza-dC against
cHL.
Materials and methods
Antibodies and inhibitors
The primary monoclonal mouse antibodies against BCL2
(#sc-509) and ACTB (#sc-130300), as well as the primary polyclonal
rabbit antibody against p65 (#sc-372), and the secondary goat
anti-rabbit (#sc-2004) and anti-mouse (#sc-2005) antibodies were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
The primary monoclonal mouse antibodies against CDKN1A (#556431)
and GAGE (#611746) were acquired from BD Bioscience (Heidelberg,
Germany). The primary monoclonal rabbit antibodies against BCL2L1
(#2764), and p44/42 MAPK (ERK1/2) (#4695), as well as the primary
polyclonal rabbit antibody against phospho-p44/42 MAPK (pERK1/2)
(#9101) were purchased from Cell Signaling Technology (Leiden, The
Netherlands). The primary monoclonal mouse anti-SSX antibody
(#OP196) was acquired from Oncogene Research Products (Boston, MA,
USA) and the primary polyclonal rabbit anti-TUBB antibody (#ab6046)
from Abcam (Cambridge, UK).
JQ1 was kindly provided by J.E. Bradner (Division of
Hematologic Neoplasia, Dana-Faber Cancer Institute, Boston, MA,
USA). 5-aza-2′-deoxycytidine (5-Aza-dC) was purchased from
Calbiochem (Darmstadt, Germany) and KP372-1 from MoBiTec GmbH
(Göttingen, Germany). ABT263, fedratinib, QNZ (EVP4593) and SH-4-54
were acquired from Selleckchem (Munich, Germany). UO126 was
purchased from Cell Signaling Technology.
Cell lines and short tandem repeat (STR)
analysis
The cHL cell lines L428, and L1236 were cultured in
complete RPMI-1640 medium as described earlier (6). The cell lines were obtained from DSMZ
(Braunschweig, Germany). The authenticity of the cell lines was
proved by STR DNA typing using GenomeLab GexP™ Genetic Analysis
System (Sciex, Darmstadt, Germany) and GenomeLabHuman STR primer
set (Beckman Coulter, Krefeld, Germany) as described in user's
manual. The obtained STR profiles were compared with comprehensive
DSMZ database of STR cell line profiles using 'Online STR Analysis'
(www.dsmz.de, 13.08.2015). 5-Aza-dC, if not mentioned
differently, was used at a concentration of 1 µM. The cells
were seeded at a density of 0.5×106/ml and incubated
with 5-Aza-dC for 24 h mimicking clinical relevant bolus dose and
time schedule. After complete exchange of medium the cells were
incubated for another 3 days before harvesting. The cells were
counted by a haemocytometer.
Gene expression profiling (GEP)
L428, and L1236 cell lines were seeded at a density
of 0.5×106/ml and incubated with 5-Aza-dC at a
concentration of 1 µM for 24 h mimicking clinical relevant
bolus dose and time schedule. After complete change of medium the
cells were incubated for another 3 days before harvesting.
Isolation of the RNA was performed using the RNeasy mini kit
(Qiagen, Hilden, Germany) according to the user's manual. Two
biological replicates were analyzed for each cell line. Labeling of
RNA (cRNA synthesis) was performed by The Microarray Facility,
University of Tübingen (Tübingen, Germany) according to a standard
Affymetrix® procedure (One-Cycle Target Labeling kit;
Affymetrix UK Ltd., High Wycombe, UK). The biotinylated
complementary RNA (cRNA) was cleaned and fragmented by a 35 min
incubation at 95°C. GeneChip® Human genome U133 Plus 2.0
array (spotted with 1,300,000 oligonucleotides informative for
47,000 transcripts originated from 39,000 genes, Affymetrix UK
Ltd.) was used as described in user's manual. We used GeneSifter
software for data processing and analysis (www.genesifter.net; 28.09.2015; PerkinElmer, Waltham,
MA, USA). Global background correction, across array normalization,
and log2 transformation of PM values were done by robust
multi-array average (RMA) method. The 3623 probe sets, modulated by
5-Aza-dC at least >3-fold in one of the cell lines, were
filtered (threshold X3, Benjamini and Hochberg correction, adjusted
P<0.05). The microarray data were deposited in the Gene
Expression Omnibus (GEO) database under accession number
GSE86068.
Gene set enrichment analysis (GSEA)
We used the Broad Institute's Molecular Signatures
Database gene set collections 'C2-curated gene sets', and
'C6-oncogenic signatures' for GSEA (http://www.broadinstitute.org/gsea/index.jsp,02.10.2015)
(22,23).
Immunoblot
Cells (1×106) were lysed in 100 µl
Laemmli buffer containing 6 M urea. Then 25 µl of the
protein extracts were separated on 10% polyacrylamide gels by SDS
page using the Mini-PROTEAN® Tetra vertical
Electrophoresis (Bio-Rad Laboratories, Munich, Germany) and blotted
to a nitrocellulose membrane using the Mini-Electrophoretic
Blotting system (CBS Scientific, Del Mar, CA, USA). The membranes
were blocked with 5% dry milk in PBS and then incubated with the
primary antibodies at 4°C overnight. After several washing steps
the membranes were incubated for 1 h with the secondary antibody.
Signals were detected with the Super Signal West Dura Extended
Duration Substrate (Thermo Scientific, Rockford, IL, USA). The
representative immunoblots were quantified with help of ImageJ
software (imagej.net; 14.01.2016).
Cell viability assay
For measurement of cell viability 50 µl
medium containing decreasing concentrations of the specific
inhibitors were pipetted in triplicate into flat bottomed 96-well
plates creating serial dilutions. Cells/well (2×104)
were seeded and cultured for 6 days. Then 25 µl
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
solution from Sigma-Aldrich (Darmstadt, Germany) (5 mg/ml in PBS)
per well were added and incubated for 2 h. After dissolving the
formazan crystals with MTT-lysis-buffer the absorbance was detected
with the Microplate Reader SpectraMax 190 (Molecular Devices,
Biberach, Germany) at a wavelength of 570 nm. All experiments were
repeated at least two times. The IC50 values were
generated by using the software CompuSyn (http://www.combosyn.com/, 16.03.2016) with
IC50 being the concentration where 50% of the maximum
possible inhibition was reached (24,25).
Combination plot
5-Aza-dC was combined with the different inhibitors
using decreasing concentrations of the two drugs at a ratio of
IC50 (5-Aza-dC)/IC50 (inhibitor). The drugs
were added simultaneously to the cells. The experiment was
performed as described in 'Cell viability assay'. The data were
analyzed applying the combination index (CI) theorem and plot of
Chou and Talalay (24,25) using the CompuSyn software
(http://www.combosyn.com/16.03.2016).
Dual luciferase reporter assay and
electrophoretic mobility shift assay (EMSA)
Nuclear extraction and EMSA were performed as
described earlier (26). The
experiment was performed twice. For the reporter assay
1×107 cells were transfected by electroporation with the
reporter vectors containing binding sides either for STAT3
(Panomics; Gentaur Molecular Products, Aachen, Germany), STAT6
(27) or NF-κB (28) using the Bio-Rad GenePulser II
(Bio-Rad Laboratories). The luciferase activity was normalized by
cotransfection of the ubi-Renilla vector. After 24 h of
incubation the luciferase activity was measured with help of the
Dual Luciferase Reporter assay system (Promega, Mannheim, Germany)
using a luminometer (Berthold Technologies, Bad Wildbach, Germany).
This experiment was performed in duplicate and repeated three
times.
Statistical analysis
The values of the Dual Luciferase reporter assay and
the measurement of cell number are shown as means ± SD. For the
statistical analysis the unpaired two-tailed Student's t-test was
used. P<0.05 were considered statistically significant. The data
were analyzed with help of GraphPad Prism 5 (GraphPad Software, San
Diego, CA, USA).
Results
5-Aza-dC is toxic for cHL cell lines and
reactivates epigenetically silenced genes at clinically achievable
concentrations
In the present study we used a 5-Aza-dC incubation
schedule aimed to obtain optimal reactivation of epigenetically
silenced genes as we described earlier for cHL (6) and for Burkitt lymphoma (28). Maximal demethylation induced by
5-Aza-dC is achieved after 72 h (29). We incubated the cells for only 24 h
with 5-Aza-dC and after complete change of the medium incubated the
cells for additional 72 h without drug, to minimize direct
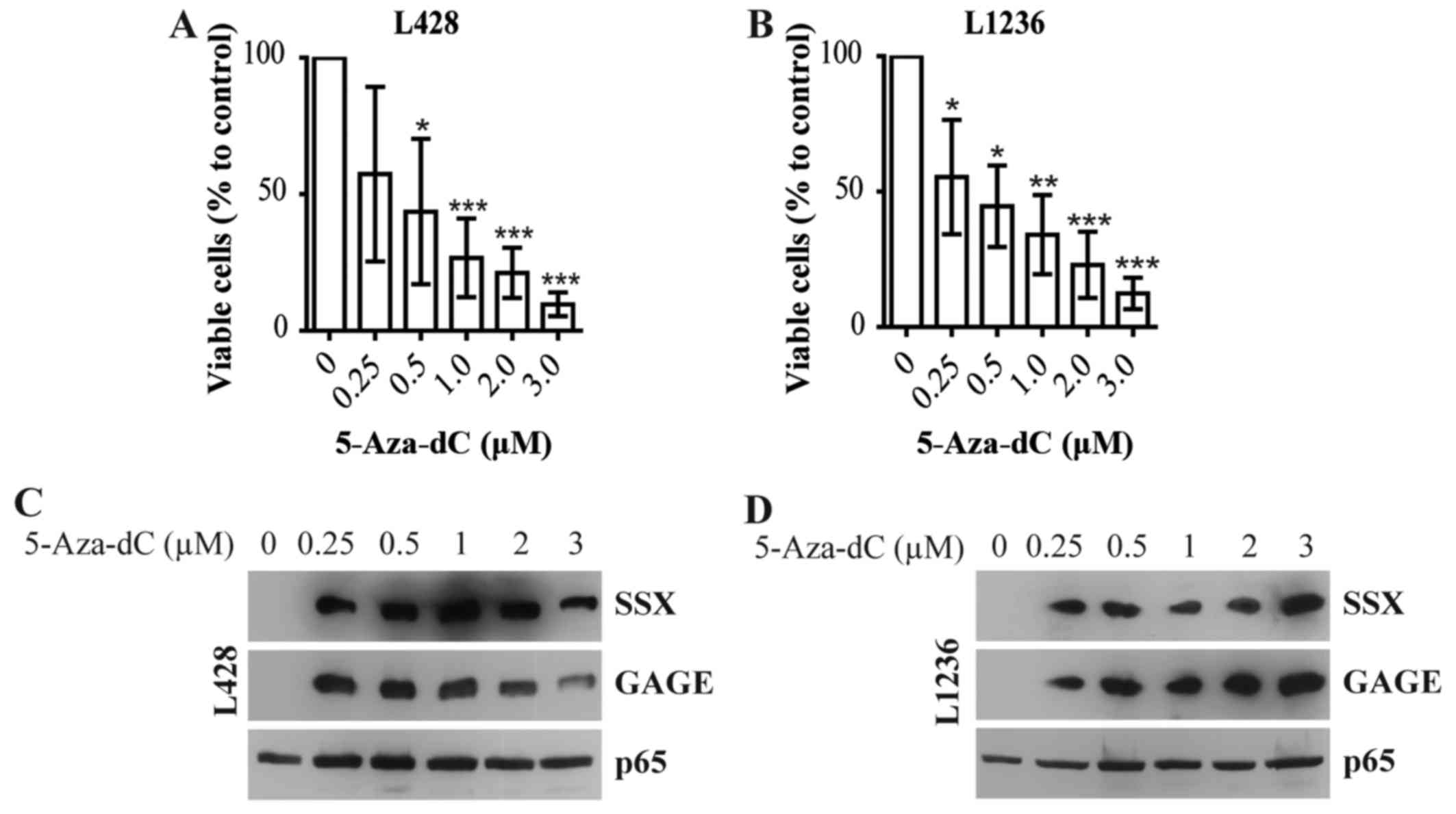
toxicity. Using this experimental setting, 5-Aza-dC strongly
reduced cell numbers of L428 (Fig.
1A) and L1236 (Fig. 1B) cHL
cell lines. Even concentrations 4–5-fold lower than peak plasma
concentration in patients [0.93–2.01 µM (30)] showed significant antitumor
effects.
To control the efficacy of the demethylating
activity of 5-Aza-dC we analyzed the expression of cancer/testis
(CT) antigens SSX and GAGE, a class of genes epigenetically
silenced by promoter hypermethylation in differentiated cells,
which can be reactivated by 5-Aza-dC in cHL cell lines (31). 5-Aza-dC induced expression of SSX
and GAGE in L428 (Fig. 1C) and
L1236 (Fig. 1D) cell lines at all
concentrations used.
Thus, 5-Aza-dC shows antitumor effects against cHL
cell lines when used at clinically relevant concentrations.
5-Aza-dC activates main oncogenic
pathways of cHL
To characterize the molecular mechanisms involved in
the 5-Aza-dC antitumor effect, we used GEP. With help of Genesifter
software we filtered out 3623 probe sets modulated more than 3-fold
in at least one of the two cell lines. To identify modulated
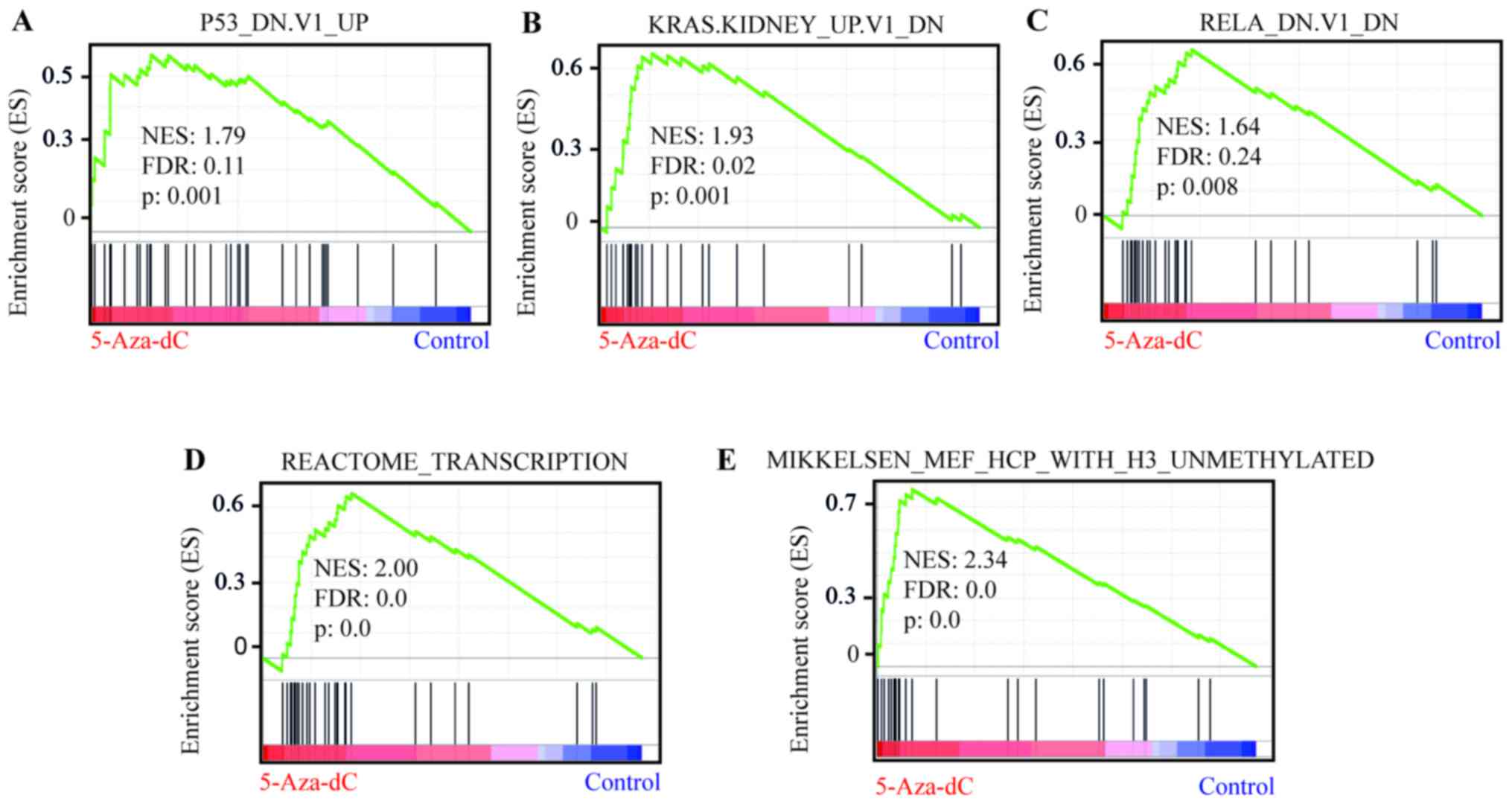
pathways we first used Gene Set Enrichment Analysis (GSEA)
(Table I). Among the significantly
enriched signatures (FDR<0.25; nominal P<0.05) were
P53_DN.V1_UP (Fig. 2A and Table I), comprising 194 genes upregulated
in cell lines with mutated TP53, KRAS.KIDNEY_UP.V1_DN (Fig. 2B and Table I), containing 142 genes induced in
epithelial kidney cancer cells by overexpression of KRAS, and
RELA_DN.V1_DN (Fig. 2C and
Table I), consisting of 141 genes
upregulated in kidney fibroblasts with mutated RELA. Applying the
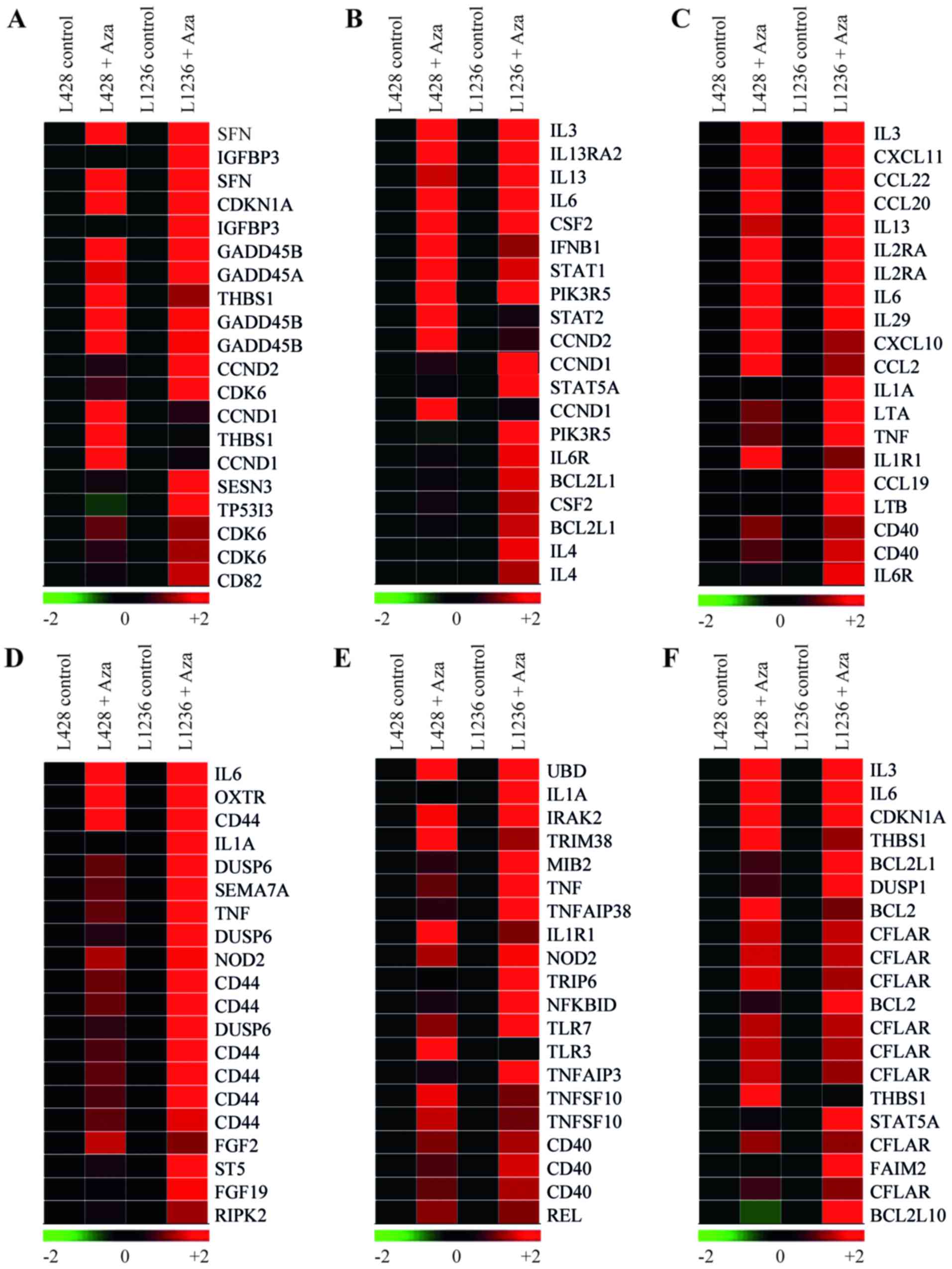
utility 'Gene function' of the Genesifter software we showed that
among the genes of the TP53 signature X57348 (SFN), a TP53
stabilizer and a negative regulator of cell cycling, was on top of
the list (Fig. 3A) (32). The TP53 associated gene list also
included CDKN1A, GADD45A and GADD45B, as well
as apoptosis-associated TP53 target gene TP53I3 (Fig. 3A) (33). Another potential tumor suppressor
gene in TP53 annotation was thrombospondin 1 (THBS1), that
facilitates RAS-induced senescence by repressing ERK signaling
(Fig. 3A) (34). Surprisingly, among the upregulated
genes we also found repression targets of TP53 including genes that
support cell cycle progression (CCND1, CCND2 and
CDK6). Among the upregulated genes of KRAS signature were
also the positive regulator of transcription BRDT (35) and protooncogene SRC.
Moreover, we found that signatures of the gene sets
REACTOME_TRANSCRIPTION (Fig. 2D
and Table I) and
MIKKELSEN_MEF_WITH_H3_UNMETHYLATED (Fig. 2E and Table I) were significantly enriched,
suggesting that 5-Aza-dC not only demethylates epigenetically
silenced genes, but raises global transcription levels in cHL cell
lines.
 | Table ISelected significantly correlated
gene sets out of the Curated Gene Sets (C2) and Oncogenic
signatures (C6) gene set collections. |
Table I
Selected significantly correlated
gene sets out of the Curated Gene Sets (C2) and Oncogenic
signatures (C6) gene set collections.
| Gene sets | NES | NOM P-value | FDR |
|---|
| C2 - Curated Gene
Sets | | | |
|
MIKKELSEN_MEF_HCP_WITH
H3_UNMETHYLATED | 2.34 | 0.00 | 0.00 |
|
REACTOME_RNA_POL_I_PROMOTER_OPENING | 2.14 | 0.00 | 0.00 |
|
REACTOME_RNA_POL_I_TRANSCRIPTION | 2.09 | 0.00 | 0.00 |
|
REACTOME_MEIOSIS | 2.05 | 0.00 | 0.00 |
|
KIM_RESPONSE_TO_TSA_AND_DECITABINE_UP | 2.03 | 0.00 | 0.00 |
|
REACTOME_TRANSCRIPTION | 2.00 | 0.00 | 0.00 |
| C6 - Oncogenic
signature | | | |
|
KRAS.KIDNEY_UP.V1_DN | 1.94 | 0.00 | 0.02 |
| P53_DN.V1_UP | 1.79 | 0.00 | 0.11 |
|
BRCA1_DN.V1_UP | 1.73 | 0.00 | 0.18 |
| RELA_DN.V1_DN | 1.64 | 0.01 | 0.24 |
We further applied Kyoto Encyclopedia of Genes and
Genomes (KEGG) database analysis to identify additional
significantly upregulated signatures (Table II). Two of the pathways with
highest enrichment scores were 'JAK-STAT signaling pathway'
(z-score, 3.93) (Fig. 3B and
Table II), and 'Cytokine-cytokine
receptor interaction' (z-score, 5.18) (Fig. 3C and Table II). Among upregulated genes of
'Cytokine-cytokine receptor interaction' signature were
CXCL10, IL-6, IL-6R and IL-13, that
provide growth and survival signals to the HRS cells (Fig. 3C) (36,37).
In addition, among the upregulated genes of JAK-STAT signature were
positive regulators of cell cycling CCND1 and CCND2
and critical for cHL survival anti-apoptotic gene BCL2L1
(Fig. 3B). The NF-κB target gene
STAT5A, which is constitutively active in cHL (38), was upregulated as well (Fig. 3B). Then we applied the utility
'Gene function' of the Genesifter software to identify other
pro-survival genes using the ontologies 'ERK1 and ERK2 cascade'
(Fig. 3D), 'NF-κB/IκB signaling'
(Fig. 3E) and 'negative regulation
of apoptosis' (Fig. 3F). This
helped us to identify several other important upregulated
proto-oncogenes including BCL2.
| Table IISignificantly enriched pathways of
the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway
enrichment analysis. |
Table II
Significantly enriched pathways of
the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway
enrichment analysis.
| KEGG pathway | Gene |
|---|
| List | Set | Z-score |
|---|
| Cytokine-cytokine
receptor interaction | 55 | 264 | 5.18 |
| JAK-STAT signaling
pathway | 32 | 153 | 3.93 |
| Toll-like receptor
signaling pathway | 21 | 101 | 3.14 |
| Cell adhesion
molecules (CAMs) | 24 | 126 | 2.89 |
| NOD-like receptor
signaling pathway | 15 | 60 | 2.31 |
| Chemokine signaling
pathway | 29 | 182 | 2.13 |
We suggested that, although 5-Aza-dC has antitumor
effect in cHL cell lines, it may be hampered by activation of
pro-survival pathways including JAK-STAT, NF-κB and KRAS.
Validation of GEP results
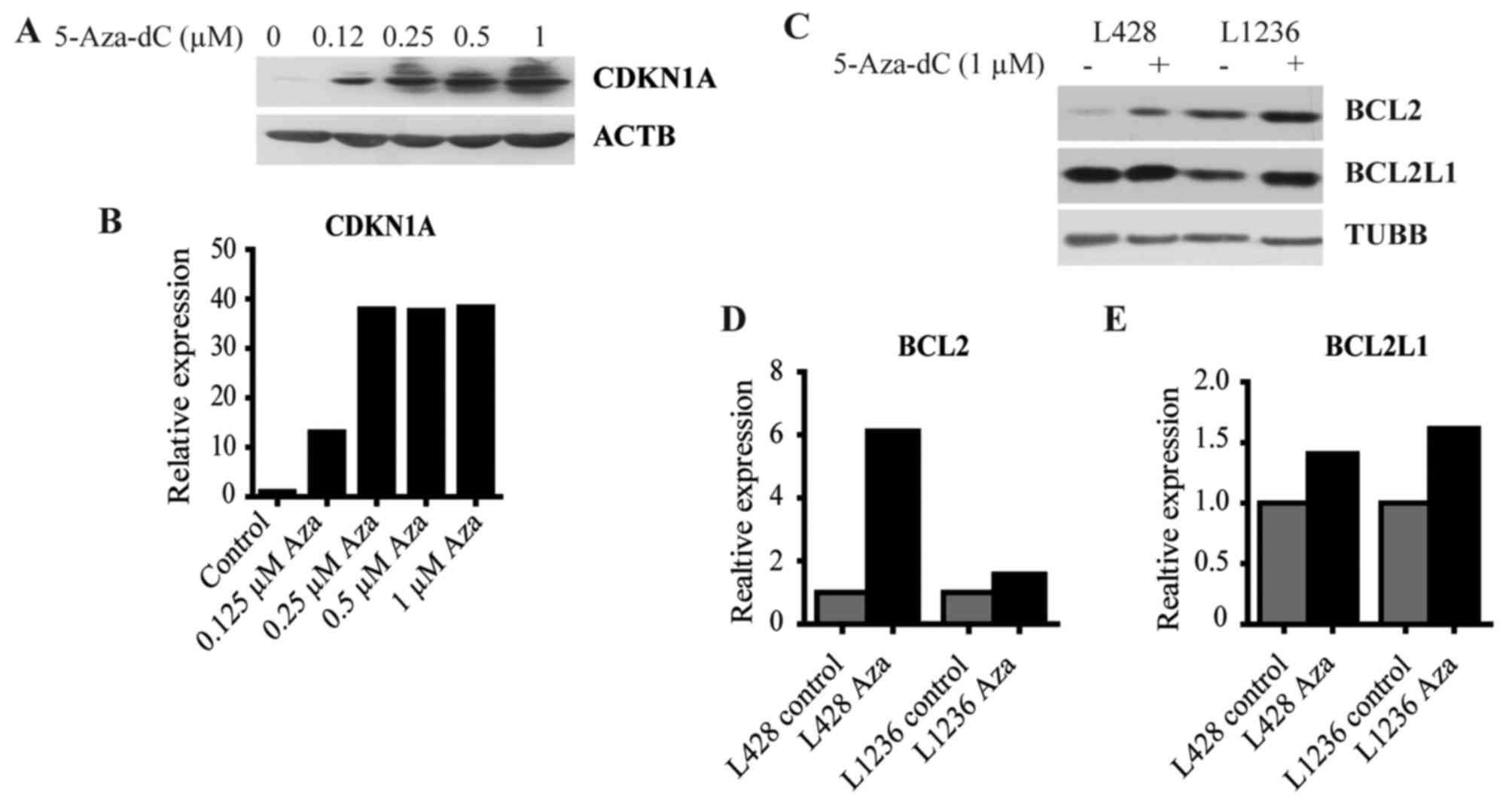
To validate results of the GEP we measured the
expression of selected genes by immunoblot. We found that 5-Aza-dC
strongly induced CDKN1A protein synthesis at a broad range of
concentrations (Fig. 4A and B).
One of the main functions of CDKN1A is the inhibition of CDK2,
which is among other proteins responsible for cell cycle
progression through S phase (39).
Moreover, CDKN1A is an antiapoptotic protein that induces cell
cycle arrest and downregulates pro-apoptotic genes such as
procaspase-3 or caspase-8 (39).
We also proved the upregulation of the antiapoptotic proteins BCL2
(Fig. 4C and D) and BCL2L1
(Fig. 4C and E).
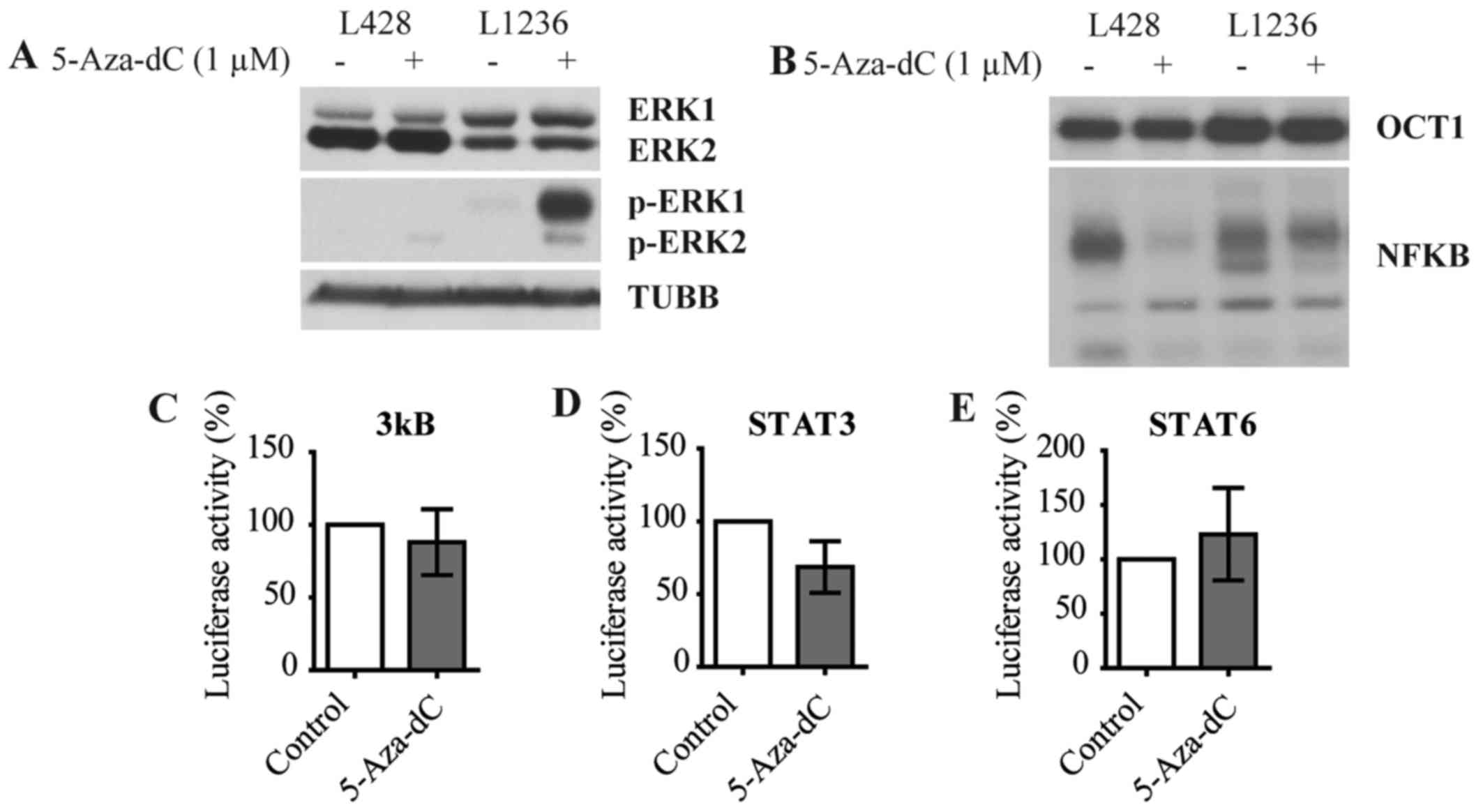
5-Aza-dC activates MEK/ERK signaling
Since GEP and subsequent enrichment analysis
indicated induction of critical oncogenic pathways in cHL, we
assessed activity of MEK/ERK, JAK-STAT and NF-κB pathways. We first
assessed the phosphorylation status of ERK1/2 using immunoblot.
5-Aza-dC strongly induced the phosphorylation of ERK1 in one the
two cell lines and phosphorylation of ERK2 in both cell lines with
no changes seen in the levels of unphosphorylated ERK1/2 protein
expression (Fig. 5A). Next using
EMSA we investigated NF-κB pathway activation. No significant
increase but rather decrease of NF-κB binding could be detected in
treated cells compared to control cells in both cell lines
(Fig. 5B). In addition we analyzed
NF-κB activation by 5-Aza-dC in L428 cells with help of luciferase
reporter assay using a vector containing three κB binding sites.
The luciferase reporter assay did also not reveal induction of
NF-κB-dependent transcription (Fig.
5C). Finally we analyzed the activity of the JAK-STAT pathway
in L428 cells using luciferase-expression vectors harboring STAT3
(Fig. 5D) and STAT6 (Fig. 5E) promoters. We did not see an
increase of luciferase activity in L428 cells after treatment with
5-Aza-dC.
Thus, although 5-Aza-dC significantly enriched gene
expression signatures of main oncogenic pathways of cHL, it did not
directly facilitate nuclear translocation of STATs and NF-κB. This
implies that other mechanisms, like transcription-activating
chromatin modifications of STAT and NF-κB target genes, may
contribute to the 5-Aza-dC induced activation of these
pathways.
Inhibition of the upregulated oncogenic
pathways increases the antitumor effect of 5-Aza-dC
We assumed that targeting of the activated
pro-survival pathways may potentiate the anti-tumor effect of
5-Aza-dC. Given that 5-Aza-dC also induced genes activating
transcription, we also included the BET inhibitor JQ1 which blocks
transcriptional activation through interaction with acetylated
chromatin.
First we assessed the antitumor effects of the
single treatments by calculating their IC50 values
(Tables III and IV). L428 and L1236 showed different
sensitivity to the inhibitors. The BCL2/BCL2L1 inhibitor ABT263 had
the highest cytotoxic activity in both cell lines (Tables III and IV). The least toxic were the MEK
inhibitor UO126, and the STAT3/STAT5 inhibitor SH-4-54, although
UO126 had somehow stronger activity in L1236 and SH-4-54 in L428
cell lines (Tables III and
IV). The JAK inhibitor fedratinib
had strong antitumor activity against L1236, whereas L428 cells
were less sensitive (Tables III
and IV). The NF-κB pathway
inhibitor QNZ, AKT inhibitor KP372-1, the bromodomain and
extra-terminal (BET) family proteins inhibitor JQ1 also differed in
their antitumor effect towards cHL cell lines. L428 was more
sensitive to KP372-1 and QNZ (Table
III), whereas JQ1 showed a stronger antitumor effect in L1236
(Table IV).
| Table IIIResults of IC50
calculation and combination plot in L428. |
Table III
Results of IC50
calculation and combination plot in L428.
| Cell line L428
Drugs | Parameters
| CI values
|
|---|
| IC50
(nM) | m | r |
ED35 |
ED50 |
ED90 |
|---|
| Aza | 57.2 | 0.89 | 0.96 | | | |
| Fedratinib | 710.5 | 3.06 | 0.97 | | | |
| Fedratinib + Aza
(12.4:1) | 217.7 | 1.37 | 1.00 | 0.58 | 0.57 | 0.80 |
| Aza | 57.2 | 0.89 | 0.96 | | | |
| JQ1 | 182.4 | 0.23 | 0.41 | | | |
| JQ1 + Aza
(3.2:1) | 24.6 | 0.83 | 0.97 | 0.84 | 0.21 | 0.12 |
| Aza | 57.2 | 0.89 | 0.96 | | | |
| KP372-1 | 346.7 | 3.46 | 0.98 | | | |
| KP372-1 + Aza
(6.1:1) | 168.7 | 2.28 | 0.93 | 1.02 | 0.83 | 0.67 |
| Aza | 57.2 | 0.89 | 0.96 | | | |
| QNZ | 143.6 | 1.07 | 1.00 | | | |
| QNZ+ Aza
(2.5:1) | 17.15 | 2.71 | 0.88 | 0.14 | 0.07 | 0.01 |
| Aza | 57.2 | 0.89 | 0.96 | | | |
| SH-4-54 | 3925.3 | 7.62 | 1.00 | | | |
| SH-4-54 + Aza
(68.6:1) | 1402.9 | 2.81 | 0.94 | 0.88 | 0.71 | 0.64 |
| Aza | 57.2 | 0.89 | 0.96 | | | |
| UO126 | 5734.0 | 1.99 | 0.98 | | | |
| UO126 + Aza
(100.2:1) | 2753.6 | 0.74 | 0.95 | 282.27 | 475.96 | 3043.62 |
| Aza | 57.2 | 0.89 | 0.96 | | | |
| ABT263 | 14.8 | 1.34 | 0.99 | | | |
| ABT263 + Aza
(1:3.9) | 23.4 | 1.05 | 0.98 | 0.65 | 0.65 | 0.75 |
| Table IVResults of IC50
calculation and combination plot in L1236. |
Table IV
Results of IC50
calculation and combination plot in L1236.
| Cell line L1236
Drugs | Parameters
| CI values
|
|---|
| IC50
(nM) | m | r |
ED35 |
ED50 |
ED90 |
|---|
| Aza | 1956.8 | 0.84 | 0.99 | | | |
| Fedratinib | 81.1 | 1.03 | 0.99 | | | |
| Fedratinib + Aza
(1:24.1) | 2141.5 | 0.87 | 1.00 | 2.01 | 2.10 | 2.57 |
| Aza | 1956.8 | 0.84 | 0.99 | | | |
| JQ1 | 47.8 | 1.42 | 0.98 | | | |
| JQ1 + Aza
(1:41.0) | 1557.2 | 0.80 | 0.96 | 1.30 | 1.55 | 3.46 |
| Aza | 1956.8 | 0.84 | 0.99 | | | |
| KP372-1 | 611.4 | 1.72 |
1.0 | | | |
| KP372-1 + Aza
(1:3.2) | 991.7 | 1.30 | 0.94 | 0.75 | 0.77 | 1.94 |
| Aza | 1956.8 | 0.84 | 0.99 | | | |
| QNZ | 947.3 | 0.74 | 0.95 | | | |
| QNZ+ Aza
(1:2.1) | 350.6 | 0.79 | 1.00 | 0.24 | 0.24 | 0.24 |
| Aza | 1956.8 | 0.84 | 0.99 | | | |
| SH-4-54 | 5387.3 | 14.52 |
1.0 | | | |
| SH-4-54 + Aza
(2.8:1) | 4079.3 | 4.61 | 0.80 | 1.52 | 1.11 | 0.83 |
| Aza | 1956.8 | 0.84 | 0.99 | | | |
| UO126 | 4430.7 | 2.62 | 0.99 | | | |
| UO126 + Aza
(2.3:1) | 6930.8 | 0.63 | 0.99 | 1.37 | 2.17 | 17.52 |
| Aza | 1956.8 | 0.84 | 0.99 | | | |
| ABT263 | 10.0 | 1.65 | 0.99 | | | |
| ABT263 + Aza
(1:196.0) | 1381.8 | 0.61 | 0.96 | 0.90 | 1.40 | 8.60 |
To analyze the dose effect relationships of the drug
combinations we applied the method of Chou and Talaly (24,25).
With help of the CompuSyn Software we produced CI-Fa combination
plots. The CI represents the value of the effect of the combined
drugs, with CI values >1 indicating antagonism, CI values =1
additive effects, and CI values <1 synergism (24,25).
The fraction affected (Fa) values represent the magnitude of the
antitumor effect of the combination. Fa=0 indicates the absence of
therapeutic effects, and Fa=1 the maximum achievable antitumor
activity (24,25). For each Fa value the software
generates a predictive CI value, which can be seen on the plots as
the curves with the solid line. The experimental combination data,
that were used to generate the CI-Fa combination plots, are
represented as dots (24,25).
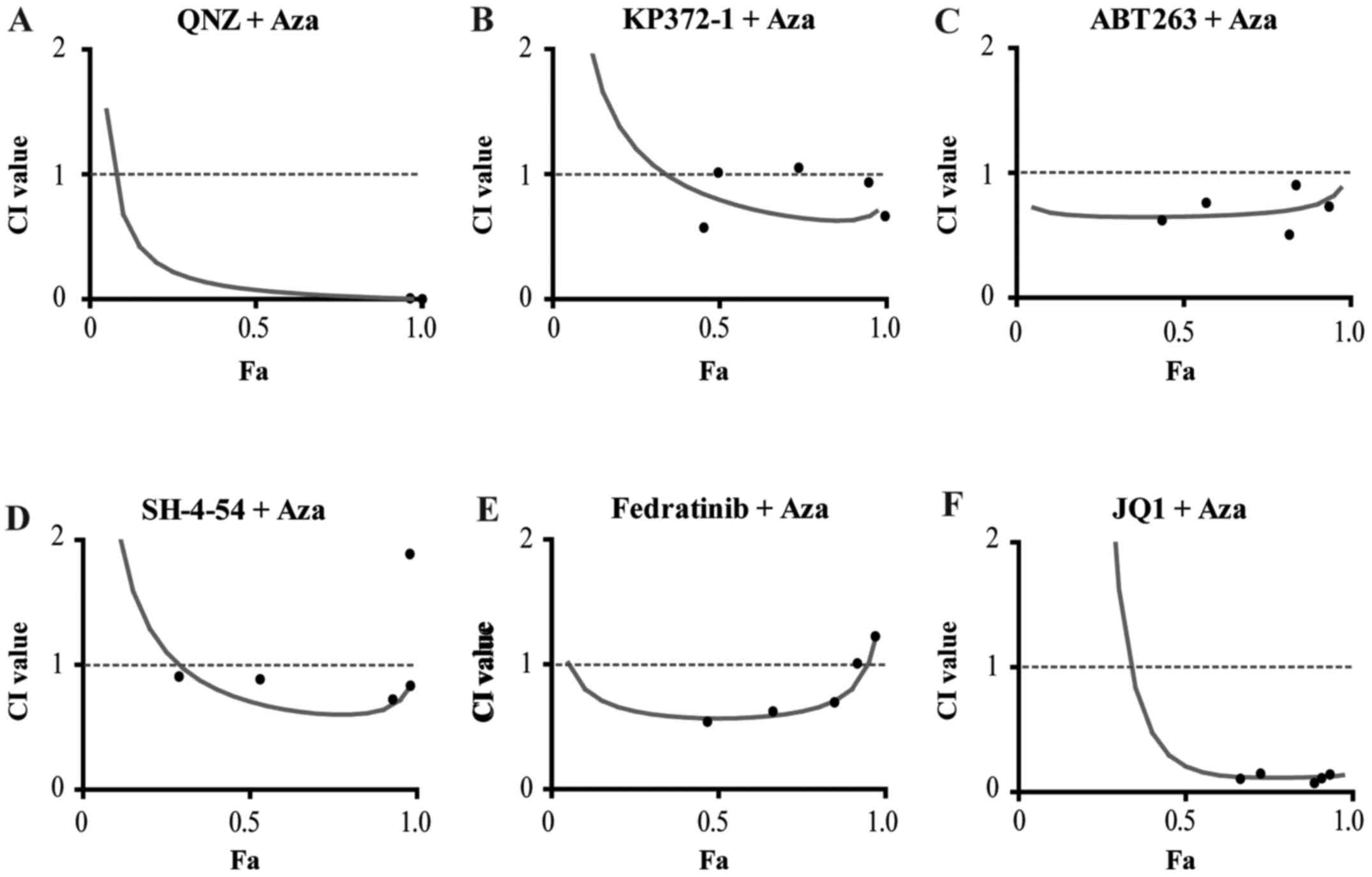
Similar to the single treatments, cHL cell lines
varied in terms of sensitivity to the drug combinations. In
general, L428 cells were more sensitive than L1236 cells. The most
effective in both cell lines was the combination with the NF-κB
inhibitor QNZ (Fig. 6A and
Table IV). Targeting AKT, one of
the downstream molecules in NF-κB signaling, with the AKT-inhibitor
KP372-1, resulted in synergistic antitumor effects in both cell
lines (Fig. 6B and Table IV).
Combination of 5-Aza-dC with the BCL2/BCL2L1
inhibitor ABT263 resulted in very strong synergy especially in L428
(Fig. 6C). In L1236 a synergistic
effect could only be observed at low Fa values (Table IV). Targeting the JAK-STAT pathway
by combining 5-Aza-dC with the STAT3/STAT5 inhibitor SH-4-54
(Fig. 6D) or JAK2 inhibitor
fedratinib (Fig. 6E) showed
synergistic effects in L428 at almost all concentrations. In L1236
only SH-4-54 showed a slight synergistic effect at intermediate Fa
values (Table IV). Fedratinib did
not increase the antitumor effect of 5-Aza-dC in L1236 (Table IV).
Among the gene sets significantly enriched by
5-Aza-dC we identified REACTOME_TRANSCRIPTION, that comprises genes
involved in transcription. We applied the concept of dual
epigenetic therapy to inhibit the 5-Aza-dC-induced transcription
activation. The inhibitor JQ1 inhibits BRDs binding to acetylated
histones (35). We observed
synergism at high concentration in L428 cells (Fig. 6F) and at low concentrations in
L1236 cells (Table IV).
Given the positive effect of 5-Aza-dC on ERK
activation we expected to observe strong synergy with MEK
inhibitors. Against our expectations, combination with the MEK
inhibitor UO126 showed strong antagonistic effects in both cell
lines (Tables III and IV).
Thus, inhibition of pro-survival pathways induced by
5-Aza-dC in cHL cell lines potentiates the antitumor effect.
Discussion
Here we provide experimental evidence that the
demethylating agent 5-Aza-dC has antitumor effects on cHL. However,
the magnitude of the effect is hampered by simultaneous induction
of pro-survival pathways. The inhibition of these pathways
significantly improved the antitumor activity of 5-Aza-dC.
5-Aza-dC was successfully used for treatment of a
wide spectrum of tumors (40). In
cHL there is only one anecdotic evidence on clinical efficacy of
5-Aza-dC (19). In addition it was
suggested that 5-Aza-dC could be used for increasing immunogenicity
of HRS cells and to make them a more attractive target for T-cell
therapy (41). This study on
antitumor effects of 5-Aza-dC at clinically relevant concentrations
supports the idea on the feasibility of demethylating therapy for
cHL.
At the same time we revealed a serious drawback of
5-Aza-dC treatment. We detected a significant upregulation of the
pathways, that are known to contribute to the oncogenic program of
cHL (MEK/ERK, NF-κB and JAK-STAT) (1). The ability of 5-Aza-dC to modulate
MEK/ERK activity is cell type specific and depends on the
activation status of other pathways. In ovarian cancer cell lines,
5-Aza-dC strongly activated ERK phosphorylation only in cell lines
harboring activating KRAS mutations (42). In the human AML cell line NB4 and
in freshly isolated AML cells, 5-Aza-dC also stimulated ERK
phosphorylation (43). In other
normal and malignant cell types including osteoclasts (44), acute lymphoblastic leukemia
(45), and lung cancer cell lines
(46) 5-Aza-dC attenuated MEK/ERK
activity, inter alia by reactivating epigenetically silenced
negative regulators. JAK-STAT signaling is inhibited by 5-Aza-dC,
in particular by the reactivation of JAK-STAT inhibitors (47,48).
The effect of 5-Aza-dC on NF-κB activity is
cell-type dependent. 5-Aza-dC activated NF-κB signaling in Burkitt
lymphoma (28) and in AML
(49), but inhibited NF-κB
activity in osteoclasts (44). Of
note, although in our experiments we observed significant
upregulation of NF-κB and STAT target genes, this was not mediated
by increased nuclear translocation of these transcription factors.
Most probably the increasing transcription of NF-κB and STAT
targets reflects solely general stimulating effects of 5-Aza-dC on
chromatin structure. It was shown that 5-Aza-dC increases global
histones acetylation independent of DNA methylation (50). Thus, 5-Aza-dC not only induces
activation of hypermethylated promoters but also stimulates
transcriptional activity of already active genes, i.e. the genes
constituting tumor survival and proliferation programs.
Consequently we have shown that repression of the
activated pro-survival pathways can potentiate the antitumor effect
of 5-Aza-dC. So far, 5-Aza-dC was used in combination with
chemotherapeutics to prevent or reverse epigenetic silencing of
genes mediating the cytotoxic effect (51). Combination of 5-Aza-dC with
epidermal growth factor receptor inhibitor gefitinib has
synergistic antitumor effects against colon carcinoma cells
(52).
To potentiate the antitumor effect of 5-Aza-dC on
cHL cell lines, we targeted JAK-STAT and NF-κB pathways. As a
single treatment the small molecular weight inhibitor of JAK2
fedratinib already showed strong antitumor activity against cHL
in vitro and in vivo (53). Our findings provide a rationale for
combination of this inhibitor with 5-Aza-dC. Targeting NF-κB with
the inhibitor QNZ also significantly increased the antitumor effect
of 5-Aza-dC. Of note, other NF-κB inhibitors like the proteasome
inhibitor bortezomib were considered as an attractive approach to
treat cHL (54), however clinical
trials did not meet the expectations (55). We hope that combination with
5-Aza-dC may be a way to improve the efficacy of NF-κB
inhibitors.
We found that MEK/ERK signaling, which is
constitutively active in most of the HRS cells (1), was activated by 5-Aza-dC.
Unexpectedly, we revealed antagonistic interactions between
5-Aza-dC and MEK1/2 inhibitor U0126. Interestingly, Stewart et
al described a decrease in the sensitivity of an ovarian cancer
cell line to the ERK inhibitor trametinib by treatment with
5-Aza-dC and attributed it to 5-Aza-dC-induced ERK hyper-activation
(42). It is conceivable that
ERK-dependent induction of BCL2 and BCL2L1 (56) contributed to the protective effect
of 5-Aza-dC. Our data on strong synergistic effects of BCL2/BCL2L1
inhibitor ABT263 with 5-Aza-dC supports this assumption. Synergism
of 5-Aza-dC with BCL2 inhibitors was described earlier in ovarian
cancer models (42).
We could also observe strong synergism of 5-Aza-dC
with the BET inhibitor JQ1. JQ1 predominantly inhibits
transcription of oncogenes highly expressed in tumor cells,
including MYC, by preventing recruitment of transcription
activating BET proteins to acetylated histones (57). Considering a functional connection
between histone modifications and DNA methylation it is not
surprising that treatment with 5-Aza-dC induces histone acetylation
and activation of transcription (50). It is therefore conceivable, that
JQ1 blocks this 5-Aza-dC-induced transcriptional activation.
Interestingly, the synergistic interaction between JQ1 and
hypomethylating drugs has been predicted earlier (58).
Due to its low toxicity 5-Aza-dC is an attractive
alternative for classical chemotherapy especially in elderly
patients and for palliative care (59). Combination of epigenetic therapy
with matched pathway inhibition may therefore be a promising
approach to treat elderly and multi-morbid patients with cHL.
Further in vitro and in vivo studies will be required
to fully understand the potential of the different combinations and
to evaluate their toxicity profile.
Acknowledgments
We thank Anita Kick for excellent technical
assistance, Harald J. Maier and Uta Manfras for their help with
EMSA, Elena Kelsch and Michaela Buck (Institute of Pathology,
University of Ulm, Germany) for STR analysis of the cell lines, and
Beatrix Schwarz for help in preparation of the manuscript. We are
thankful to J.E. Bradner (Division of Hematologic Neoplasia,
Dana-Faber Cancer Institute, Boston, MA, USA) for donation of JQ1.
This study was supported in part by grant 110564 from the Deutsche
Krebshilfe e.V. (to T.W. and A.U.). T.S. was supported by grant
111817 from the Deutsche Krebshilfe e.V.
References
|
1
|
Küppers R: The biology of Hodgkin's
lymphoma. Nat Rev Cancer. 9:15–27. 2009. View Article : Google Scholar
|
|
2
|
Gobbi PG, Ferreri AJ, Ponzoni M and Levis
A: Hodgkin lymphoma. Crit Rev Oncol Hematol. 85:216–237. 2013.
View Article : Google Scholar
|
|
3
|
Küppers R and Hansmann ML: The Hodgkin and
Reed/Sternberg cell. Int J Biochem Cell Biol. 37:511–517. 2005.
View Article : Google Scholar
|
|
4
|
Doerr JR, Malone CS, Fike FM, Gordon MS,
Soghomonian SV, Thomas RK, Tao Q, Murray PG, Diehl V, Teitell MA,
et al: Patterned CpG methylation of silenced B cell gene promoters
in classical Hodgkin lymphoma-derived and primary effusion lymphoma
cell lines. J Mol Biol. 350:631–640. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ehlers A, Oker E, Bentink S, Lenze D,
Stein H and Hummel M: Histone acetylation and DNA demethylation of
B cells result in a Hodgkin-like phenotype. Leukemia. 22:835–841.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ushmorov A, Leithäuser F, Sakk O,
Weinhaüsel A, Popov SW, Möller P and Wirth T: Epigenetic processes
play a major role in B-cell-specific gene silencing in classical
Hodgkin lymphoma. Blood. 107:2493–2500. 2006. View Article : Google Scholar
|
|
7
|
Yuki H, Ueno S, Tatetsu H, Niiro H, Iino
T, Endo S, Kawano Y, Komohara Y, Takeya M, Hata H, et al: PU.1 is a
potent tumor suppressor in classical Hodgkin lymphoma cells. Blood.
121:962–970. 2013. View Article : Google Scholar
|
|
8
|
García JF, Villuendas R, Algara P, Sáez
AI, Sánchez-Verde L, Martínez-Montero JC, Martínez P and Piris MA:
Loss of p16 protein expression associated with methylation of the
p16INK4A gene is a frequent finding in Hodgkin's disease. Lab
Invest. 79:1453–1459. 1999.
|
|
9
|
Sánchez-Aguilera A, Delgado J, Camacho FI,
Sánchez-Beato M, Sánchez L, Montalbán C, Fresno MF, Martín C, Piris
MA and García JF: Silencing of the p18INK4c gene by promoter
hypermethylation in Reed-Sternberg cells in Hodgkin lymphomas.
Blood. 103:2351–2357. 2004. View Article : Google Scholar
|
|
10
|
Guan H, Xie L, Leithäuser F, Flossbach L,
Möller P, Wirth T and Ushmorov A: KLF4 is a tumor suppressor in
B-cell non-Hodgkin lymphoma and in classic Hodgkin lymphoma. Blood.
116:1469–1478. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pathania R, Ramachandran S, Elangovan S,
Padia R, Yang P, Cinghu S, Veeranan-Karmegam R, Arjunan P,
Gnana-Prakasam JP, Sadanand F, et al: DNMT1 is essential for
mammary and cancer stem cell maintenance and tumorigenesis. Nat
Commun. 6:69102015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Völkel P, Dupret B and Le Bourhisxand
Angrand PO: Diverse involvement of EZH2 in cancer epigenetics. Am J
Transl Res. 7:175–193. 2015.PubMed/NCBI
|
|
13
|
VanderMolen KM, McCulloch W, Pearce CJ and
Oberlies NH: Romidepsin (Istodax, NSC 630176, FR901228, FK228,
depsipeptide): A natural product recently approved for cutaneous
T-cell lymphoma. J Antibiot (Tokyo). 64:525–531. 2011. View Article : Google Scholar
|
|
14
|
Subbiah V, Brown RE, McGuire MF, Buryanek
J, Janku F, Younes A and Hong D: A novel immunomodulatory
molecularly targeted strategy for refractory Hodgkin's lymphoma.
Oncotarget. 5:95–102. 2014.PubMed/NCBI
|
|
15
|
Oki Y, Copeland A and Younes A: Clinical
development of panobinostat in classical Hodgkin's lymphoma. Expert
Rev Hematol. 4:245–252. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jóna A, Khaskhely N, Buglio D, Shafer JA,
Derenzini E, Bollard CM, Medeiros LJ, Illés A, Ji Y and Younes A:
The histone deacetylase inhibitor entinostat (SNDX-275) induces
apoptosis in Hodgkin lymphoma cells and synergizes with Bcl-2
family inhibitors. Exp Hematol. 39:1007–1017.e1. 2011. View Article : Google Scholar :
|
|
17
|
Kim TK, Gore SD and Zeidan AM: Epigenetic
therapy in acute myeloid keukemia: Current and future directions.
Semin Hematol. 52:172–183. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Saba HI: Decitabine in the treatment of
myelodysplastic syndromes. Ther Clin Risk Manag. 3:807–817.
2007.
|
|
19
|
D'Alò F, Leone G, Hohaus S, Teofili L,
Bozzoli V, Tisi MC, Rufini V, Calcagni ML and Voso MT: Response to
5-azacytidine in a patient with relapsed Hodgkin lymphoma and a
therapy-related myelodysplastic syndrome. Br J Haematol.
154:141–143. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Derissen EJ, Beijnen JH and Schellens JH:
Concise drug review: Azacitidine and decitabine. Oncologist.
18:619–624. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ushmorov A, Ritz O, Hummel M, Leithäuser
F, Möller P, Stein H and Wirth T: Epigenetic silencing of the
immunoglobulin heavy-chain gene in classical Hodgkin
lymphoma-derived cell lines contributes to the loss of
immunoglobulin expression. Blood. 104:3326–3334. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mootha VK, Lindgren CM, Eriksson KF,
Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E,
Ridderstråle M, Laurila E, et al: PGC-1alpha-responsive genes
involved in oxidative phosphorylation are coordinately
downregulated in human diabetes. Nat Genet. 34:267–273. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES, et al: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maier HJ, Schips TG, Wietelmann A, Krüger
M, Brunner C, Sauter M, Klingel K, Böttger T, Braun T and Wirth T:
Cardiomyocyte-specific IκB kinase (IKK)/NF-κB activation induces
reversible inflammatory cardiomyopathy and heart failure. Proc Natl
Acad Sci USA. 109:11794–11799. 2012. View Article : Google Scholar
|
|
27
|
Ritz O, Guiter C, Dorsch K, Dusanter-Fourt
I, Wegener S, Jouault H, Gaulard P, Castellano F, Möller P and
Leroy K: STAT6 activity is regulated by SOCS-1 and modulates BCL-XL
expression in primary mediastinal B-cell lymphoma. Leukemia.
22:2106–2110. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guan H, Xie L, Klapproth K, Weitzer CD,
Wirth T and Ushmorov A: Decitabine represses translocated MYC
oncogene in Burkitt lymphoma. J Pathol. 229:775–783. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bender CM, Gonzalgo ML, Gonzales FA,
Nguyen CT, Robertson KD and Jones PA: Roles of cell division and
gene transcription in the methylation of CpG islands. Mol Cell
Biol. 19:6690–6698. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Atallah E, Kantarjian H and Garcia-Manero
G: The role of decitabine in the treatment of myelodysplastic
syndromes. Expert Opin Pharmacother. 8:65–73. 2007. View Article : Google Scholar
|
|
31
|
Shafer JA, Cruz CR, Leen AM, Ku S, Lu A,
Rousseau A, Heslop HE, Rooney CM, Bollard CM and Foster AE:
Antigen-specific cytotoxic T lymphocytes can target chemoresistant
side-population tumor cells in Hodgkin lymphoma. Leuk Lymphoma.
51:870–880. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hermeking H and Benzinger A: 14-3-3
proteins in cell cycle regulation. Semin Cancer Biol. 16:183–192.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Polyak K, Xia Y, Zweier JL, Kinzler KW and
Vogelstein B: A model for p53-induced apoptosis. Nature.
389:300–305. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Baek KH, Bhang D, Zaslavsky A, Wang LC,
Vachani A, Kim CF, Albelda SM, Evan GI and Ryeom S:
Thrombospondin-1 mediates oncogenic Ras-induced senescence in
premalignant lung tumors. J Clin Invest. 123:4375–4389. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Muller S, Filippakopoulos P and Knapp S:
Bromodomains as therapeutic targets. Expert Rev Mol Med.
13:e292011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mazur G, Jaskuła E, Kryczek I, Dłubek D,
Butrym A, Wróbel T, Lange A and Kuliczkowski K: Proinflammatory
chemokine gene expression influences survival of patients with
non-Hodgkin's lymphoma. Folia Histochem Cytobiol. 49:240–247. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ouaguia L, Mrizak D, Renaud S, Moralès O
and Delhem N: Control of the inflammatory response mechanisms
mediated by natural and induced regulatory T-cells in HCV-,
HTLV-1-, and EBV-associated cancers. Mediators Inflamm.
2014:5642962014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hinz M, Lemke P, Anagnostopoulos I, Hacker
C, Krappmann D, Mathas S, Dörken B, Zenke M, Stein H and
Scheidereit C: Nuclear factor kappaB-dependent gene expression
profiling of Hodgkin's disease tumor cells, pathogenetic
significance, and link to constitutive signal transducer and
activator of transcription 5a activity. J Exp Med. 196:605–617.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Brown R and Plumb JA: Demethylation of DNA
by decitabine in cancer chemotherapy. Expert Rev Anticancer Ther.
4:501–510. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cruz CR, Gerdemann U, Leen AM, Shafer JA,
Ku S, Tzou B, Horton TM, Sheehan A, Copeland A, Younes A, et al:
Improving T-cell therapy for relapsed EBV-negative Hodgkin lymphoma
by targeting upregulated MAGE-A4. Clin Cancer Res. 17:7058–7066.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Stewart ML, Tamayo P, Wilson AJ, Wang S,
Chang YM, Kim JW, Khabele D, Shamji AF and Schreiber SL: KRAS
genomic status predicts the sensitivity of ovarian cancer cells to
decitabine. Cancer Res. 75:2897–2906. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nishioka C, Ikezoe T, Yang J, Komatsu N,
Koeffler HP and Yokoyama A: Blockade of MEK signaling potentiates
5-Aza-2′-deoxycytidine-induced apoptosis and upregulation of
p21(waf1) in acute myelogenous leukemia cells. Int J Cancer.
125:1168–1176. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Guan H, Mi B, Li Y, Wu W, Tan P, Fang Z,
Li J, Zhang Y and Li F: Decitabine represses osteoclastogenesis
through inhibition of RANK and NF-κB. Cell Signal. 27:969–977.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Stevenson WS, Best OG, Przybylla A, Chen
Q, Singh N, Koleth M, Pierce S, Kennedy T, Tong W, Kuang SQ, et al:
DNA methylation of membrane-bound tyrosine phosphatase genes in
acute lymphoblastic leukaemia. Leukemia. 28:787–793. 2014.
View Article : Google Scholar
|
|
46
|
Lin JC, Wu YY, Wu JY, Lin TC, Wu CT, Chang
YL, Jou YS, Hong TM and Yang PC: TROP2 is epigenetically
inactivated and modulates IGF-1R signalling in lung adenocarcinoma.
EMBO Mol Med. 4:472–485. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Han Y, Amin HM, Frantz C, Franko B, Lee J,
Lin Q and Lai R: Restoration of shp1 expression by
5-AZA-2′-deoxycytidine is associated with downregulation of
JAK3/STAT3 signaling in ALK-positive anaplastic large cell
lymphoma. Leukemia. 20:1602–1609. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Galm O, Yoshikawa H, Esteller M, Osieka R
and Herman JG: SOCS-1, a negative regulator of cytokine signaling,
is frequently silenced by methylation in multiple myeloma. Blood.
101:2784–2788. 2003. View Article : Google Scholar
|
|
49
|
Laurenzana A, Petruccelli LA, Pettersson
F, Figueroa ME, Melnick A, Baldwin AS, Paoletti F and Miller WH Jr:
Inhibition of DNA methyltransferase activates tumor necrosis factor
alpha-induced monocytic differentiation in acute myeloid leukemia
cells. Cancer Res. 69:55–64. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Takebayashi S, Nakao M, Fujita N, Sado T,
Tanaka M, Taguchi H and Okumura K: 5-Aza-2′-deoxycytidine induces
histone hyperacetylation of mouse centromeric heterochromatin by a
mechanism independent of DNA demethylation. Biochem Biophys Res
Commun. 288:921–926. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Plumb JA, Strathdee G, Sludden J, Kaye SB
and Brown R: Reversal of drug resistance in human tumor xenografts
by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene
promoter. Cancer Res. 60:6039–6044. 2000.PubMed/NCBI
|
|
52
|
Lou YF, Zou ZZ, Chen PJ, Huang GB, Li B,
Zheng DQ, Yu XR and Luo XY: Combination of gefitinib and DNA
methylation inhibitor decitabine exerts synergistic anti-cancer
activity in colon cancer cells. PLoS One. 9:e977192014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hao Y, Chapuy B, Monti S, Sun HH, Rodig SJ
and Shipp MA: Selective JAK2 inhibition specifically decreases
Hodgkin lymphoma and mediastinal large B-cell lymphoma growth in
vitro and in vivo. Clin Cancer Res. 20:2674–2683. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jost PJ and Ruland J: Aberrant NF-kappaB
signaling in lymphoma: Mechanisms, consequences, and therapeutic
implications. Blood. 109:2700–2707. 2007.
|
|
55
|
Blum KA, Johnson JL, Niedzwiecki D,
Canellos GP, Cheson BD and Bartlett NL: Single agent bortezomib in
the treatment of relapsed and refractory Hodgkin lymphoma: Cancer
and leukemia Group B protocol 50206. Leuk Lymphoma. 48:1313–1319.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Boucher MJ, Morisset J, Vachon PH, Reed
JC, Lainé J and Rivard N: MEK/ERK signaling pathway regulates the
expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of
human pancreatic cancer cells. J Cell Biochem. 79:355–369. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chapuy B, McKeown MR, Lin CY, Monti S,
Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al:
Discovery and characterization of super-enhancer-associated
dependencies in diffuse large B cell lymphoma. Cancer Cell.
24:777–790. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Godley LA and Le Beau MM: The histone code
and treatments for acute myeloid leukemia. N Engl J Med.
366:960–961. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Malik P and Cashen AF: Decitabine in the
treatment of acute myeloid leukemia in elderly patients. Cancer
Manag Res. 6:53–61. 2014.PubMed/NCBI
|