1. Introduction
Gastrointestinal (GI) cancer, which refers to the
malignancies of the GI tract and accessory organs of digestion,
represents the most common cancer and the leading cause of
cancer-related death worldwide (1). According to the American Cancer
Society, a total of 304,930 new GI cancer cases and an approximate
deaths of 153,030 are estimated to occur during 2016 (2). In China, cancers of stomach, liver,
esophagus and colorectum are among the 5 leading causes of cancer
death (3). Most patients with GI
cancer have advanced tumors with regional or distant metastasis
upon presentation, which precludes them for radical resection.
Among those who have received intentionally curable surgery, some
still failed to survive due to the occult dissemination of cancer
cells.
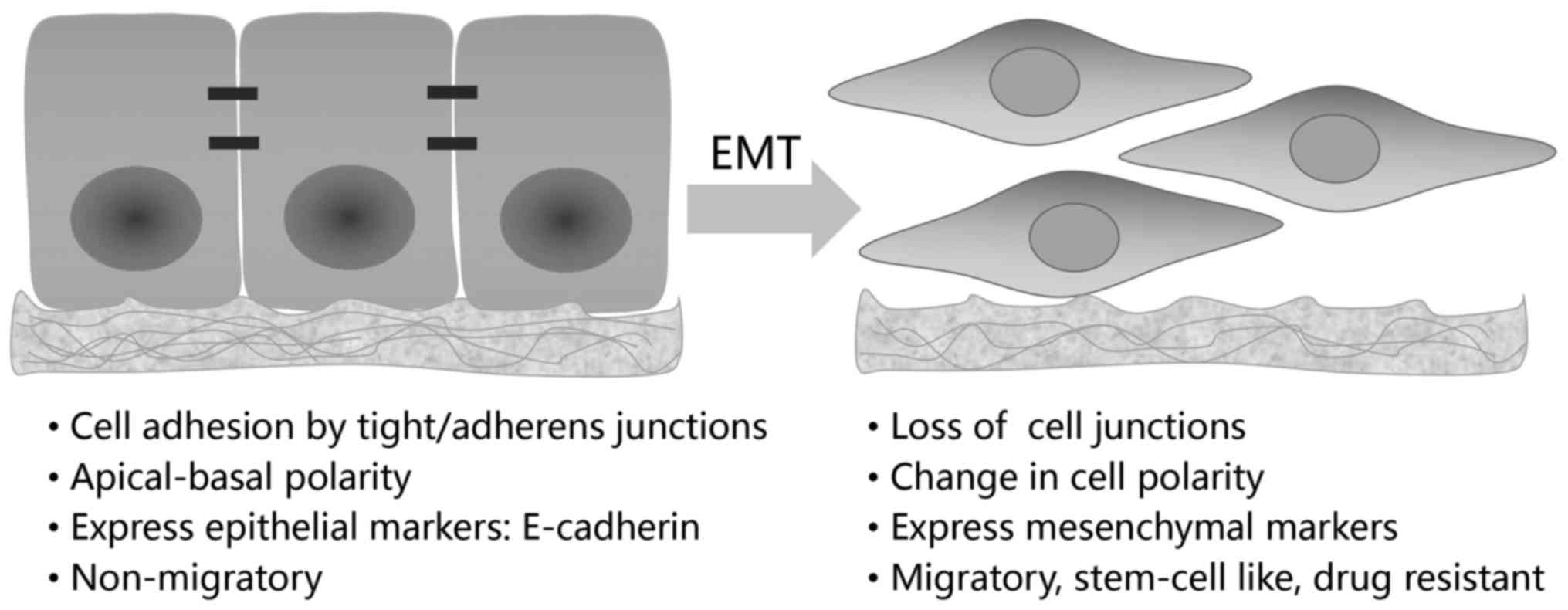
The epithelial-mesenchymal transition (EMT), first
described by Elizabeth Hay in 1980s using a model of chick
primitive streak formation, is integral in embryonic development,
tissue repairing and also, occurs as an unintentional behavior of
cells during fibrosis and cancer progression (4,5).
According to these distinct biological settings, EMT has been
proposed to be categorized into three subtypes (6). The first, termed 'type 1', is
associated with embryogenesis and organ development, such as
gastrulation, neural crest formation and heart valve formation. The
type 2 EMT, set in a context of trauma or inflammatory injury, is
required for wound healing and tissue repairing. If the
inflammation is persistent, however, this type of EMT also cause
undesirable result-organ fibrosis. Different from these two types,
the type 3 EMT, which exclusively occurs in neoplastic cells, is
completely detrimental. In tumors of epithelial origin, immotile
epithelial cells undergoing type 3 EMT are converted to
mesenchymal-like cells with migratory ability (Fig. 1). By conferring cancer cells with
capacity to invade, EMT drives the progression of indolent cancer
in situ to an aggressive metastatic one (4–6). In
addition, emerging evidence has shown that type 3 EMT also
contributes to induction of cancer stem cells (CSCs), drug
resistance and immune escape during GI cancer progression (4,7,8).
Signal transducer and activator of transcription 3
(STAT3), which belongs to the STAT family, is in general
transiently activated in normal cells but constitutively activated
in a wide variety of blood malignancies and solid tumors, including
breast cancer, prostate cancer, head and neck cancer, melanoma,
brain cancer, as well as GI cancers (9–13).
As one of the limited transcription factors that converge multiple
oncogenic signaling pathways, STAT3 acts as the critical 'switch'
and controls the expression program of tumor-associated genes,
whereby STAT3 is extensively involved in biological processes of GI
malignancies varying from cell cycle, apoptosis, angiogenesis,
stemness, metastasis to immune evasion (9).
In this review, we aim to discuss the molecular
processes of STAT3-mediated EMT in GI cancer. We first describe the
basic mechanisms that regulate the EMT process, then outlines the
extensive oncogenic function of STAT3 in GI cancer, in particular
its potential role in tumorigenesis, metastasis and generation of
CSCs. Based on the theory that EMT is the early step of metastasis
and is intimately associated with tumor stemness, we hypothesized
that STAT3 may also exert an effect in EMT of GI cancer. Therefore,
in the third section, we focus on the possible mechanisms by which
STAT3 contributes to the initiation and resolution of EMT program.
Through investigating into its interactions with specific
transcription factors, miRNAs and signaling pathways, a critical
role of STAT3 in GI cancer EMT has been fundamentally established,
which makes STAT3 a candidate for preventing and reversing the EMT
process in GI cancer.
2. Molecular mechanism of EMT
Hallmark of EMT is the loss of E-cadherin, a
dominant constituent of the adhesion junctions (4–6,14).
E-cadherin functions to maintain an intact cell-cell interaction
and a stabilized cytoskeleton, thus preventing tumor cell mobility,
invasion and subsequent dissemination. In GI cancer, reduced
expression of E-cadherin is significantly correlated with
poorly-differentiated phenotype, lymph node and distant organ
metastasis (15,16). Therefore, repression of this
determinant molecule is considered to be a central event during
EMT. As such, the EMT-transcription factors (EMT-TFs) are basically
classified according to their direct or indirect repression on
E-cadherin. The zinc-finger binding transcription factor Snail1 was
first discovered to downregulate E-cadherin gene expression by
directly binding to its promoter in epithelial tumor cells
(17). After the initial
identification of the interaction between Snail1 and CDH1 gene
(which encodes E-cadherin), many other EMT-TFs such as Snail2 (also
known as Slug), zinc finger E-box-binding homeobox 1 (ZEB1) and
ZEB2 (also know as SIP1), the basic helix-loop-helix (bHLH) factor
E47 (also known as TCF3), Krüppel-like factor 8 (KLF8) were
successively discovered (4). While
other factors, including Twist, fork-head box protein C2 (FOXC2),
goosecoid and E2-2 (also known as TCF4), are demonstrated to induce
EMT without direct binding to the promoter of CDH1 (4). Except for a dramatic change in
E-cadherin expression, EMT is characterized by the reduction of
other epithelial molecules such as claudins, occludin, zona
occludens-1 (ZO-1) and cytokeratins, as well as the concomitant
increase in mesenchymal markers, including vimentin, N-cadherin,
fibronectin, fibroblast specific protein-1 (FSP-1), α-smooth muscle
actin (α-SMA) (4–6).
The mechanisms of EMT has been studied for decades
and it is now generally thought to be transcriptionally regulated
(4,5,14).
These EMT-associated transcription factors, as referred to above,
are regulated by various signaling pathways, including those
activated by transforming growth factor-β (TGF-β), Wnt, Notch,
epidermal growth factor (EGF), fibroblast growth factor (FGF),
hypoxia inducible factor (HIF), NF-κB and Sonic Hedgehog (Shh)
signaling (5,14,18).
These pathways signal through triggering intracellular kinase
cascades, which then operate in crosstalk to form a regulator
network of EMT (5). In addition to
the EMT-inducing transcription factors and their upstream signaling
pathways, recent studies have further illustrated three additional
regulatory mechanisms of EMT, including the expression of small
non-coding RNAs (ncRNAs), alternative splicing and translational
and post-translational modification (14,18).
Intriguingly, a recent study by Rhim et al
(19) has proposed that the EMT
process seems to occur much earlier in tumor than expected. The
lineage tracing system adopted by Rhim et al enables them to
specifically label and track pancreatic epithelial cells in
genetically engineered mouse model of pancreatic intraepithelial
neoplasia (PanIN). To their surprise, the labeled pancreatic cells
are detected in adjacent tissues and circulating system
unexpectedly early, even before the formation of an identifiable
primary tumor (19). The theory
that EMT and the breach of basement membrane occurs prior to tumor
formation probably shed some light on the aggressiveness of
pancreatic cancer and other GI cancers, where many patients have
already progressed to late stage upon presentation of the disease.
This speculation underscores the urgency to better understand the
mechanisms of EMT so as to manage the aggressiveness of GI
cancer.
3. Oncogenic role of STAT3 in GI cancer
STAT3 was originally discovered as a latent
cytoplasmic transcription factor that was activated by
interleukin-6 (IL-6) and EGF (20). Generally, activation of STAT3
relies on the phosphorylation of a conserved tyrosine residue
(Y705) by upstream tyrosine kinases, including growth factor
receptors, cytokine receptor-associated Janus kinase (JAK), as well
as cytoplasmic tyrosine kinases, such as Src and Abelson kinase
(ABL) (9,21,22).
Specifically, STAT3 can be activated by growth factor receptors
that have intrinsic tyrosine kinase activity, such as epidermal
growth factor receptor (EGFR) and platelet-derived growth factor
receptor (PDGFR). Unlike receptor tyrosine kinases (RTKs), many
cytokine receptors like IL-6 family cytokines, do not have
intrinsic tyrosine kinase activity. In that case, the
receptor-associated tyrosine kinases, typically JAK, are recruited
upon ligand engagement and then activated to phosphorylate STAT3
(9,21). In addition to IL-6 receptors,
recent studies have identified Toll-like receptors (TLRs) and
G-protein-coupled receptors (GPCRs) as novel activators of
JAK-STAT3 pathway (22), which
also exert tumor-promoting effects. Once phosphorylated, these
STAT3 monomers form dimers through the reciprocal interactions of
Src homology2 (SH2) domain and then translocate into the nucleus,
where the dimer can directly regulate expression of the target
genes by binding to specific DNA sequences (9).
Activation of STAT3 in normal cells is rapid and
transient, mainly due to the negative regulation of STAT3 by
suppressor of cytokine signaling (SOCS) and protein inhibitor of
activated STAT (PIAS) (9).
However, in contrast to that of normal cells, persistent activation
of STAT3 has been frequently detected in a variety of human cancer
cell lines and tissues, especially those of GI cancer (Table I), including gastric cancer
(10,23), colorectal cancer (11), pancreatic cancer (12) and hepatocellular carcinoma (HCC)
(13). Despite the undefined
mechanism of STAT3 constitutive activation in tumor, strong
biological bases have supported this protein as an oncogenic
driver, and consequently, have validated STAT3 as a promising
target for cancer therapy (21,24).
 | Table IDeregulated activation of STAT3 in GI
cancer cell lines and tumor specimens. |
Table I
Deregulated activation of STAT3 in GI
cancer cell lines and tumor specimens.
| GI cancer | Cancer cell
lines | Tumor
specimens | Correlated
genes | Refs. |
|---|
| Pancreatic
cancer | SW1990, PANC-1,
BxPc-3, FG, Capan-1, Capan-2, MiaPaCa | Pancreatic ductal
adenocarcinoma | Cyclin-D2, VEGF,
MMP-2, ZEB1, E-cadherin | (12,38,41,46,131) |
| Gastric cancer | AGS, MKN1, MKN7,
MKN28, HCG 27 | Gastric
adenocarcinoma | Survivin, VEGF,
Bcl-2 | (10,23) |
| Colorectal
cancer | HT-29, CoGa-1,
SW480, SW1116, LoVo | Colorectal
carcinoma | MMP1, β-catenin,
ZEB1, E-cadherin, | (11,49,121,159) |
| Liver cancer | HCCLM3 | Hepatocellular
carcinoma | VEGF, survivin,
MMP-2, MMP-9 | (13,39) |
| Esophageal
cancer | EC9706, EC 109,
KYSE80, 150, 410, 510 | Esophageal squamous
cell carcinoma | β-catenin | (161) |
Constitutive activation of STAT3 is required for
tumorigenesis of many GI cancers, including pancreatic cancer
(25), HCC (26), gastric cancer and colon cancer
(27). One of the earliest clues
for the role of STAT3 in tumorigenesis was its association with
malignant transformation. Compelling evidence was given when
STAT3C, a constitutively activated mutant form of STAT3,
transformed immortalized fibroblasts in vitro and caused
tumor formation in mice (28).
Other clues for its link with cancer came from the subsequent
findings that STAT3 contributed to tumor growth and survival. As an
oncoprotein, aberrant activation of STAT3 prevented apoptosis by
upregulating the anti-apoptotic Bcl-2 family genes Bcl-xL and MCL1,
as well as survivin, a member of the inhibitor of apoptosis (IAP)
family (9). Also, STAT3 signaling
has been implicated in the regulation of cellular proliferation,
which is dependent on STAT3-induced expression of c-Myc and cyclin
D1 (9). As a consequence, blocking
STAT3 signaling was often sufficient to induce growth arrest and
apoptosis in many different cancer types, in turn demonstrating the
oncogenic properties of STAT3 (21,24).
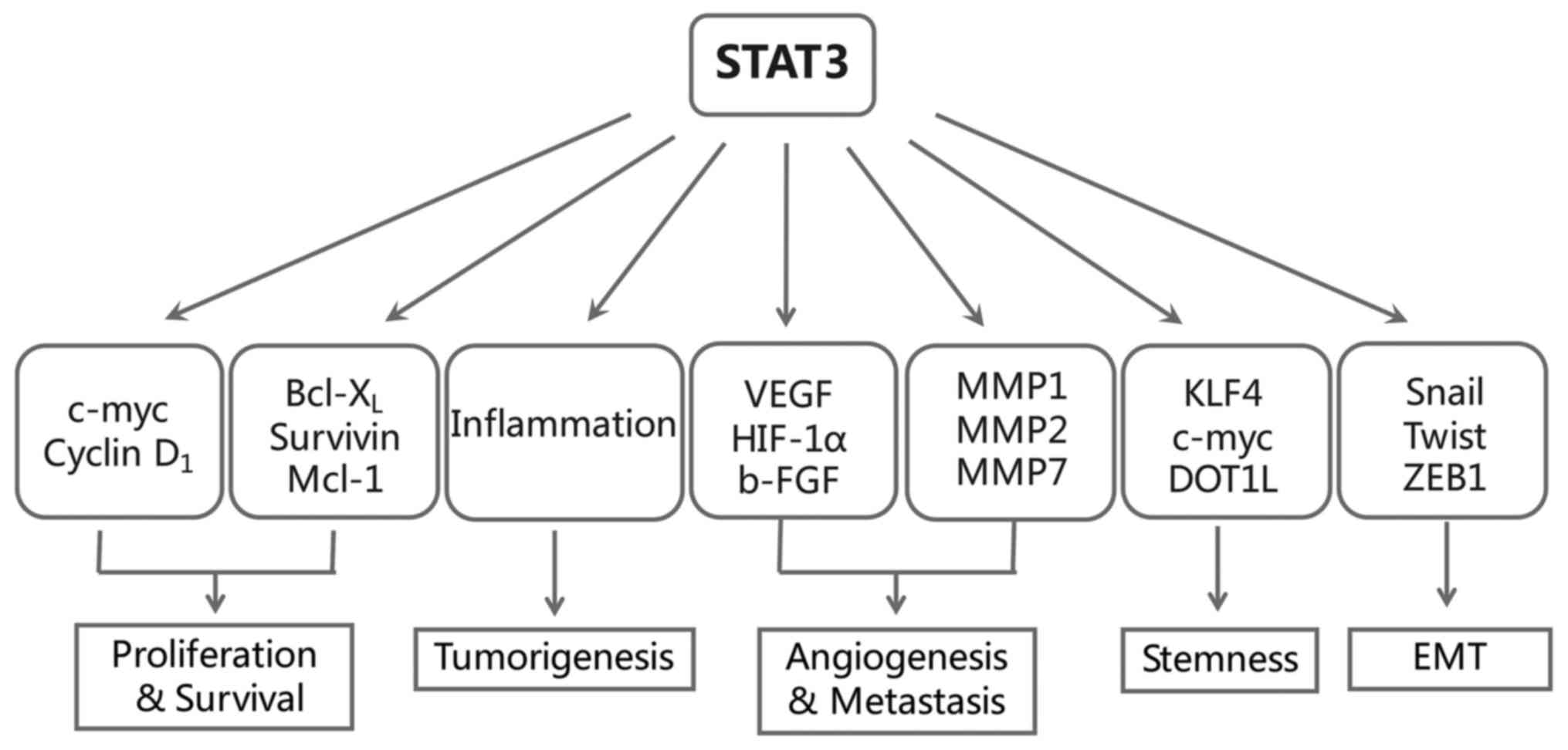
Notably, except for the classic oncogenic functions
of STAT3 mentioned above, in recent years, the role of STAT3 in
inflammatory-associated tumorigenesis, cancer metastasis and
stemness have become topics of particular interest (Fig. 2). We then take closer look into
these three parts.
Role of STAT3 in inflammatory-associated
tumorigenesis of GI cancer
Inflammation virtually exists in every neoplastic
lesion and contributes to tumorigenesis and progression by
supplying immune factors that promote proliferation, survival,
angiogenesis, invasion and metastasis. In particular, chronic
inflammation is typically involved in tumorigenesis of
gastrointestinal and hepatobiliary organs, including
Helicobacter pylori (H. pylori) infection being a
risk factor for gastric cancer and MALT lymphoma, inflammatory
bowel diseases (IBD) for colorectal cancer, chronic virus hepatitis
for HCC, Barrett's esophagus for esophageal cancer and chronic
pancreatitis for pancreatic ductal adenocarcinoma (PDAC).
STAT3 is a transcription factor aberrantly activated
in most GI cancers and has both potent pro-inflammation and
oncogenic properties. It is therefore conceivable that STAT3 plays
an important role in facilitating a tumor-promoting inflammatory
microenvironment in GI cancer (29). For example, chronic pancreatitis is
a well-known risk factor for PDAC. The inflammatory mediator STAT3
has been demonstrated to be an essential component of the
permissive environment provided by pancreatitis to drive the
formation of KRAS-dependent PDAC precursor and its subsequent
progression (30,31). In the setting of both acute and
chronic pancreatitis, deletion of STAT3 in transgenic mice
expressing KRASG12D interferes with the acinar to ductal
de-differentiation, resulting in fewer PanINs formation (30). Corresponding to the previous study
that IL-17, mainly produced by Th17 and IL-17+/γδT cells
recruited to the stroma of pancreatic cancer, can promote PanIN
initiation and progression in cooperation with IL-6/STAT3 signaling
(32), Loncle et al
(33) have recently discovered
that STAT3 is also involved in a novel REG3β-JAK2-STAT3
inflammatory signaling triggered by IL-17 and propels transitional
process from chronic pancreatitis to PDAC.
However, this linking role played by STAT3 appears
to be ubiquitous in GI cancer, not restricted solely to pancreatic
cancer. Sphingosine-1-phosphate receptor 1 (S1PR1), a GPCR that
responds to the lipid metabolic signaling, is upregulated by STAT3
in tumors and reciprocally activates STAT3. Recently, Liang et
al (34) elegantly
demonstrated that sphin-gosine-1-phosphate (S1P) drives a malicious
amplification loop involving SIPR1 and NF-κB/IL-6/STAT3, which
links chronic intestinal inflammation to the occurring of
colitis-associated cancer (CAC). A crucial role of STAT3 in the
development of CAC is also supported by the finding that IL-6/STAT3
functions as a pro-tumorigenic signaling in the model of CAC
(35). In the CAC model induced by
dextran sulphate sodium (DSS), IL-6 activates STAT3 and promotes
the survival of pre-malignant intestinal epithelial cells, which
curbs their chance to further mutate or to subsequently form tumors
(35). However, in gastric cancer,
Ernst et al (36)
demonstrated that it is IL-11, but not IL-6, that leads to abnormal
activation of STAT3 and selectively triggers gastric adenoma
formation in gp130Y757F mice.
In conclusion, STAT3 may assume a central node and a
checkpoint during inflammation-associated tumorigenesis, which has
been demonstrated to be one of the most important pathogenic
mechanisms of GI cancers.
Role of STAT3 in angiogenesis, invasion
and metastasis of GI cancer
Tumors cannot sustain their growth or survive at a
second site unless they are supplied with enough nutrients and
oxygen from newly formed blood vessels. The role of STAT3 in tumor
angiogenesis was first identified when vascular endothelial growth
factor (VEGF), one of the most potent angiogenic factor, was
demonstrated to be a target of STAT3 in various cancers including
pancreatic cancer, gastric cancer and HCC (23,37–39).
An activated STAT3 mutant (STAT3C) was found to upregulate VEGF
expression and stimulate tumor angiogenesis in pancreatic cancer,
while interrupting STAT3 signaling with dominant-negative Stat3
protein significantly abrogated this effect and suppressed tumor
growth and metastasis in vivo (38). In support of this, we have shown
that silencing STAT3 by RNAi led to decrease of VEGF and matrix
metalloproteinase-2 (MMP-2) at both mRNA and protein levels in
pancreatic cancer cells (40).
Besides, by analyzing 71 pancreatic adenocarcinoma specimens, we
found that the expression of p-STAT3 was clinically correlated with
microvascular density (MVD), tumor size, TNM stage and lymphatic
metastasis of pancreatic cancer, which may be partly attributed to
its relationship with VEGF and VEGF-C (41). Here we propose that via
upregulating VEGF-C, which acts on lymphatic endothelial cells to
promote their proliferation and migration, STAT3 also contributes
to the early lymphatic metastasis (41). In addition to directly binding to
the VEGF promoter, STAT3 promotes tumor vascularization indirectly
by controlling the expression of HIF-1α (42), which acts as the final switch of
VEGF expression. Furthermore, tumor-associated myeloid cells that
display activated STAT3 also contribute to the production of VEGF
and bFGF (43). By inducing these
angiogenic factors, STAT3 activated in stroma cells functions to
facilitate endothelium proliferation, migration and microtube
formation (43).
Proteolytic enzymes such as MMPs are required for
the degradation and remodeling of the extracellular matrix (ECM)
and the basement membrane, which is a key step of tumor invasion
and metastasis. It has been shown that STAT3 signaling enforces
MMP-7 expression in pancreatic cancer cells and that MMP-7 deletion
limits tumor size and metastasis in mice (30,44).
Except for MMP-7, MMP-2 has been identified as another target of
STAT3 and also contributes to tumor invasion and metastasis
(45). In line with this, we
reported a reduced expression of MMP-2 in pancreatic cancer cells
caused by STAT3 inhibition, which may account for the impaired
invasion ability observed in vitro and in vivo
(46–48). Similarly, p-STAT3 was also shown
co-localized with MMP-1 in colorectal cancer and was later
demonstrated to experimentally regulate the expression of MMP-1
(49). Therefore, by inducing
diverse MMPs such as MMP-1, MMP-2 and MMP-7, STAT3 plays a major
role during the invasion and infiltration of GI cancers.
As we discussed above, a crucial role of STAT3 in
cancer cell proliferation, survival, angiogenesis and invasion has
been well documented. It is reasonable to speculate that STAT3 also
contributes to cancer metastasis since metastatic potential depends
on multiple factors that determine the growth, apoptosis,
angiogenesis, and invasion of cancer cells. For example, gain- and
loss- of function model demonstrated that STAT3 promoted brain
metastasis in melanoma via dysregulated expression of bFGF, VEGF
and MMP-2 (50). A study using an
orthotropic mouse model of pancreatic cancer showed that blockade
of STAT3 via ectopic expression of dominant-negative STAT3 markedly
reduced the incidence of liver metastasis as well as angiogenesis
(38). Similarly, in mice bearing
orthotopically implanted HCC cells, inhibition of STAT3 with
anti-sense oligonucleotide resulted in decreased vascularization,
local transmission and lung metastasis, along with an impaired
expression of VEGF, bFGF, MMP-2 and MMP-9 (39).
Furthermore, STAT3 abnormally activated in cancer
microenvironment also contributes to the metastasis cascade. For
example, STAT3 activated in immune cells has been well illustrated
to play an immunosuppressive role during cancer development, which
facilitates the dissemination and colonization of cancer cells
(51,52). Additionally, a recent study by Deng
et al (53) suggests an
involvement of S1PR1-JAK2-STAT3 signaling in establishing
pre-metastatic niches in various cancers. These pre-metastatic
niches, are mainly comprised of immune cells including myeloid
cells, provide a sanctuary for disseminated cancer cells to
colonize and form metastases at the hostile distant sites. Of note,
both the migration and outgrowth of myeloid cells at distant organs
require the signaling of S1PR1-STAT3. As expected, inhibiting STAT3
in myeloid compartment disrupts the existing pre-metastatic niches,
as well as the subsequent metastasis (53). Taken together, constitutive
activation of STAT3 in GI cancer functions to promote cancer
progression by facilitating angiogenesis, invasion and
metastasis.
Role of STAT3 in CSC generation of GI
cancer
CSCs are defined as a subpopulation of tumor cells
that sustain self-renewal and are particularly resistant to
conventional therapies. Several lines of evidence have demonstrated
that STAT3 plays an essential role in promoting and maintaining the
stemness of GI cancers (22).
STAT3 is constitutively activated in colon
cancer-initiating cells marked with aldehyde dehydrogenase (ALDH)
and CD133 positive (54). Except
for these two molecular signatures, CD44 has also been recognized
as a marker for CSC of colon cancer which potentially links with
STAT3. A recent study (55) has
found that CD44, when internalized and translocated to the nucleus,
interacts with acetylated STAT3 and together binds to the promoters
of target genes, such as c-myc and twist. This nuclear
CD44/acetylated-STAT3 complex then functions to reprogram cancer
cells to CSC-like cells (55).
Similarly, in liver cancer, CD24+ HCC cells possess
characteristics of stem cells and CD24 has been found to drive
tumor initiation and self-renewal through STAT3-mediated
upregulation of NANOG (56). In
addition, it has also been shown that IL-6/STAT3 signaling
upregulated expression of another CSC marker CD133 and promoted
liver carcinogenesis (57).
Leukemia inhibitory factor (LIF)/STAT3 pathway has
been extensively studied as a potent inducer of mouse embryonic
stem cell self-renewal (22,58).
Efforts to delineate the downstream effector of LIF signaling has
identified Krüppel-like factor 4 (KLF4) as a direct target of STAT3
(58), while KLF4 is known as a
reprogramming factor important for stem cell maintenance and
prevention of differentiation. Except for LIF, IL-6 is also
involved in promoting STAT3-mediated CSC expansion in several types
of malignancies (59,60). Furthermore, immunosuppressive
cells, including myeloid derived suppressor cells (MDSCs) and
tumor-associated macrophages (TAMs), enhanced CSC subpopulation and
promoted tumorigenesis in pancreatic cancer and HCC mainly through
IL-6/STAT3 signaling (61,62). More recently, Kryczek et al
(63) have revealed a novel
mechanism by which the IL-22/STAT3 signaling operates to increase
cancer stemness as well as tumorigenic potential in colorectal
cancer. DOT1L, histone 3 lysine 79 (H3K79) methyltransferase, was
induced by STAT3 activation and then operative to upregulate the
expression of three core stem cell genes, namely NANOG, Sox2 and
Oct4, through methylation of H3K79 (63). Thus, STAT3-mediated epigenetic
regulation has also been implicated in STAT3 induced CSC
generation.
Collectively, these findings confirm that STAT3
indeed plays a critical role in cancer stemness during GI cancer
development, though many mechanisms are still undefined. In recent
years, it has been proposed that cancer cells enforced to undergo
EMT process can simultaneously acquire CSC-like properties
(8). Under that circumstance, our
team has been seeking to determine whether STAT3 may assume one of
the potential links between EMT and stemness in GI cancer, based on
our findings and those of others of the extensive involvement of
STAT3 in GI cancer EMT.
4. Role of STAT3 in regulating EMT of GI
cancer
While STAT3 plays a critical role in the initiation
and progression of GI cancer, it remains elusive whether the
aberrant signaling also contributes to EMT, the early step of tumor
invasion and metastasis. Recent studies have shed some light on the
puzzle, suggesting that STAT3 plays a role in stimulating and
controlling the rapid transition of cells between epithelial and
mesenchymal phenotype, both in physiological and pathological
conditions. For example, during wound healing, keratinocytes at the
border of the wound recapitulate part of the EMT process. Deficient
in STAT3, cell migration of keratinocytes in response to injury is
severely compromised (64). By
upregulating zinc transporter LIV-1, STAT3 is essential for the
migration of zebrafish gastrula organizer cells during its
gastrulation (65).
Importantly, STAT3 has been cumulatively associated
with the type 3 EMT, where it acts as a transcription activator of
EMT-related genes in human cancers, especially in GI cancers
(Table II). In this regard, we
have recently uncovered the function of STAT3 in regulating EMT of
pancreatic cancer (66). Treatment
of IL-6 resulted in STAT3 abnormal activation and surprisingly,
forced pancreatic cancer cells to experience typical EMT
morphological changes, accompanied by an increased invasion ability
and reduced E-cadherin expression (66,67).
Targeting STAT3 signaling either by siRNA or JAK inhibitor AG490
counteracted this effect (46,66,67),
indicating that activation of STAT3 is one of the prerequisites of
IL-6-induced EMT in pancreatic cancer and it may comprise a target
to combat EMT. Additionally, Liu et al (68) have recently demonstrated that
aberrant activation of STAT3, but not the Akt or ERK, mediated
Fos-related antigen-1 (Fra-1) upregulation in response to IL-6 in
colon cancer cells. Fra-1 has been emerging as a central node of
EMT and stemness in various cancer (69). By inducing Snail, Slug, ZEB1, as
well as MMP-2 and MMP-9, the innovative IL-6/STAT3/Fra-1 signaling
axis is responsible for EMT and metastasis of colorectal cancer
(68).
| Table IIMolecular changes associated with
STAT3-mediated EMT in GI cancer. |
Table II
Molecular changes associated with
STAT3-mediated EMT in GI cancer.
| GI cancer | Epithelial
markers↑ | Mesenchymal
markers↓ | Downstream
factors | Refs. |
|---|
| Pancreatic
cancer | E-cadherin | N-cadherin,
vimentin, fibronectin | Snail | (66,88,96) |
| Colorectal
cancer | E-cadherin,
ZO-1 | N-cadherin,
vimentin, fibronectin | Fra-1, Slug, ZEB1,
Snail | (67,93,121,129) |
| Liver cancer | E-cadherin,
β-catenin | N-cadherin,
vimentin | Snail, Twist,
LncTCF7 | (92,95,110,143) |
| Gastric cancer | E-cadherin, ZO-1,
claudin-1 | Vimentin | Twist, ZEB1,
Slug | (149,155) |
Despite the above observations, the underlying
mechanisms of STAT3-induced EMT are not fully understood as yet. To
explore this, in the following sections, we will first discuss the
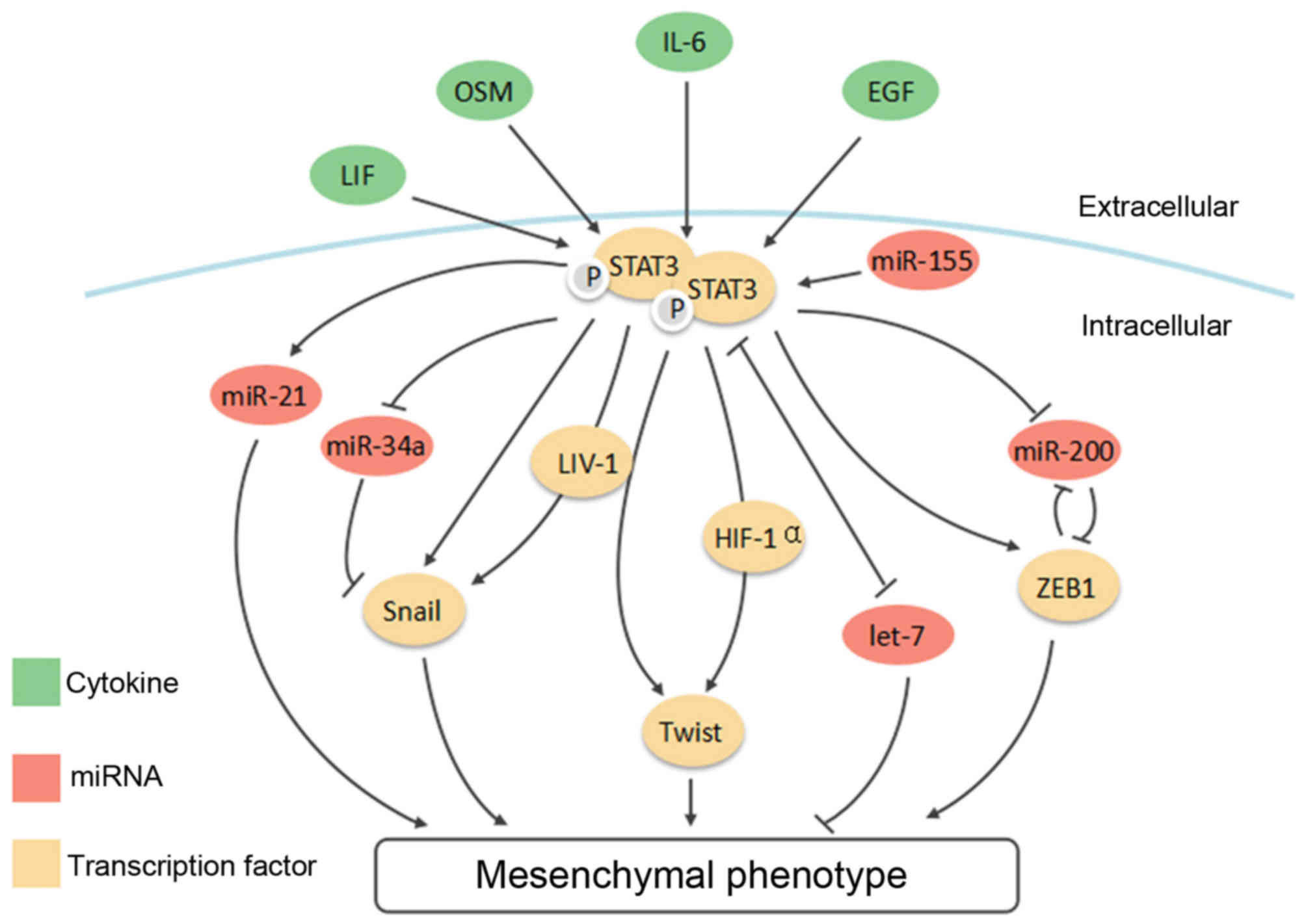
ever-expanding connections between STAT3 and a list of 'master' EMT
transcription factors, such as Snail, Twist1 and ZEB1, then unveil
its interaction with the EMT new player, non-coding RNAs (Fig. 3). Since the regulatory network of
EMT is rather complex and choreographed by multiple signaling
pathways, we will finally look into crosstalk between STAT3 and
other selected signaling pathways, including TGF-β, Notch and Wnt
signaling.
STAT3 and EMT-transcription factors
STAT3 and Snail
Snail, a zinc finger protein encoded by SNAI1, is
one of the master governors of EMT during embryo-genesis, fibrosis
as well as cancer progression (17). The role of Snail has been defined
in tumor invasion, stemness, recurrence and immune suppression
(70–72). Thus, overexpression of Snail is
correlated with lymph node metastases and poor prognosis of
patients with GI cancer, such as pancreatic cancer (73), gastric cancer (74), colorectal cancer (75) and liver cancer (76). Repression of epithelial genes by
Snail requires its C-terminal directly binding to the 5′-CACCTG-3′
elements present in the E-box of epithelial genes and its further
recruitment and interactions with other co-repressors.
Specifically, the SIN3A, histone deacetylase 1 (HDAC1), HDAC2 and
polycomb repressive complex 2 (PRC2) are recruited by Snail upon
its binding at E-cadherin promoter and then downregulate E-cadherin
syngergistically through interacting with the SNAG sequence located
at the N-terminal of Snail (77,78).
Ubiquitin E3 ligase Ring1B and its paralog Ring1A also form
repression complex with Snail, thereby promoting mesenchymal
transformation of pancreatic cells through the C-terminal zinc
finger domains of Snail (79).
As a master regulator of EMT, Snail is regulated by
various signaling pathways, either at transcription or
post-transcription level. For example, SMAD3 and SMAD4 interact and
form a complex with Snail to repress Occludin and E-cadherin during
TGF-β-driven EMT (80). Notch
signaling is required for hypoxia-induced tumor cells EMT and
invasion by controlling the expression of Snail (81). An important modification mechanism
of Snail is the phosphorylation by glycogen synthase kinase-3β
(GSK-3β), which leads to both nuclear export and ubiquitin-mediated
degradation of Snail (82).
Accordingly, several pathways, such as Wnt, PI3K/Akt and NF-κB,
increase Snail activity by alleviating the phosphorylation by
GSK-3β or disrupting the interaction between these two molecules
(82,83).
Apart from the classic signaling pathways mentioned
above, STAT3 has been emerging as a novel regulator of Snail in GI
cancer. As mentioned before, STAT3 transactivates LIV-1 during
gastrulation in zebrafish, which then mediates the nuclear
localization of Snail and the repression of E-cadherin (62). In addition to embryonic
development, 76.4% pancreatic cancer tissues (84) and 61% liver cancer tissues
(85) showed abnormal expression
of LIV-1, which was clinically correlated with tumor size and
lymphatic metastases. An investigation (86) into breast cancer cell lines has
further confirmed the association between STAT3 and LIV-1. It
proposed that the Zn2+ influx triggered by the zinc
transporter LIV-1 and STAT3 functioned to inactivate GSK-3β,
rendering it unable to phosphorylate Snail, which in turn
stabilizes the Snail protein (86). However, whether the same mechanism
exists in GI cancer has not been reported yet. Therefore, our team
has been working on it, hoping to further explain the mechanism by
which STAT3 enhances the expression of Snail.
Additionally, Snail is also directly targeted by
transcription factor STAT3 in diverse epithelial cancers (66,87,88).
IL-6 promotes head and neck tumor EMT via JAK/STAT3/Snail signaling
(87). Consistently, we have
previously shown that activation of STAT3 in response to IL-6
caused a series of EMT-related changes in pancreatic cancer, mainly
by targeting Snail (66). STAT3
can also act as a mediator that converges the signals of TGF-β and
Ras and is thus indispensable for the induction of Snail during
pancreatic cancer progression (88). NDRG2, a member of N-myc downstream
regulated gene 2 (NDRG) family, has been reported as a tumor
suppressor gene in various GI cancers including colon, liver and
pancreatic cancer (89). It was
shown that upregulation of NDRG2 inactivated STAT3-Snail signaling
and thus impaired the EMT potential of cancer cells (90,91),
in turn demonstrating that STAT3/Snail signaling is critical for
the invasion of cancer cells.
Furthermore, this malicious STAT3/Snail axis is also
associated with CSC genes (92,93).
For example, STAT3/Snail signaling could be operative in HCC cells
co-expressing Oct4 and NANOG, to empower HCC cells with mesenchymal
phenotype as well as CSC properties (92). Yao et al (93) recently identified insulin-like
growth factor (IGF) and its downstream STAT3 as signaling pathway
controlling the expression of NANOG in colorectal cancer (CRC)
cells, which then activate Slug to impinge upon the EMT program.
Inhibition of STAT3 in CRC cells not only attenuated migration and
invasion abilities, but also impaired the self-renewal of CRCs due
to the reduced expression of NANOG (93). The potential link of the key stem
cell genes and STAT3/Snail or STAT3/Slug established here may
further confirm the obscure association between stemness and EMT in
GI cancer.
Except for tumor cell itself, the surrounding stroma
functions and constantly communicates with tumor cells to promote
an invasive and drug-resistant phenotype. In particular, the
release of interleukins by immune cells, endothelial cells and
fibroblasts instruct tumor cells to undergo the EMT program
(94). Recently, an attempt to
characterize the role of immune-related cytokines in tumor
microenvironment has identified IL-8 derived from macrophage as the
chief one that dominates EMT process in HCC (95). The JAK2/STAT3 lies downstream of
this IL-8 signaling, conveys the extracellular stimuli into the
nuclear, and switches the epithelial phenotype of HCC cells into
mesenchymal one via activating Snail (95). It has also been shown that
pancreatic cancer cells treated with conditioned medium of
pancreatic stellate cells (PSCs) exhibited enhanced migration
ability and expression of mesenchymal markers (96,97).
This effect was attributed to the enriched IL-6 secreted by PSCs
and subsequent activation of STAT3 and Snail within cancer cells
(96), whereas a possible role of
TGF-β in this process was excluded (97).
STAT3 and Twist
As a bHLH transcription factor, Twist forms homo- or
hetero-dimers to repress epithelial genes and activate mesenchymal
genes that define the EMT phenotype, leading to disassembly of
epithelial junctions and disruption of cellular polarity (98). Overexpression of Twist is
responsible for the poor prognosis of GI tumors, including
hepatocellular carcinoma (76),
esophageal squamous cell carcinoma (99), gastric cancer (100), colorectal cancer (75) and pancreatic cancer (101). Recently, EMT and its reverse
process, mesenchymal-epithelial transition (MET) has received much
attention. Interestingly, this reversible EMT pattern contributing
to different stages of metastasis was implemented by dynamic
expression of Twist (102).
Turning on Twist conferred cancer cells with invasive properties,
facilitating delamination and intravasation, while shutdown of
Twist and the resultant initiation of MET program allowed the
disseminated cells to restore an epithelial phenotype, acquiring
capacities to proliferate and further form macrometastases in
distant sites (102).
Among the multiple upstream signaling pathways,
HIF-1α is a critical one that binds to the hypoxia-response element
in the Twist promoter and enhances its expression, thereby
initiating the metastatic cascade in response to intratumoral
hypoxia (103). Twist is also
subject to posttranscriptional modification. Activation of MAPK,
either by TGF-β or Ras signaling, has been shown to phosphorylate
Twist at Ser68, preventing it from ubiquitination and degradation
(104). Moreover, phosphorylated
Twist by Akt/protein kinase B (PKB) can target TGF-β2
transcriptionally and further promotes metastasis by enhancing
TGF-β receptor signaling (105).
As with Snail, Twist interacts with several components of the
Mi2/nucleosome remodeling and deacetylase (Mi2/NuRD) complex to
repress E-cadherin synergistically (106). Of note, the stemness factor Bmi1
has been identified as a transcriptional target of Twist (107). Once activated by Twist, Bmi1 acts
in a concerted fashion with Twist to repress E-cadherin and the
cell cycle inhibitor p16 (also known as INK4α) simultaneously, thus
conferring tumor with migrating and self-renewing abilities
(107).
Except for the signaling pathways mentioned above,
the STAT3/Twist signaling has also been recognized to orchestrate
EMT in diverse malignancies including GI cancer. The interaction
was first discovered in breast cancer cells (108,109). Activation of STAT3 induced Twist
expression at mRNA and protein levels and promoted migration,
invasion and colony formation of breast cancer cells.
Mechanistically, activated STAT3 can directly bind to the second
proximal STAT3-binding site on Twist promoter and transcriptionally
upregulate its expression (108).
IL-6 functions as an inducer of EMT phenotype in breast cancer
through activating STAT3 and Twist (109). Furthermore, enforced expression
of Twist augments the production of IL-6, giving rise to an
autocrine activation of STAT3 and positive feedback of EMT
(109). Conceivably, this
cooperation between STAT3 and Twist also exists in GI cancer. For
example, Twist has been found to be transcriptionally activated by
STAT3 in HCC (110). Furthermore,
STAT3-mediated Twist expression and EMT process can be triggered by
EGF/EGFR signaling as well (111,112). During this process, STAT3 induces
Twist directly or indirectly via stabilizing HIF-1α protein, which
is another important stimulator of Twist expression (103). Clinical evidence has shown the
level of Twist correlates strongly with that of p-STAT3 in late
stage tumor tissues (108,112),
underscoring the significance of STAT3/Twist signaling during tumor
progression.
STAT3 and ZEB1
Abnormal expression of ZEB1 has been observed in
many GI cancers, such as pancreatic cancer, gastric cancer, colon
cancer, and liver cancer (113).
The two zinc-finger clusters contained by ZEB1 are highly conserved
and critical for its binding ability at the promoter of target
genes, such as CDH1, the gene encoding E-cadherin (113). Like Snail and Twist,
ZEB1-mediated transcription repression of epithelial genes also
involves recruitment of co-repressors, such as C-terminal-binding
protein (CtBP) (114) and SWI/SNF
chromatin remodeling protein BRGI (115). Additionally, ZEB1 has been shown
to recruit HDAC1 and HDAC2 specifically to the CDH1 promoter in
pancreatic cancer, resulting in histone deacetylation and
repression of the gene (116).
In the hierarchical structure of EMT-TFs, Snail
controls ZEB1 expression at different levels and cooperates with
Twist during the induction of ZEB1 (117). Additionally, diverse signaling
pathways have already been shown to activate ZEB1 expression during
embryonic development and tumorigenesis, such as TGF-β, Wnt, NF-κB,
HIF signaling (113). Of note,
the reciprocal regulation between ZEB proteins and miR-200 family
has been well established in recent years (118,119). miR-200 members were first found
to maintain an epithelial phenotype of tumor cells by directly
targeting ZEB1 and ZEB2 (118).
Later study, however, identified miR-200 as targets of ZEB1 as well
(119). Thus miR-200 and ZEB1
form a double-negative feedback loop that finely tunes the EMT
process. Strikingly, via suppressing stemness-inhibiting miRNA such
as miR-200c, miR-203 and miR-183, the EMT activator ZEB1 also
contributes to the stem cell properties of tumor cells indirectly
by rescuing the expression of stem cell factors, primarily Bmil,
Sox2 and KLF4, which are otherwise inhibited by these miRNAs
(120).
Likewise, ZEB1 has also been shown to be
transcriptionally regulated by STAT3. A STAT3/ZEB1/E-cadherin
cascade is uncovered in several malignancies, particularly in
colorectal cancer (CRC) (121–123). Xiong et al (121) found that activation of STAT3
dramatically increased CRC cell invasiveness and resistance to
apoptosis. Mechanistically, STAT3 enhance the expression of ZEB1
and as a result, triggered the EMT program in CRC. In support of
this, a recent study has confirmed the existence of STAT3/ZEB1 axis
in colorectal cancer, proposing that blocking STAT3/ZEB1 signaling
by IL-32θ can inhibit EMT as well as stemness in tumor cells
(123). Although IL-32 was
previously shown to promote gastric cancer metastasis (124), IL-32θ, the newly discovered
isoform of IL-32, was demonstrated to reduce the metastatic
potential by directly binding to STAT3 and interfering with its
nuclear translocation. An impaired self-renewal ability has also
been observed in the IL-32θ-overexpressing cells, which was then
attributed to the downregulation of ZEB1 and Bmi1 (123). The expression of p-STAT3 and ZEB1
are significantly correlated with tumor size and metastasis stages
of CRC patients (121), further
supporting the clinical association between STAT3 and ZEB1.
However, research in this field is relatively
insufficient, and most efforts are restricted to colorectal cancer.
Whether this collaboration between STAT3 and ZEB1 also exists in
other GI cancers needs to be further explored.
Interactions between STAT3 and
non-coding RNA
Non-coding RNAs (ncRNAs) refer to a group of RNAs
that are not translated into proteins. ncRNAs are highly abundant
and contain many functionally important RNAs. For example, transfer
RNA (tRNA), ribosomal RNA (rRNA), small nuclear RNAs (snRNAs), and
small nucleolar (snoRNAs) are required for critical biological
processes, such as protein synthesis, RNA splicing and nuclear
organization. Importantly, some other ncRNAs, specifically
microRNAs (miRNAs), long non-coding RNAs (lncRNAs) and circular
RNAs (circRNAs), have been reported to play regulatory roles in
cancer pathogenesis, including cancer proliferation, apoptosis,
angiogenesis and invasion.
Compelling evidence has shown the interactions
between STAT3 and the regulatory ncRNAs during GI cancer initiation
and progression (125). Herein we
mainly focus on their contribution to EMT and metastasis, by
summarizing which we further conclude to have the role of STAT3 in
coordinating the regulatory circuit of EMT-related transcription
factors and ncRNAs.
Interaction with microRNA
miRNAs are now commonly recognized as potent
modifiers of gene expression profiles in many biological and
pathological processes. These small non-coding RNA molecules,
generally single strands of nucleotides <22, regulate genes
expression by binding to the 3′-untranslated regions (3′-UTR) of
their target mRNAs and then inducing translation repression and/or
mRNAs degradation. In particular, emerging evidence has shown that
some miRNAs are able to define the cellular phenotype through
interactions with transcription regulators of EMT (18). Among these miRNAs, miR-200 family
and miR-34 are undoubtedly the most characteristic ones. As
mentioned above, miR-200 family members are described as
gatekeepers of epithelial phenotype by reciprocally repressing ZEB
family of transcription factors (118,119). Similarly, miR-34 and Snail also
form a double negative feedback loop to control cellular plasticity
(126). Both miR-200s and miR-34
are positively modulated by p53 (127,128). Therefore, loss of p53 function,
which occurs frequently during cancer development, leads to
repression of these miRNAs and further facilitates the transition
of epithelial cells into mesenchymal, migratory ones (127).
Interaction with tumor suppressing
microRNA miR-34
Recently, Rokavec et al (129) proposed an IL-6/STAT3/miR-34a
feedback loop that drives mesenchymal phenotype in various
carcinomas, including CRC. Upon exposure to IL-6, CRC cells
underwent a typical EMT process, which was mechanistically
attributed to the activation of STAT3 and its direct repression on
miR-34a, one of the most characteristic miRNAs that impeded the EMT
process. It was then found that miR-34a also targeted the IL-6
receptor and thus interrupted the IL-6/STAT3/miR34a signaling.
Therefore, the repression of miR-34 by STAT3 activation in turn
reinforced the feedback loop (129). Rokavec et al then provided
in vivo evidence by knocking down miR-34a in a mouse model
of colitis-associated cancer. Deficiency in miR-34a further
facilitated the IL-6-STAT3-mediating tumorigenesis and strikingly,
enabled the tumor progress to an invasive one, which has not been
observed in the same CAC model with intact expression of miR-34a
(35).
Let-7
In addition to miR-34, STAT3 has also been shown to
downregulate miR-200 and let-7 (130). STAT3 was activated upon
oncostatin M (OSM) treatment and mediated the phenotypic transition
by driving two circuits, namely STAT3/lin-28/let-7/HMGA2 and
STAT3/miR-200/ZEB1 (130). While
the relationship between miR-200 and ZEB1 has been well interpreted
above, let-7 is another tumor suppressor shown to interact with
STAT3 extensively. STAT3 inhibited let-7 by transactivating lin-28
(125,130), whereas let-7 downregulated STAT3
by increasing SOCS3 expression (131), one of the negative regulators of
STAT3. Studies showed that upregulation of let-7 restored the
sensitivity to cisplatin in esophageal squamous cell carcinoma
(132) and impaired migration of
pancreatic cancer cells (131),
both due to the interrupted activation of STAT3. Notably,
re-expressing let-7 and miR-200 reversed the EMT phenotype of
gemcitabine-resistant pancreatic cancer cells (133). Since STAT3 has been shown to
induce EMT via repressing let-7 and miR-200 (130), it is tempting to posit that
targeting STAT3 in GI cancer may restore the expression of these
two miRNAs and achieve the same favorable effect.
Interaction with oncogenic microRNA
miR-21
miR-21 is overexpressed in a variety of GI tumors,
including pancreatic cancer, esophageal cancer, colon cancer and
cholangiocarcinoma (134,135). It is generally considered to be
oncogenic miRNA that targets tumor suppressor genes, such as
programmed cell death 4 (PDCD4) and tissue inhibitor of
metalloproteinase 3 (TIMP3) (135). Importantly, miR-21, containing 2
conserved STAT3 binding sites in its enhancer, is a typical miRNA
lying downstream of STAT3 signaling (136). It is recently reported that
miR-21 mediates the promoting effect of LIF/STAT3 on EMT and
metastasis (137). LIF, via
binding to its receptor complex LIF-R/gp130, can trigger distinct
signaling pathways including JAK/STAT3, MAPK, ERK and AKT. Herein,
LIF was shown to promote tumor cell acquisition of mesenchymal
features depending on the induction of miR-21 by STAT3 (137). In line with this, STAT3/miR-21
activation by IL-6 was also shown to be required for
arsenite-induced EMT of human bronchial epithelial cells (138), highlighting the association
between STAT3 and its oncogenic partner miR-21 during EMT
process.
miR-155
Previous study has indicated that miR-155 is
overexpressed in PDAC cell lines and acts as an oncogenic miRNA by
repressing tumor protein 53-induced nuclear protein 1 (TP53INP1)
(139). We have recently found
that the expression of miR-155 was correlated with lymph node
metastases and clinical stages of pancreatic cancer (140). Furthermore, overexpression of
miR-155 in pancreatic cancer cells played a causable role in the
downregulation of SOCS1 and subsequent upregulation of STAT3, which
then promoted emergence of EMT-related features, as enhanced
invasion and migration. This is in tandem with another study
showing that miR-155 can promote STAT3-mediated tumorigenesis in
breast cancer by targeting SOCS1, one of the major suppressing
factors of STAT3 activation (141).
Interaction with long non-coding
RNA
lncRNAs are defined as non-protein coding
transcripts longer than 200 nucleotides. Recently, an lncRNA
activated by TGF-β (lncRNA-ATB) was demonstrated to promote
invasion and metastasis in HCC. By competitively binding miR-200
family, lncRNA-ATB upregulated ZEB1 and ZEB2, and thus induced the
EMT process (142). Subsequently,
an aberrant IL-6/STAT3/IncTCF7 signaling was observed in HCC, which
also contributes to the aggressiveness of the HCC via triggering
EMT (143). These studies well
present a new direction for future study by illustrating the novel
involvement of lncRNA in EMT process and their possible association
with EMT key regulators such as TGF-β and STAT3.
Interaction with circular RNA
circRNAs are a novel class of endogenous non-coding
RNAs that also participates in the gene expression regulation.
Unlike miRNAs and lncRNAs that are terminated with 5′ caps and 3′
tails, circRNAs form a covalently closed loop without accessible
termini, rendering it stable and resistant to exonuclease-mediated
degradation (144). Despite
circRNAs being abundant in eukaryocytes, it was not noted until
recent reports revealed that circRNAs could act as a miRNA sponge
to inhibit its activity and thus regulate gene expression (145).
ciRS-7, presented with >70 conserved miR-7
binding sites, was able to bind to miR-7 efficiently and suppress
its activity, which in turn increased levels of miR-7 targets
(145). As a tumor-suppressing
miRNA, miR-7 has been shown to reverse EMT and impair metastasis in
gastric cancer by targeting insulin-like growth factor receptor 1
(IGFR1) and indirectly downregulating Snail (146). In addition, miR-7 is severely
depressed in colorectal cancer and serves as a tumor suppressor by
targeting the oncogenic transcription factor Yin Yang 1 (YY1)
(147). Therefore, it is tempting
to speculate that ciRS-7 or some unknown circRNAs contribute to the
silencing of tumor-suppressing miRNAs like miR-7 in GI cancer, thus
giving rise to the expression of oncogenic transcription factors.
For example, circRNA_001569 was recently identified as a sponge of
miR-145 and functioned to upregulate miR-145 targets E2F5, BAG4 and
FMNL2, resulting in enhanced proliferation and invasion of
colorectal cancer cells (148).
From this perspective, we have recently noted a
correlative expression of STAT3 and one of the circRNAs in diverse
pancreatic cancer cell lines and tissues (data not shown). Our
following experiments are aimed to find the possible link miRNAs
between these two factors and to further define their exact
function in EMT process of pancreatic cancer. Obviously, the
emerging circRNAs and their mysterious interactions with miRNAs and
transcription factors have prompted new and promising avenues to
uncover the highly complex network of EMT regulation.
Crosstalk with other signaling
pathways
Crosstalk between STAT3 and TGF-β
signaling pathways
TGF-β is a poten inducer of EMT through both
Smad-dependent transcriptional events and Smad-independent
signaling (149). Given the
bi-directional role of TGF-β in carcinogenesis, alterations in
TGF-β signaling that shift the tumor-promoting and
tumor-suppressing balance is rather critical. Deletion of Smad4,
one of the Smad family of signal transducers from TGF-β, occurs in
up to 50% advanced pancreatic cancer, making it candidate for
further investigation. Zhao et al (150) restored Smad4 expression in
pancreatic cancer cells and observed an impaired ability in
invasion and metastasis, which was later attributed to a
Smad4-dependent suppression of STAT3Tyr705 phosphorylation. The
researchers first established cross-talk between Smad4-independent
TGF-β signaling and STAT3 in pancreatic cancer where the persistent
activation of STAT3 due to the loss of Smad4 cooperated with TGF-β
to promote an invasive lineage (150). The same synergistic effect of
IL-6/STAT3 and TGF-β in inducing EMT was also observed in lung
carcinomas in vitro, albeit here STAT3 was shown necessary
for the canonical TGF-β/Smad signaling (151).
Another study has shown that Snail expression was
selectively induced by TGF-β in pancreatic cells harboring mutated
KRAS (88). Through this process,
STAT3 acted as a crucial node that TGF-β and Ras signals converged
on. Treatment of TGF-β relieved the interaction between STAT3 and
its antagonist PIAS3, which instead bound to Smad3 and
consequently, intensified the TGF-β signaling. Interestingly, the
dissociated STAT3 enhanced Snail expression and triggered EMT in
some manner without binding to its canonical DNA-binding sites at
Snail promoter (88). Taken
together, there is tight crosstalk between STAT3 and the classic
EMT-inducing signaling TGF-β, which may contribute to the EMT and
invasion of GI cancers.
Crosstalk between STAT3 and Notch
signaling pathways
Notch, one of the embryonic pathways of epithelial
plasticity, has been associated with tumor recurrence and stemness
partially by inducing EMT (152).
Similar to TGF-β, Notch signaling is tightly involved in initiation
and progression of GI cancer. Investigation on elevated Notch-2 and
its ligand Jagged-1 in gemcitabine-resistant cancer cells uncovered
that Notch signaling was mechanistically linked with acquisition of
a mesenchymal phenotype in pancreatic cancer (153). Targeting both Notch and
JAK2/STAT3 pharmacologically showed efficacy on cell growth and
epithelial plasticity in pancreatic cancer, suggesting a favorable
interplay between these two oncogenic pathways during cancer
progression (154). Of note,
in vitro and in vivo studies demonstrated the
involvement of a novel Notch/STAT3/Twist cascade in gastric cancer
cell growth and metastasis, where the interaction between p-STAT3
and the promoter of Twist was enforced by Notch1 receptor
intracellular domain (N1IC) (155). Therefore, the Notch/STAT3/Twist
cascade has been demonstrated to promote colony formation,
migration and invasion in gastric cancer (155).
How does the activated Notch1 receptor promote
STAT3 phosphorylation? One mechanism may be explained by the direct
phosphorylation of STAT3 in the presence of Hes1 (155,156), the most characteristic target of
Notch signaling. Alternatively, Notch1 signaling may promote
expression of cytokines that can subsequently activate STAT3. For
example, Notch was reported to stimulate IL-6 expression in breast
cancer cells and then drive both autocrine and paracrine JAK/STAT3
signaling via a non-canonical mode, independent of Hes1 (157). Notably, the activation of
jagged1/Notch by IL-6/STAT3 was also identified in the development
of trastuzumab resistance in gastric cancer cells (158). Prolonged treatment by trastuzumab
induced EMT-like and drug-resistant phenotype in gastric cancer,
which was attributed to the compensatory activation of STAT3 and
its reciprocal activation of Notch signaling (158). Collectively, this reciprocal
interaction between STAT3 and Notch signaling may be responsible
for the invasion, stemness as well as drug resistance of GI
cancers.
Crosstalk between STAT3 and Wnt
signaling pathways
Wnt signaling is another signaling that potentially
interacts with STAT3 to elicit the EMT process in GI cancer. In the
absence of Wnt signals, β-catenin is phosphorylated by a complex of
GSK-3β, Axin and adenomatous polyposis coli (APC), which renders
β-catenin sequestered in the cytoplasm. However, in canonical Wnt
signaling, activation of the receptor Frizzled by Wnt ligands
disrupts the formation of the complex and thus enables β-catenin to
translocate into the nucleus, where β-catenin forms complex with T
cell factor/lymphoid enhancing factor (TCF/LEF) family
transcription factors and jointly binds to the promoter of their
target genes, such as Snail, Slug and Twist (5).
Intriguingly, it has been suggested that activation
of STAT3 may participate in the canonical Wnt signaling in GI
cancer. For example, co-expression of p-STAT3 and β-catenin was
observed in colorectal cancer tissues and was associated with worse
prognosis (159). Subsequent
in vitro experiment demonstrated that STAT3 activation is
required for the nuclear accumulation of β-catenin, which is the
commonly recognized key event for the development of colorectal
cancer (159). Furthermore, a
recent study has shown that STAT3 overexpression significantly
increased the levels of β-catenin and TCF1, and further promoted
the proliferation and survival of pancreatic cancer cells (160). Additionally, STAT3 was also
regulated by β-catenin/TCF signaling (161). A functional TCF binding element
was detected in the promoter of STAT3, and transfected β-catenin in
esophageal cancer cells enhanced the expression of STAT3. It was
then proposed that STAT3 is a target of β-catenin/TCF signaling and
by upregulating STAT3, β-catenin/TCF promoted the esophageal
tumorigenesis (161).
Apart from the mechanisms mentioned above, STAT3
has been recently implicated in a noncanonical Wnt signaling that
regulates EMT in a wide range of solid tumors (162). Wnt 5a/b and their cognate
receptor Fzd2 were found to be generally elevated in late-stage
mesenchymal type malignancies including HCC and colon cancer.
Following study revealed an unconventional mechanism of STAT3
activation process in these Wnt5-Fzd2-expressing tumor cells. Wnt
receptor Frizzled2, when activated by its ligands, recruited
tyrosine kinase Fyn, a Src family kinase, to phosphorylate STAT3 on
Tyr705, and thus initiated the transcriptional program that
ultimately drove the process of EMT, cellular migration and
invasion (162). In brief,
despite it is evident that STAT3 interacts with Wnt signaling in
various GI cancers, it remains largely unknown how this crosstalk
contributes to the cancer invasion and metastasis. Apparently, this
needs to be further investigated.
5. Conclusion and outlook
Aberrant activation of STAT3 has been detected at
high frequency in a majority of epithelial malignancies, including
those of GI cancer. However, excessive STAT3 activation usually
occurs in the absence of genetic mutation. Instead it often results
from the autocrine and paracrine production of IL-6 family
cytokines derived from tumor cells and stroma cells. These multiple
oncogenic signaling pathways are responsible for the abnormal
activation of STAT3 and are targets for further investigation.
However, regardless of the undefined mechanisms of STAT3 activation
in tumor cells, strong biological bases have supported it as an
oncogenic driver involved in the initiation and progression of GI
cancer.
In this review, we first summarized the role of
STAT3 in GI cancer pathogenesis, particularly focusing on the
processes of inflammatory-associated tumorigenesis, angiogenesis,
invasion and metastasis, and CSCs generation. After that, we looked
into the possible mechanisms of STAT3-mediated EMT in GI cancer. On
the basis of its extensive interaction with EMT-inducing
transcription factors, miRNAs and other signaling pathways, a
critical role of STAT3 in GI cancer EMT has been fundamentally
established. However, studies of this part are not abundant. New
players in EMT regulatory network such as miRNAs, lncRNAs and
circRNAs are emerging quickly and more importantly, some of them
can be therapeutically manipulated. Therefore, a comprehensive
understanding of the aberrant STAT3 signaling cascade in GI
malignancies and its association with these classic and novel
regulators of EMT may hold great promise for the identification and
validation of therapeutic targets that can effectively repress and
control the aggressiveness of GI cancer.
Acknowledgments
This study was supported by grants from the
Shanghai Health and Family Planning Commission (no. XYQ2013092),
the Shanghai Municipal Human Resources and Social Security Bureau
(nos. 2012040 and 13PJD024), the Shanghai Municipal Science and
Technology Commission (no. 144119668) and the Shanghai Municipal
Education Commission-Gaofeng Clinical Medicine Grant Support (no.
20161425).
References
|
1
|
Bjelakovic G, Nikolova D, Simonetti RG and
Gluud C: Antioxidant supplements for preventing gastrointestinal
cancers. Cochrane Database Syst Rev. 3:CD0041832008.
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:re82014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zheng X, Carstens JL, Kim J, Scheible M,
Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R:
Epithelial-to-mesenchymal transition is dispensable for metastasis
but induces chemoresistance in pancreatic cancer. Nature.
527:525–530. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu H and Jove R: The STATs of cancer--new
molecular targets come of age. Nat Rev Cancer. 4:97–105. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kanda N, Seno H, Konda Y, Marusawa H,
Kanai M, Nakajima T, Kawashima T, Nanakin A, Sawabu T, Uenoyama Y,
et al: STAT3 is constitutively activated and supports cell survival
in association with survivin expression in gastric cancer cells.
Oncogene. 23:4921–4929. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Corvinus FM, Orth C, Moriggl R, Tsareva
SA, Wagner S, Pfitzner EB, Baus D, Kaufmann R, Huber LA, Zatloukal
K, et al: Persistent STAT3 activation in colon cancer is associated
with enhanced cell proliferation and tumor growth. Neoplasia.
7:545–555. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Scholz A, Heinze S, Detjen KM, Peters M,
Welzel M, Hauff P, Schirner M, Wiedenmann B and Rosewicz S:
Activated signal transducer and activator of transcription 3
(STAT3) supports the malignant phenotype of human pancreatic
cancer. Gastroenterology. 125:891–905. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang SF, Wang SN, Wu CF, Yeh YT, Chai CY,
Chunag SC, Sheen MC and Lee KT: Altered p-STAT3 (tyr705) expression
is associated with histological grading and intratumour microvessel
density in hepatocellular carcinoma. J Clin Pathol. 60:642–648.
2007. View Article : Google Scholar
|
|
14
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pignatelli M, Ansari TW, Gunter P, Liu D,
Hirano S, Takeichi M, Klöppel G and Lemoine NR: Loss of membranous
E-cadherin expression in pancreatic cancer: Correlation with lymph
node metastasis, high grade, and advanced stage. J Pathol.
174:243–248. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee SJ, Choi SY, Kim WJ, Ji M, Lee TG, Son
BR, Yoon SM, Sung R, Lee EJ, Youn SJ, et al: Combined aberrant
expression of E-cadherin and S100A4, but not β-catenin is
associated with disease-free survival and overall survival in
colorectal cancer patients. Diagn Pathol. 8:992013. View Article : Google Scholar
|
|
17
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rhim AD, Mirek ET, Aiello NM, Maitra A,
Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK,
Vonderheide RH, et al: EMT and dissemination precede pancreatic
tumor formation. Cell. 148:349–361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhong Z, Wen Z and Darnell JE Jr: Stat3: A
STAT family member activated by tyrosine phosphorylation in
response to epidermal growth factor and interleukin-6. Science.
264:95–98. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Turkson J: STAT proteins as novel targets
for cancer drug discovery. Expert Opin Ther Targets. 8:409–422.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu H, Lee H, Herrmann A, Buettner R and
Jove R: Revisiting STAT3 signalling in cancer: New and unexpected
biological functions. Nat Rev Cancer. 14:736–746. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Choi JH, Ahn MJ, Park CK, Han HX, Kwon SJ,
Lee YY and Kim IS: Phospho-Stat3 expression and correlation with
VEGF, p53, and Bcl-2 in gastric carcinoma using tissue microarray.
APMIS. 114:619–625. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wake MS and Watson CJ: STAT3 the oncogene
- still eluding therapy? FEBS J. 282:2600–2611. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Corcoran RB, Contino G, Deshpande V,
Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA
and Bardeesy N: STAT3 plays a critical role in KRAS-induced
pancreatic tumorigenesis. Cancer Res. 71:5020–5029. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rebouissou S, Amessou M, Couchy G, Poussin
K, Imbeaud S, Pilati C, Izard T, Balabaud C, Bioulac-Sage P and
Zucman-Rossi J: Frequent in-frame somatic deletions activate gp130
in inflammatory hepatocellular tumours. Nature. 457:200–204. 2009.
View Article : Google Scholar :
|
|
27
|
Putoczki TL, Thiem S, Loving A, Busuttil
RA, Wilson NJ, Ziegler PK, Nguyen PM, Preaudet A, Farid R, Edwards
KM, et al: Interleukin-11 is the dominant IL-6 family cytokine
during gastrointestinal tumorigenesis and can be targeted
therapeutically. Cancer Cell. 24:257–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bromberg JF, Wrzeszczynska MH, Devgan G,
Zhao Y, Pestell RG, Albanese C and Darnell JE Jr: Stat3 as an
oncogene. Cell. 98:295–303. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: A leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fukuda A, Wang SC, Morris JP IV, Folias
AE, Liou A, Kim GE, Akira S, Boucher KM, Firpo MA, Mulvihill SJ, et
al: Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma
initiation and progression. Cancer Cell. 19:441–455. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lesina M, Kurkowski MU, Ludes K, Rose-John
S, Treiber M, Klöppel G, Yoshimura A, Reindl W, Sipos B, Akira S,
et al: Stat3/Socs3 activation by IL-6 transsignaling promotes
progression of pancreatic intraepithelial neoplasia and development
of pancreatic cancer. Cancer Cell. 19:456–469. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
McAllister F, Bailey JM, Alsina J, Nirschl
CJ, Sharma R, Fan H, Rattigan Y, Roeser JC, Lankapalli RH, Zhang H,
et al: Oncogenic Kras activates a hematopoietic-to-epithelial IL-17
signaling axis in preinvasive pancreatic neoplasia. Cancer Cell.
25:621–637. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Loncle C, Bonjoch L, Folch-Puy E,
Lopez-Millan MB, Lac S, Molejon MI, Chuluyan E, Cordelier P, Dubus
P, Lomberk G, et al: IL17 functions through the novel
REG3b-JAK2-STAT3 inflammatory pathway to promote the transition
from chronic pancreatitis to pancreatic Cancer. Cancer Res.
75:4852–4862. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liang J, Nagahashi M, Kim EY, Harikumar
KB, Yamada A, Huang WC, Hait NC, Allegood JC, Price MM, Avni D, et
al: Sphingosine-1-phosphate links persistent STAT3 activation,
chronic intestinal inflammation, and development of
colitis-associated cancer. Cancer Cell. 23:107–120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Grivennikov S, Karin E, Terzic J, Mucida
D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H,
Eckmann L, et al: IL-6 and Stat3 are required for survival of
intestinal epithelial cells and development of colitis-associated
cancer. Cancer Cell. 15:103–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ernst M, Najdovska M, Grail D,
Lundgren-May T, Buchert M, Tye H, Matthews VB, Armes J, Bhathal PS,
Hughes NR, et al: STAT3 and STAT1 mediate IL-11-dependent and
inflammation-associated gastric tumorigenesis in gp130 receptor
mutant mice. J Clin Invest. 118:1727–1738. 2008.PubMed/NCBI
|
|
37
|
Niu G, Wright KL, Huang M, Song L, Haura
E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, et al:
Constitutive Stat3 activity upregulates VEGF expression and tumor
angiogenesis. Oncogene. 21:2000–2008. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wei D, Le X, Zheng L, Wang L, Frey JA, Gao
AC, Peng Z, Huang S, Xiong HQ, Abbruzzese JL, et al: Stat3
activation regulates the expression of vascular endothelial growth
factor and human pancreatic cancer angiogenesis and metastasis.
Oncogene. 22:319–329. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li WC, Ye SL, Sun RX, Liu YK, Tang ZY, Kim
Y, Karras JG and Zhang H: Inhibition of growth and metastasis of
human hepatocellular carcinoma by antisense oligonucleotide
targeting signal transducer and activator of transcription 3. Clin
Cancer Res. 12:7140–7148. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang C, Jiang T, Zhu L, Liu J, Cao J,
Huang KJ and Qiu ZJ: STAT3-targeting RNA interference inhibits
pancreatic cancer angiogenesis in vitro and in vivo. Int J Oncol.
38:1637–1644. 2011.PubMed/NCBI
|
|
41
|
Huang C, Huang R, Chang W, Jiang T, Huang
K, Cao J, Sun X and Qiu Z: The expression and clinical significance
of pSTAT3, VEGF and VEGF-C in pancreatic adenocarcinoma. Neoplasma.
59:52–61. 2012. View Article : Google Scholar
|
|
42
|
Xu Q, Briggs J, Park S, Niu G, Kortylewski
M, Zhang S, Gritsko T, Turkson J, Kay H, Semenza GL, et al:
Targeting Stat3 blocks both HIF-1 and VEGF expression induced by
multiple oncogenic growth signaling pathways. Oncogene.
24:5552–5560. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kujawski M, Kortylewski M, Lee H, Herrmann
A, Kay H and Yu H: Stat3 mediates myeloid cell-dependent tumor
angiogenesis in mice. J Clin Invest. 118:3367–3377. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li HD, Huang C, Huang KJ, Wu WD, Jiang T,
Cao J, Feng ZZ and Qiu ZJ: STAT3 knockdown reduces pancreatic
cancer invasiveness and matrix metalloproteinase-7 expression in
nude mice. PLoS One. 6:e259412011. View Article : Google Scholar
|
|
45
|
Xie TX, Wei D, Liu M, Gao AC, Ali-Osman F,
Sawaya R and Huang S: Stat3 activation regulates the expression of
matrix metalloproteinase-2 and tumor invasion and metastasis.
Oncogene. 23:3550–3560. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Huang C, Cao J, Huang KJ, Zhang F, Jiang
T, Zhu L and Qiu ZJ: Inhibition of STAT3 activity with AG490
decreases the invasion of human pancreatic cancer cells in vitro.
Cancer Sci. 97:1417–1423. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Qiu Z, Huang C, Sun J, Qiu W, Zhang J, Li
H, Jiang T, Huang K and Cao J: RNA interference-mediated signal
transducers and activators of transcription 3 gene silencing
inhibits invasion and metastasis of human pancreatic cancer cells.
Cancer Sci. 98:1099–1106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yang G, Huang C, Cao J, Huang KJ, Jiang T
and Qiu ZJ: Lentivirus-mediated shRNA interference targeting STAT3
inhibits human pancreatic cancer cell invasion. World J
Gastroenterol. 15:3757–3766. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tsareva SA, Moriggl R, Corvinus FM,
Wiederanders B, Schütz A, Kovacic B and Friedrich K: Signal
transducer and activator of transcription 3 activation promotes
invasive growth of colon carcinomas through matrix
metalloproteinase induction. Neoplasia. 9:279–291. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xie TX, Huang FJ, Aldape KD, Kang SH, Liu
M, Gershenwald JE, Xie K, Sawaya R and Huang S: Activation of stat3
in human melanoma promotes brain metastasis. Cancer Res.
66:3188–3196. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jones LM, Broz ML, Ranger JJ, Ozcelik J,
Ahn R, Zuo D, Ursini-Siegel J, Hallett MT, Krummel M and Muller WJ:
Stat3 establishes an immunosuppressive microenvironment during the
early stages of breast carcinogenesis to promote tumor growth and
metastasis. Cancer Res. 76:1416–1428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yu H, Kortylewski M and Pardoll D:
Crosstalk between cancer and immune cells: Role of STAT3 in the
tumour microenvironment. Nat Rev Immunol. 7:41–51. 2007. View Article : Google Scholar
|
|
53
|
Deng J, Liu Y, Lee H, Herrmann A, Zhang W,
Zhang C, Shen S, Priceman SJ, Kujawski M, Pal SK, et al:
S1PR1-STAT3 signaling is crucial for myeloid cell colonization at
future metastatic sites. Cancer Cell. 21:642–654. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lin L, Liu A, Peng Z, Lin HJ, Li PK, Li C
and Lin J: STAT3 is necessary for proliferation and survival in
colon cancer-initiating cells. Cancer Res. 71:7226–7237. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Su YJ, Lai HM, Chang YW, Chen GY and Lee
JL: Direct reprogramming of stem cell properties in colon cancer
cells by CD44. EMBO J. 30:3186–3199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lee TK, Castilho A, Cheung VC, Tang KH, Ma
S and Ng IO: CD24(+) liver tumor-initiating cells drive
self-renewal and tumor initiation through STAT3-mediated NANOG
regulation. Cell Stem Cell. 9:50–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Won C, Kim BH, Yi EH, Choi KJ, Kim EK,
Jeong JM, Lee JH, Jang JJ, Yoon JH, Jeong WI, et al: Signal
transducer and activator of transcription 3-mediated CD133
upregulation contributes to promotion of hepatocellular carcinoma.
Hepatology. 62:1160–1173. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Niwa H, Ogawa K, Shimosato D and Adachi K:
A parallel circuit of LIF signalling pathways maintains
pluripotency of mouse ES cells. Nature. 460:118–122. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Marotta LL, Almendro V, Marusyk A,
Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ,
Choudhury SA, Maruyama R, et al: The JAK2/STAT3 signaling pathway
is required for growth of CD44+CD24− stem
cell-like breast cancer cells in human tumors. J Clin Invest.
121:2723–2735. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Schroeder A, Herrmann A, Cherryholmes G,
Kowolik C, Buettner R, Pal S, Yu H, Müller-Newen G and Jove R: Loss
of androgen receptor expression promotes a stem-like cell phenotype
in prostate cancer through STAT3 signaling. Cancer Res.
74:1227–1237. 2014. View Article : Google Scholar
|
|
61
|
Panni RZ, Sanford DE, Belt BA, Mitchem JB,
Worley LA, Goetz BD, Mukherjee P, Wang-Gillam A, Link DC, Denardo
DG, et al: Tumor-induced STAT3 activation in monocytic
myeloid-derived suppressor cells enhances stemness and mesenchymal
properties in human pancreatic cancer. Cancer Immunol Immunother.
63:513–528. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wan S, Zhao E, Kryczek I, Vatan L,
Sadovskaya A, Ludema G, Simeone DM, Zou W and Welling TH:
Tumor-associated macrophages produce interleukin 6 and signal via
STAT3 to promote expansion of human hepatocellular carcinoma stem
cells. Gastroenterology. 147:1393–1404. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kryczek I, Lin Y, Nagarsheth N, Peng D,
Zhao L, Zhao E, Vatan L, Szeliga W, Dou Y, Owens S, et al:
IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3
transcription factor activation and induction of the
methyltransferase DOT1L. Immunity. 40:772–784. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sano S, Itami S, Takeda K, Tarutani M,
Yamaguchi Y, Miura H, Yoshikawa K, Akira S and Takeda J:
Keratinocyte-specific ablation of Stat3 exhibits impaired skin
remodeling, but does not affect skin morphogenesis. EMBO J.
18:4657–4668. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yamashita S, Miyagi C, Fukada T, Kagara N,
Che YS and Hirano T: Zinc transporter LIVI controls
epithelial-mesenchymal transition in zebrafish gastrula organizer.
Nature. 429:298–302. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Huang C, Yang G, Jiang T, Zhu G, Li H and
Qiu Z: The effects and mechanisms of blockage of STAT3 signaling
pathway on IL-6 inducing EMT in human pancreatic cancer cells in
vitro. Neoplasma. 58:396–405. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Huang C, Yang G, Jiang T, Huang K, Cao J
and Qiu Z: Effects of IL-6 and AG490 on regulation of Stat3
signaling pathway and invasion of human pancreatic cancer cells in
vitro. J Exp Clin Cancer Res. 29:512010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Liu H, Ren G, Wang T, Chen Y, Gong C, Bai
Y, Wang B, Qi H, Shen J, Zhu L, et al: Aberrantly expressed Fra-1
by IL-6/STAT3 transactivation promotes colorectal cancer
aggressiveness through epithelial-mesenchymal transition.
Carcinogenesis. 36:459–468. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E,
Reinhardt F, Wu ZJ, Krall JA, Bierie B, Guo W, et al: Protein
kinase Cα is a central signaling node and therapeutic target for
breast cancer stem cells. Cancer Cell. 24:347–364. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wang Y, Shi J, Chai K, Ying X and Zhou BP:
The Role of Snail in EMT and Tumorigenesis. Curr Cancer Drug
Targets. 13:963–972. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ota I, Li XY, Hu Y and Weiss SJ: Induction
of a MT1-MMP and MT2-MMP-dependent basement membrane transmigration
program in cancer cells by Snail1. Proc Natl Acad Sci USA.
106:20318–20323. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kudo-Saito C, Shirako H, Takeuchi T and
Kawakami Y: Cancer metastasis is accelerated through
immunosuppression during Snail-induced EMT of cancer cells. Cancer
Cell. 15:195–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hotz B, Arndt M, Dullat S, Bhargava S,
Buhr HJ and Hotz HG: Epithelial to mesenchymal transition:
Expression of the regulators snail, slug, and twist in pancreatic
cancer. Clin Cancer Res. 13:4769–4776. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Shin NR, Jeong EH, Choi CI, Moon HJ, Kwon
CH, Chu IS, Kim GH, Jeon TY, Kim DH, Lee JH, et al: Overexpression
of Snail is associated with lymph node metastasis and poor
prognosis in patients with gastric cancer. BMC Cancer. 12:5212012.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Kim YH, Kim G, Kwon CI, Kim JW, Park PW
and Hahm KB: TWIST1 and SNAI1 as markers of poor prognosis in human
colorectal cancer are associated with the expression of ALDH1 and
TGF-β1. Oncol Rep. 31:1380–1388. 2014.PubMed/NCBI
|
|
76
|
Yang MH, Chen CL, Chau GY, Chiou SH, Su
CW, Chou TY, Peng WL and Wu JC: Comprehensive analysis of the
independent effect of twist and snail in promoting metastasis of
hepatocellular carcinoma. Hepatology. 50:1464–1474. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
von Burstin J, Eser S, Paul MC, Seidler B,
Brandl M, Messer M, von Werder A, Schmidt A, Mages J, Pagel P, et
al: E-cadherin regulates metastasis of pancreatic cancer in vivo
and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex.
Gastroenterology. 137:361–371. 371.e1–5. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Herranz N, Pasini D, Díaz VM, Francí C,
Gutierrez A, Dave N, Escrivà M, Hernandez-Muñoz I, Di Croce L,
Helin K, et al: Polycomb complex 2 is required for E-cadherin
repression by the Snail1 transcription factor. Mol Cell Biol.
28:4772–4781. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Chen J, Xu H, Zou X, Wang J, Zhu Y, Chen
H, Shen B, Deng X, Zhou A, Chin YE, et al: Snail recruits Ring1B to
mediate transcriptional repression and cell migration in pancreatic
cancer cells. Cancer Res. 74:4353–4363. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Vincent T, Neve EP, Johnson JR, Kukalev A,
Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL,
et al: A SNAIL1-SMAD3/4 transcriptional repressor complex promotes
TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol.
11:943–950. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Sahlgren C, Gustafsson MV, Jin S,
Poellinger L and Lendahl U: Notch signaling mediates
hypoxia-induced tumor cell migration and invasion. Proc Natl Acad
Sci USA. 105:6392–6397. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim
NH, Cha SY, Ryu JK, Choi YJ, Kim J, et al: A Wnt-Axin2-GSK3β
cascade regulates Snail1 activity in breast cancer cells. Nat Cell
Biol. 8:1398–1406. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM
and Zhou BP: Stabilization of snail by NF-kappaB is required for
inflammation-induced cell migration and invasion. Cancer Cell.
15:416–428. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Unno J, Satoh K, Hirota M, Kanno A, Hamada
S, Ito H, Masamune A, Tsukamoto N, Motoi F, Egawa S, et al: LIV-1
enhances the aggressive phenotype through the induction of
epithelial to mesenchymal transition in human pancreatic carcinoma
cells. Int J Oncol. 35:813–821. 2009.PubMed/NCBI
|
|
85
|
Shen R, Xie F, Shen H, liu Q, Zheng T, Kou
X, Wang D and Yang J: Negative correlation of LIV-1 and E-cadherin
expression in hepatocellular carcinoma cells. PLoS One.
8:e565422013. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hogstrand C, Kille P, Ackland ML, Hiscox S
and Taylor KM: A mechanism for epithelial-mesenchymal transition
and anoikis resistance in breast cancer triggered by zinc channel
ZIP6 and STAT3 (signal transducer and activator of transcription
3). Biochem J. 455:229–237. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Yadav A, Kumar B, Datta J, Teknos TN and
Kumar P: IL-6 promotes head and neck tumor metastasis by inducing
epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling
pathway. Mol Cancer Res. 9:1658–1667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Saitoh M, Endo K, Furuya S, Minami M,
Fukasawa A, Imamura T and Miyazawa K: STAT3 integrates cooperative
Ras and TGF-β signals that induce Snail expression. Oncogene.
35:1049–1057. 2016. View Article : Google Scholar
|
|
89
|
Lee DC, Kang YK, Kim WH, Jang YJ, Kim DJ,
Park IY, Sohn BH, Sohn HA, Lee HG, Lim JS, et al: Functional and
clinical evidence for NDRG2 as a candidate suppressor of liver
cancer metastasis. Cancer Res. 68:4210–4220. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kim MJ, Lim J, Yang Y, Lee MS and Lim JS:
N-myc downstream-regulated gene 2 (NDRG2) suppresses the
epithelial-mesenchymal transition (EMT) in breast cancer cells via
STAT3/Snail signaling. Cancer Lett. 354:33–42. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Wang J, Yin D, Xie C, Zheng T, Liang Y,
Hong X, Lu Z, Song X, Song R, Yang H, et al: The iron chelator
Dp44mT inhibits hepatocellular carcinoma metastasis via N-Myc
downstream-regulated gene 2 (NDRG2)/gp130/STAT3 pathway.
Oncotarget. 5:8478–8491. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Yin X, Zhang BH, Zheng SS, Gao DM, Qiu SJ,
Wu WZ and Ren ZG: Coexpression of gene Oct4 and Nanog initiates
stem cell characteristics in hepatocellular carcinoma and promotes
epithelial-mesenchymal transition through activation of Stat3/Snail
signaling. J Hematol Oncol. 8:232015. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Yao C, Su L, Shan J, Zhu C, Liu L, Liu C,
Xu Y, Yang Z, Bian X, Shao J, et al: IGF/STAT3/NANOG/Slug signaling
axis simultaneously controls epithelial-mesenchymal transition and
stemness maintenance in colorectal cancer. Stem Cells. 34:820–831.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Jung HY, Fattet L and Yang J: Molecular
pathways: Linking tumor microenvironment to epithelial-mesenchymal
transition in metastasis. Clin Cancer Res. 21:962–968. 2015.
View Article : Google Scholar
|
|
95
|
Fu XT, Dai Z, Song K, Zhang ZJ, Zhou ZJ,
Zhou SL, Zhao YM, Xiao YS, Sun QM, Ding ZB, et al:
Macrophage-secreted IL-8 induces epithelial-mesenchymal transition
in hepatocellular carcinoma cells by activating the
JAK2/STAT3/Snail pathway. Int J Oncol. 46:587–596. 2015.
|
|
96
|
Hamada S, Masamune A, Yoshida N, Takikawa
T and Shimosegawa T: IL-6/STAT3 plays a regulatory role in the
interaction between pancreatic stellate cells and cancer cells. Dig
Dis Sci. 61:1561–1571. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Kikuta K, Masamune A, Watanabe T, Ariga H,
Itoh H, Hamada S, Satoh K, Egawa S, Unno M and Shimosegawa T:
Pancreatic stellate cells promote epithelial-mesenchymal transition
in pancreatic cancer cells. Biochem Biophys Res Commun.
403:380–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Yang J, Mani SA, Donaher JL, Ramaswamy S,
Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A and
Weinberg RA: Twist, a master regulator of morphogenesis, plays an
essential role in tumor metastasis. Cell. 117:927–939. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Sasaki K, Natsugoe S, Ishigami S,
Matsumoto M, Okumura H, Setoyama T, Uchikado Y, Kita Y, Tamotsu K,
Sakamoto A, et al: Significance of Twist expression and its
association with E-cadherin in esophageal squamous cell carcinoma.
J Exp Clin Cancer Res. 28:1582009. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Feng MY, Wang K, Song HT, Yu HW, Qin Y,
Shi QT and Geng JS: Metastasis-induction and apoptosis-protection
by TWIST in gastric cancer cells. Clin Exp Metastasis.
26:1013–1023. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Satoh K, Hamada S, Kimura K, Kanno A,
Hirota M, Umino J, Fujibuchi W, Masamune A, Tanaka N, Miura K, et
al: Upregulation of MSX2 enhances the malignant phenotype and is
associated with twist 1 expression in human pancreatic cancer
cells. Am J Pathol. 172:926–939. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Tsai JH, Donaher JL, Murphy DA, Chau S and
Yang J: Spatiotemporal regulation of epithelial-mesenchymal
transition is essential for squamous cell carcinoma metastasis.
Cancer Cell. 22:725–736. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Yang MH, Wu MZ, Chiou SH, Chen PM, Chang
SY, Liu CJ, Teng SC and Wu KJ: Direct regulation of TWIST by HIF-1α
promotes metastasis. Nat Cell Biol. 10:295–305. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Hong J, Zhou J, Fu J, He T, Qin J, Wang L,
Liao L and Xu J: Phosphorylation of serine 68 of Twist1 by MAPKs
stabilizes Twist1 protein and promotes breast cancer cell
invasiveness. Cancer Res. 71:3980–3990. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Xue G, Restuccia DF, Lan Q, Hynx D,
Dirnhofer S, Hess D, Rüegg C and Hemmings BA: Akt/PKB-mediated
phosphorylation of Twist1 promotes tumor metastasis via mediating
cross-talk between PI3K/Akt and TGF-β signaling axes. Cancer
Discov. 2:248–259. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Fu J, Qin L, He T, Qin J, Hong J, Wong J,
Liao L and Xu J: The TWIST/Mi2/NuRD protein complex and its
essential role in cancer metastasis. Cell Res. 21:275–289. 2011.
View Article : Google Scholar
|
|
107
|
Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY,
Yang WH, Huang CH, Kao SY, Tzeng CH, Tai SK, et al: Bmi1 is
essential in Twist1-induced epithelial-mesenchymal transition. Nat
Cell Biol. 12:982–992. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola
D, Mansour M, Xu LM, Costanzo C, Cheng JQ and Wang LH: Twist is
transcrip-tionally induced by activation of STAT3 and mediates
STAT3 oncogenic function. J Biol Chem. 283:14665–14673. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Sullivan NJ, Sasser AK, Axel AE, Vesuna F,
Raman V, Ramirez N, Oberyszyn TM and Hall BM: Interleukin-6 induces
an epithelial-mesenchymal transition phenotype in human breast
cancer cells. Oncogene. 28:2940–2947. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Zhang C, Guo F, Xu G, Ma J and Shao F:
STAT3 cooperates with Twist to mediate epithelial-mesenchymal
transition in human hepatocellular carcinoma cells. Oncol Rep.
33:1872–1882. 2015.PubMed/NCBI
|
|
111
|
Cho KH, Choi MJ, Jeong KJ, Kim JJ, Hwang
MH, Shin SC, Park CG and Lee HY: A ROS/STAT3/HIF-1α signaling
cascade mediates EGF-induced TWIST1 expression and prostate cancer
cell invasion. Prostate. 74:528–536. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei
Y, Abbruzzese JL, Hortobagyi GN and Hung MC: Epidermal growth
factor receptor cooperates with signal transducer and activator of
transcription 3 to induce epithelial-mesenchymal transition in
cancer cells via upregulation of TWIST gene expression. Cancer Res.
67:9066–9076. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Zhang P, Sun Y and Ma L: ZEB1: At the
crossroads of epithelial-mesenchymal transition, metastasis and
therapy resistance. Cell Cycle. 14:481–487. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Shi Y, Sawada J, Sui G, Affar B, Whetstine
JR, Lan F, Ogawa H, Luke MP, Nakatani Y and Shi Y: Coordinated
histone modifications mediated by a CtBP co-repressor complex.
Nature. 422:735–738. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Sánchez-Tilló E, Lázaro A, Torrent R,
Cuatrecasas M, Vaquero EC, Castells A, Engel P and Postigo A: ZEB1
represses E-cadherin and induces an EMT by recruiting the SWI/SNF
chromatin-remodeling protein BRG1. Oncogene. 29:3490–3500. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Aghdassi A, Sendler M, Guenther A, Mayerle
J, Behn CO, Heidecke CD, Friess H, Büchler M, Evert M, Lerch MM, et
al: Recruitment of histone deacetylases HDAC1 and HDAC2 by the
transcriptional repressor ZEB1 downregulates E-cadherin expression
in pancreatic cancer. Gut. 61:439–448. 2012. View Article : Google Scholar
|
|
117
|
Dave N, Guaita-Esteruelas S, Gutarra S,
Frias À, Beltran M, Peiró S and de Herreros AG: Functional
cooperation between Snail1 and twist in the regulation of ZEB1
expression during epithelial to mesenchymal transition. J Biol
Chem. 286:12024–12032. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Gregory PA, Bert AG, Paterson EL, Barry
SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y and Goodall GJ:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Burk U, Schubert J, Wellner U, Schmalhofer
O, Vincan E, Spaderna S and Brabletz T: A reciprocal repression
between ZEB1 and members of the miR-200 family promotes EMT and
invasion in cancer cells. EMBO Rep. 9:582–589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Wellner U, Schubert J, Burk UC,
Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D,
zur Hausen A, et al: The EMT-activator ZEB1 promotes tumorigenicity
by repressing stemness-inhibiting microRNAs. Nat Cell Biol.
11:1487–1495. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Xiong H, Hong J, Du W, Lin YW, Ren LL,
Wang YC, Su WY, Wang JL, Cui Y, Wang ZH, et al: Roles of STAT3 and
ZEB1 proteins in E-cadherin downregulation and human colorectal
cancer epithelial-mesenchymal transition. J Biol Chem.
287:5819–5832. 2012. View Article : Google Scholar
|
|
122
|
Avtanski DB, Nagalingam A, Bonner MY,
Arbiser JL, Saxena NK and Sharma D: Honokiol inhibits
epithelial-mesenchymal transition in breast cancer cells by
targeting signal transducer and activator of transcription
3/Zeb1/E-cadherin axis. Mol Oncol. 8:565–580. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Bak Y, Kwon T, Bak IS, Hong J, Yu DY and
Yoon DY: IL-32θ inhibits stemness and epithelial-mesenchymal
transition of cancer stem cells via the STAT3 pathway in colon
cancer. Oncotarget. 7:7307–7317. 2016.PubMed/NCBI
|
|
124
|
Tsai CY, Wang CS, Tsai MM, Chi HC, Cheng
WL, Tseng YH, Chen CY, Lin CD, Wu JI, Wang LH, et al:
Interleukin-32 increases human gastric cancer cell invasion
associated with tumor progression and metastasis. Clin Cancer Res.
20:2276–2288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Cao Q, Li YY, He WF, Zhang ZZ, Zhou Q, Liu
X, Shen Y and Huang TT: Interplay between microRNAs and the STAT3
signaling pathway in human cancers. Physiol Genomics. 45:1206–1214.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Siemens H, Jackstadt R, Hünten S, Kaller
M, Menssen A, Götz U and Hermeking H: miR-34 and SNAIL form a
double-negative feedback loop to regulate epithelial-mesenchymal
transitions. Cell Cycle. 10:4256–4271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Chang CJ, Chao CH, Xia W, Yang JY, Xiong
Y, Li CW, Yu WH, Rehman SK, Hsu JL, Lee HH, et al: p53 regulates
epithelial-mesenchymal transition and stem cell properties through
modulating miRNAs. Nat Cell Biol. 13:317–323. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Kim NH, Kim HS, Li XY, Lee I, Choi HS,
Kang SE, Cha SY, Ryu JK, Yoon D, Fearon ER, et al: A p53/miRNA-34
axis regulates Snail1-dependent cancer cell epithelial-mesenchymal
transition. J Cell Biol. 195:417–433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Rokavec M, Öner MG, Li H, Jackstadt R,
Jiang L, Lodygin D, Kaller M, Horst D, Ziegler PK, Schwitalla S, et
al: IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated
colorectal cancer invasion and metastasis. J Clin Invest.
124:1853–1867. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Guo L, Chen C, Shi M, Wang F, Chen X, Diao
D, Hu M, Yu M, Qian L and Guo N: Stat3-coordinated
Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain
oncostatin M-driven epithelial-mesenchymal transition. Oncogene.
32:5272–5282. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Patel K, Kollory A, Takashima A, Sarkar S,
Faller DV and Ghosh SK: MicroRNA let-7 downregulates STAT3
phos-phorylation in pancreatic cancer cells by increasing SOCS3
expression. Cancer Lett. 347:54–64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Sugimura K, Miyata H, Tanaka K, Hamano R,
Takahashi T, Kurokawa Y, Yamasaki M, Nakajima K, Takiguchi S, Mori
M, et al: Let-7 expression is a significant determinant of response
to chemotherapy through the regulation of IL-6/STAT3 pathway in
esophageal squamous cell carcinoma. Clin Cancer Res. 18:5144–5153.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Li Y, VandenBoom TG II, Kong D, Wang Z,
Ali S, Philip PA and Sarkar FH: Upregulation of miR-200 and let-7
by natural agents leads to the reversal of
epithelial-to-mesenchymal transition in gemcitabine-resistant
pancreatic cancer cells. Cancer Res. 69:6704–6712. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Nagao Y, Hisaoka M, Matsuyama A, Kanemitsu
S, Hamada T, Fukuyama T, Nakano R, Uchiyama A, Kawamoto M,
Yamaguchi K, et al: Association of microRNA-21 expression with its
targets, PDCD4 and TIMP3, in pancreatic ductal adenocarcinoma. Mod
Pathol. 25:112–121. 2012. View Article : Google Scholar
|
|
135
|
Selaru FM, Olaru AV, Kan T, David S, Cheng
Y, Mori Y, Yang J, Paun B, Jin Z, Agarwal R, et al: MicroRNA-21 is
overexpressed in human cholangiocarcinoma and regulates programmed
cell death 4 and tissue inhibitor of metalloproteinase 3.
Hepatology. 49:1595–1601. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Löffler D, Brocke-Heidrich K, Pfeifer G,
Stocsits C, Hackermüller J, Kretzschmar AK, Burger R, Gramatzki M,
Blumert C, Bauer K, et al: Interleukin-6 dependent survival of
multiple myeloma cells involves the Stat3-mediated induction of
microRNA-21 through a highly conserved enhancer. Blood.
110:1330–1333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Yue X, Zhao Y, Zhang C, Li J, Liu Z, Liu J
and Hu W: Leukemia inhibitory factor promotes EMT through
STAT3-dependent miR-21 induction. Oncotarget. 7:3777–3790.
2016.
|
|
138
|
Luo F, Xu Y, Ling M, Zhao Y, Xu W, Liang
X, Jiang R, Wang B, Bian Q and Liu Q: Arsenite evokes IL-6
secretion, autocrine regulation of STAT3 signaling, and miR-21
expression, processes involved in the EMT and malignant
transformation of human bronchial epithelial cells. Toxicol Appl
Pharmacol. 273:27–34. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Gironella M, Seux M, Xie MJ, Cano C,
Tomasini R, Gommeaux J, Garcia S, Nowak J, Yeung ML, Jeang KT, et
al: Tumor protein 53-induced nuclear protein 1 expression is
repressed by miR-155, and its restoration inhibits pancreatic tumor
development. Proc Natl Acad Sci USA. 104:16170–16175. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Huang C, Li H, Wu W, Jiang T and Qiu Z:
Regulation of miR-155 affects pancreatic cancer cell invasiveness
and migration by modulating the STAT3 signaling pathway through
SOCS1. Oncol Rep. 30:1223–1230. 2013.PubMed/NCBI
|
|
141
|
Jiang S, Zhang HW, Lu MH, He XH, Li Y, Gu
H, Liu MF and Wang ED: MicroRNA-155 functions as an OncomiR in
breast cancer by targeting the suppressor of cytokine signaling 1
gene. Cancer Res. 70:3119–3127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ,
Tao QF, Liu F, Pan W, Wang TT, Zhou CC, et al: A long noncoding RNA
activated by TGF-β promotes the invasion-metastasis cascade in
hepato-cellular carcinoma. Cancer Cell. 25:666–681. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Wu J, Zhang J, Shen B, Yin K, Xu J, Gao W
and Zhang L: Long noncoding RNA lncTCF7, induced by IL-6/STAT3
transactivation, promotes hepatocellular carcinoma aggressiveness
through epithelial-mesenchymal transition. J Exp Clin Cancer Res.
34:1162015. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Qu S, Yang X, Li X, Wang J, Gao Y, Shang
R, Sun W, Dou K and Li H: Circular RNA: A new star of noncoding
RNAs. Cancer Lett. 365:141–148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Hansen TB, Jensen TI, Clausen BH, Bramsen
JB, Finsen B, Damgaard CK and Kjems J: Natural RNA circles function
as efficient microRNA sponges. Nature. 495:384–388. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Zhao X, Dou W, He L, Liang S, Tie J, Liu
C, Li T, Lu Y, Mo P, Shi Y, et al: MicroRNA-7 functions as an
anti-metastatic microRNA in gastric cancer by targeting
insulin-like growth factor-1 receptor. Oncogene. 32:1363–1372.
2013. View Article : Google Scholar
|
|
147
|
Zhang N, Li X, Wu CW, Dong Y, Cai M, Mok
MT, Wang H, Chen J, Ng SS, Chen M, et al: microRNA-7 is a novel
inhibitor of YY1 contributing to colorectal tumorigenesis.
Oncogene. 32:5078–5088. 2013. View Article : Google Scholar
|
|
148
|
Xie H, Ren X, Xin S, Lan X, Lu G, Lin Y,
Yang S, Zeng Z, Liao W, Ding YQ, et al: Emerging roles of
circRNA_001569 targeting miR-145 in the proliferation and invasion
of colorectal cancer. Oncotarget. 7:26680–26691. 2016.PubMed/NCBI
|
|
149
|
Massagué J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Zhao S, Venkatasubbarao K, Lazor JW,
Sperry J, Jin C, Cao L and Freeman JW: Inhibition of STAT3 Tyr705
phosphorylation by Smad4 suppresses transforming growth factor
beta-mediated invasion and metastasis in pancreatic cancer cells.
Cancer Res. 68:4221–4228. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Liu RY, Zeng Y, Lei Z, Wang L, Yang H, Liu
Z, Zhao J and Zhang HT: JAK/STAT3 signaling is required for
TGF-β-induced epithelial-mesenchymal transition in lung cancer
cells. Int J Oncol. 44:1643–1651. 2014.PubMed/NCBI
|
|
152
|
Espinoza I and Miele L: Deadly crosstalk:
Notch signaling at the intersection of EMT and cancer stem cells.
Cancer Lett. 341:41–45. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Wang Z, Li Y, Kong D, Banerjee S, Ahmad A,
Azmi AS, Ali S, Abbruzzese JL, Gallick GE and Sarkar FH:
Acquisition of epithelial-mesenchymal transition phenotype of
gemcitabine-resistant pancreatic cancer cells is linked with
activation of the notch signaling pathway. Cancer Res.
69:2400–2407. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Palagani V, Bozko P, El Khatib M, Belahmer
H, Giese N, Sipos B, Malek NP and Plentz RR: Combined inhibition of
Notch and JAK/STAT is superior to monotherapies and impairs
pancreatic cancer progression. Carcinogenesis. 35:859–866. 2014.
View Article : Google Scholar
|
|
155
|
Hsu KW, Hsieh RH, Huang KH, Fen-Yau Li A,
Chi CW, Wang TY, Tseng MJ, Wu KJ and Yeh TS: Activation of the
Notch1/STAT3/Twist signaling axis promotes gastric cancer
progression. Carcinogenesis. 33:1459–1467. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Kamakura S, Oishi K, Yoshimatsu T,
Nakafuku M, Masuyama N and Gotoh Y: Hes binding to STAT3 mediates
crosstalk between Notch and JAK-STAT signalling. Nat Cell Biol.
6:547–554. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Jin S, Mutvei AP, Chivukula IV, Andersson
ER, Ramsköld D, Sandberg R, Lee KL, Kronqvist P, Mamaeva V, Ostling
P, et al: Non-canonical Notch signaling activates IL-6/JAK/STAT
signaling in breast tumor cells and is controlled by p53 and
IKKα/IKKβ. Oncogene. 32:4892–4902. 2013. View Article : Google Scholar
|
|
158
|
Yang Z, Guo L, Liu D, Sun L, Chen H, Deng
Q, Liu Y, Yu M, Ma Y, Guo N, et al: Acquisition of resistance to
trastuzumab in gastric cancer cells is associated with activation
of IL-6/STAT3/Jagged-1/Notch positive feedback loop. Oncotarget.
6:5072–5087. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Kawada M, Seno H, Uenoyama Y, Sawabu T,
Kanda N, Fukui H, Shimahara Y and Chiba T: Signal transducers and
activators of transcription 3 activation is involved in nuclear
accumulation of beta-catenin in colorectal cancer. Cancer Res.
66:2913–2917. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Pramanik KC, Fofaria NM, Gupta P, Ranjan
A, Kim SH and Srivastava SK: Inhibition of β-catenin signaling
suppresses pancreatic tumor growth by disrupting nuclear
β-catenin/TCF-1 complex: Critical role of STAT-3. Oncotarget.
6:11561–11574. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Yan S, Zhou C, Zhang W, Zhang G, Zhao X,
Yang S, Wang Y, Lu N, Zhu H and Xu N: beta-Catenin/TCF pathway
upregulates STAT3 expression in human esophageal squamous cell
carcinoma. Cancer Lett. 271:85–97. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Gujral TS, Chan M, Peshkin L, Sorger PK,
Kirschner MW and MacBeath G: A noncanonical Frizzled2 pathway
regulates epithelial-mesenchymal transition and metastasis. Cell.
159:844–856. 2014. View Article : Google Scholar : PubMed/NCBI
|