Introduction
Gliomas are primary solid tumors of the brain and
may arise from transformations of three major types of glial cells,
including astrocytes, oligodendrocytes, and ependymal cells
(1). According to the World Health
Organization grading system, gliomas are classed into low-grade
tumors (grades I and II) and high-grade tumors (grades III and IV).
Glioblastoma multiforme (GBM), classified as a malignant grade IV
glioma, is the most aggressive brain tumor (2). The location of tumor occurrence in
the brain and the high variability in genetic abnormalities are two
major risk factors existing in malignant gliomas (3). In addition, malignant gliomas present
unique features of rapid growth and high migration that
consequently result in poor prognoses and high mortality rates in
GBM patients (4). In the clinic,
the median overall survival time and the 5-year survival rate of
GBM patients are ~10.2–14.6 months and 5%, respectively (5). Therefore, discovering more-effective
therapeutic strategies for glioma patients remains a challenge
today.
Hypoxia is a condition in which a tissue is not
satisfactorily oxygenated. Under hypoxic stimulation, tissues/cells
can be driven either to survive by expanding the glycolysis rate or
to death via a programmed pathway (6). In contrast, hypoxic microenvironments
inside cancers are able to promote tumor growth, angiogenesis, and
invasion (7). Brain hypoxia
frequently occurs in brain neoplasms and traumatic brain injuries
and is furthermore recognized as a major potential cause of
secondary injury in neurologically critically ill patients
(8). In hypoxic conditions,
hypoxia-inducible factor (HIF)-1α, a transcription factor, is
highly induced and essentially participates in regulating cell
survival or death (9,10). HIF-1α functions by binding to
specific recognition sequences in the genome to stimulate
transcriptions of certain genes involved in various metabolic
pathways that are necessary for cells in response to an oxygen-poor
environment (11,12). Joseph et al, reported that
an HIF-1α-mediated signaling axis contributes to a hypoxia-induced
mesenchymal shift, migration, and invasion in malignant gliomas
(13). Nonetheless, a previous
study showed the positive roles of HIF-1α in hypoxia-induced
glioblastoma cell death (14).
Therefore, hypoxia has bidirectional roles in controlling glioma
growth and death.
Autophagy is a preserved progression inside cells
that uses double-membrane vesicles, called autophagosomes, to
deliver cytoplasmic contents to lysosomes for degradation of
long-lived and misfolded proteins, damaged and dysfunctional
organelles, and foreign particles (15,16).
Microtubule-associated protein light chain 3 (LC3), a consistent
marker of autophagy, is important for the progression of autophagy
because it is essential for the formation of autophagosomes
(17). Autophagy is critical for
maintaining homeostasis in certain diseases, including
neurodegeneration, cancers and aging (15,18).
Recently, induction of autophagy by drugs was considered a novel
therapeutic strategy for treating GBM cancer stem cells and
temozolomide (TMZ)-resistant glioma cells (19). Dolma et al showed that
inhibition of the D4 dopamine receptor delays autophagic flux,
proliferation, and survival of glioblastoma stem cells (20). In our lab, we proved the effects of
honokiol, a polyphenol, on initiating autophagy and subsequent cell
death in human malignant gliomas in vitro and in vivo
(21). Recently, induction of
hypoxia was investigated as an effective treatment strategy
(19,22). However, autophagic cells can
survive or undergo apoptosis (23,24).
Our previous studies showed honokiol-induced autophagic death in
neuroblastomas and gliomas (21,25).
p53, a tumor suppressor protein, is involved in regulating cell
survival, autophagy and apoptosis (26). In this study, we investigated the
effects of hypoxia on autophagy and apoptosis of human malignant
glioma cells and the possible mechanisms.
Materials and methods
Cell culture and drug treatment
Human glioma U87-MG cells and DBTRG-05MG cells
purchased from American Type Culture Collection (Manassas, VA, USA)
were cultured in minimum essential medium (MEM; Gibco-BRL Life
Technologies, Grand Island, NY, USA) supplemented with 10% fetal
bovine serum (FBS), 2 mM L-glutamine, 100 IU/ml penicillin, 100
mg/ml streptomycin, 1 mM sodium pyruvate, and 1 mM non-essential
amino acids at 37°C in a humidified atmosphere of 5%
CO2. Cells were grown to confluence before drug
treatment. Cobalt chloride (CoCl2) purchased from Sigma
(St. Louis, MO, USA) was freshly dissolved in in 1X
phosphate-buffered saline (PBS; 0.14 M NaCl, 2.6 mM KCl, 8 mM
Na2HPO4 and 1.5 mM
KH2PO4). Human U87-MG cells were exposed to
different concentrations of CoCl2 and/or other agents
indicated in the text for various intervals. Control cells received
PBS only. 3-Methyladenine (3-MA) and chloroquine, inhibitors of
cell autophagy, and rapamycin (Rapa), an inducer of cell autophagy,
were purchased from Sigma. To determine the effects of 3-MA,
chloroquine, and Rapa on CoCl2-induced autophagy,
caspase-3 activation, cell apoptosis, and cell death, the glioma
cells were pretreated with 1 mM 3-MA, 20 µM chloroquine, or
0.5 µM Rapa for 1 h and then exposed to
CoCl2.
Assays of cell morphology and cell
viability
The toxicity of hypoxia to human glioma U87-MG cells
was determined by analyses of cell morphology and viability as
described previously (12).
Briefly, U87-MG cells (104 cells/well) were seeded in
96-well tissue culture plates overnight. After drug treatment,
U87-MG cells were cultured in new medium containing 0.5 mg/ml
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide for a
further 3 h. The blue formazan products in U87-MG cells were
dissolved in dimethyl sulfoxide and spectrophotometrically measured
at a wavelength of 550 nm. Cell morphologies were observed and
photographed using a light microscope (Nikon, Tokyo, Japan).
Cell proliferation assay
The effect of hypoxia on cell proliferation was
analyzed by measuring the incorporation of bromodeoxyuridine (BrdU)
into genomic DNA as described previously (27). Human glioma U87-MG cells were
seeded at 3×103 cells/well in 96-well microtiter plates.
After drug treatment, 10 mM BrdU was added to the culture medium
for incorporation into the DNA of replicating cells. After 2 h of
incubation, cells were fixed in 4% paraformaldehyde. BrdU
incorporation was determined by a cell proliferation enzyme-linked
immunosorbent assay (ELISA) BrdU kit (Roche, Mannheim,
Germany).
Assay of cell autophagy
Cell autophagy was assayed by quantifying acidic
vesicular organelles using flow cytometry as described previously
(25). After drug treatment, human
glioma U87-MG and DBTRG-05MG cells (105) were treated
with 1 µg/ml of acridine orange (AO) for 20 min. Then, the
cells were collected in phenol red-free MEM. The green and red
fluorescence of AO in cells were measured with a flow cyto-meter
(Becton-Dickinson, San Jose, CA, USA). Fluorescent intensities were
quantified with the aid of CellQuest software (Becton-Dickinson). A
fluorescent microscope (Nikon) was used to observe and photograph
cells with fluorescent signals.
Assay of caspase-3 activity
Caspase-3 activity was determined using a
fluorometric substrate assay kit as described previously (28). Briefly, after drug administration,
human glioma U87-MG cells were lysed using a buffer containing 1%
Nonidet P-40, 200 mM NaCl, 20 mM Tris/HCl (pH 7.4), 10 mg/ml
leupeptin, 0.27 U/ml aprotinin, and 100 mm PMSF. Cell extracts (25
mg of total protein) were incubated with 50 mM of a specific
fluorogenic peptide substrate in 200 ml of a cell-free system
buffer composed of 10 mM HEPES (pH 7.4), 220 mM mannitol, 68 mM
sucrose, 2 mM NaCl, 2.5 mM KH2PO4, 0.5 mM
EGTA, 2 mM MgCl2, 5 mM pyruvate, 0.1 mM PMSF, and 1 mM
dithiothreitol. The peptide substrate for the caspase-3 enzyme
assay was DEVD. The peptide was conjugated to
7-amino-4-trifluoromethyl coumarin for fluorescence detection.
Intensities of the fluorescent products were measured with a
spectrometer.
Quantification of apoptotic cells
Apoptosis of human glioma U87-MG and DBTRG-05MG
cells was determined using propidium iodide (PI) to detect DNA
injury in nuclei according to a previously described method
(29). Briefly, after drug
administration, human glioma U87-MG cells were harvested and fixed
in cold 80% ethanol. Following centrifugation and washing, fixed
cells were stained with PI and analyzed using a FACScan flow
cytometer (Becton-Dickinson).
Immunoblot analyses
Protein analyses were performed according to a
previously described assay (30).
Briefly, after drug treatment, cell lysates were prepared in iced
radioimmunoprecipitate test buffer (25 mM Tris HCl, 0.1% sodium
dodecylsulfate (SDS). 1% Triton X-100, 1% sodium deoxycholate, 0.15
M NaCl, and 1 mM EDTA). Protein concentrations were measured using
a bicinchonic acid protein assay kit (Pierce, Rockford, IL, USA).
The protein (50 µg/well) was subjected to SDS polyacrylamide
gel electrophoresis (PAGE), and transferred to nitrocellulose
membranes. After blocking, LC3-I, LC3-II, HIF-1α, and p53 were
immunodetected using rabbit polyclonal antibodies or mouse
monoclonal antibodies against related proteins (Cell Signaling,
Danvers, MA, USA). Cellular β-actin protein was immunodetected
using a mouse monoclonal antibody against mouse β-actin (Sigma) as
the internal control. These protein bands were quantified using a
digital imaging system (UVtec, Cambridge, UK).
p53 knockdown
Translation of p53 messenger (m)RNA in human glioma
U87-MG and DBTRG-05MG cells was knocked-down using an RNA
interference (RNAi) method following a small interfering (si)RNA
transfection protocol provided by Santa Cruz Biotechnology (Santa
Cruz, CA, USA) as described previously (31). p53 siRNA was purchased from Santa
Cruz Biotechnology, and is a pool of three target-specific 20-25-nt
siRNAs designed to suppress p53 expression. Scrambled siRNA,
purchased from Santa Cruz Biotechnology, contained non-targeting
20-25-nt siRNA and was applied to control cells as a negative
standard. Briefly, after culturing U87-MG cells in antibiotic-free
MEM at 37°C in a humidified atmosphere of 5% CO2 for 24
h, the siRNA duplex solution, which was diluted in an siRNA
transfection medium (Santa Cruz Biotechnology), was added to human
glioma cells. After transfection with scrambled or p53 siRNA for 24
h, the medium was replaced with normal MEM, and the cells were
treated with CoCl2.
Statistical analysis
Statistical differences between the control and
drug-treated groups were considered significant when the p-value of
Duncan's multiple-range test was <0.05. Statistical analysis
between drug-treated groups was carried out using a two-way
analysis of variance (ANOVA).
Results
Cell morphology, cell viability, and cellular HIF-1α
levels were analyzed to determine the effects of CoCl2
on inducing of hypoxia and cytotoxicity in human glioma U87-MG
cells (Fig. 1). Exposure of human
U87-MG cells to 25 µM CoCl2 for 24 h decreased
cell numbers (Fig. 1A). After
exposure to 50, 100, 150 and 200 µM CoCl2 for 24
h, cell numbers time-dependently decreased. Treatment of human
U87-MG cells with 25 µM CoCl2 for 24 h caused a
31% reduction in cell viability (Fig.
1B). When the administered concentrations of CoCl2
reached 50, 100, 150 and 200 µM, cell viabilities were
diminished by 36, 45, 52 and 63%, respectively. Exposure of U87-MG
cells to 100 µM CoCl2 for 1 h did not influence
cell viability (Fig. 1C). In
contrast, viabilities of U87-MG cells were lowered by 19, 30 and
47% after CoCl2 treatment for 6, 12 and 24 h,
respectively. Low levels of HIF-1α were detected in untreated
U87-MG cells (Fig. 1D, top panel,
lane 1). However, exposure to 100 µM CoCl2 for 24
h obviously enhanced amounts of cellular HIF-1α (lane 2). β-actin
was analyzed as the internal control (bottom panel). These protein
bands were quantified and statistically analyzed (Fig. 1E). Treatment with 100 µM
CoCl2 for 24 h significantly increased levels of HIF-1α
in human U87-MG cells by 2.7-fold.
To decide the effects of CoCl2 treatment
on cell proliferation, a BrdU incorporation assay was conducted
(Fig. 2). Exposure of human glioma
U87-MG cells to 25 µM CoCl2 for 24 h caused a 15%
diminution in cell proliferation (Fig.
2A). In comparison, CoCl2 at 50, 100, 150 and 200
µM suppressed proliferation of human U87-MG cells by 17, 20,
28 and 42%, respectively. Exposure of human U87-MG cells to 100
µM CoCl2 for 1, 6, and 12 h did not affect cell
proliferation (Fig. 2B).
Nonetheless, treatment with CoCl2 for 24 h caused a
significant 49% decline in the proliferation of human U87-MG
cells.
Autophagic cells and cellular LC3-II levels were
examined in order to determine the effects of CoCl2
treatment on autophagy of human U87-MG cells (Fig. 3). Treatment with 25 µM
CoCl2 for 24 h induced 16% of human U87-MG cells to
undergo autophagy (Fig. 3A).
Fractions of autophagic cells in human U87-MG cells were
significantly augmented to 19, 28, 41 and 52% following respective
exposure to 50, 100, 150 and 200 µM CoCl2 for 24
h. Treatment of human U87-MG cells with 100 µM
CoCl2 for 1 and 6 h did not change cell autophagy
(Fig. 3B). In contrast, after
exposure for 12 and 24 h, CoCl2 administration induced
cell autophagy by 17 and 25%, respectively. Staining with AO
revealed that exposure of human U87-MG cells to CoCl2
for 6 h did not change the proportion of cells with acidic
vesicular organelles (Fig. 3C). In
comparison, after exposure to CoCl2 for 12 or 24 h,
proportions of human U87-MG cells stained with AO were augmented.
Exposure of human U87-MG cells to 100 µM CoCl2
for 1 or 6 h did not affect levels of the LC3-II protein (Fig. 3D, top panel, lanes 2 and 3).
However, after treatment with CoCl2 for 12 or 24 h,
amounts of LC3-II in U87-MG cells were obviously augmented (lanes 4
and 5). β-actin was analyzed as the internal control (bottom
panel). These protein bands were quantified and statistically
analyzed (Fig. 3E). Exposure of
human U87-MG cells to 100 µM CoCl2 for 12 or 24 h
led to 72 and 98% increases in amounts of the LC3-II protein.
To confirm CoCl2-induced autophagy, 3-MA,
an inhibitor of autophagy, and Rapa, an inducer of autophagy, were
applied to human U87-MG cells (Fig.
4). Treatment of human U87-MG cells with 100 µM
CoCl2 for 24 h induced 28% of glioma cells to undergo
autophagy (Fig. 4A). Pretreatment
with 3-MA alone did not influence cell autophagy but significantly
decreased hypoxia-induced cell autophagy by 68%. In contrast,
pretreatment with Rapa alone triggered 16% of human U87-MG cells to
undergo autophagy (Fig. 4B).
Interestingly, pretreatment with Rapa synergistically induced
hypoxia-induced cell autophagy by 40%.
Caspase-3 activity and apoptotic cells were analyzed
to determine the effects of CoCl2-induced autophagy on
cell apoptosis (Fig. 5). Treatment
of human U87-MG cells with 100 µM CoCl2 for 1 and
6 h did not influence caspase-3 activity (Fig. 5A). When the treatment time
intervals reached 12 and 24 h, caspase-3 activities were
significantly augmented in human U87-MG cells by 2.5- and 3.8-fold,
respectively. Administration of 100 µM CoCl2 to
human U87-MG cells for 1 and 6 h did not affect cell apoptosis
(Fig. 5B). Nevertheless, after
exposure to CoCl2 for 12 or 24 h, the fractions of human
U87-MG cells that underwent apoptosis were significantly increased
to 24 and 37%. Pretreatment of human U87-MG cells with 3-MA alone
did not change caspase-3 activity (Fig. 5C) or cell apoptosis (Fig. 5D). In contrast, pretreatment with
3-MA caused significant 61 and 58% alterations in hypoxia-induced
caspase-3 activation and cell apoptosis, respectively (Fig. 5C and D).
Chloroquine, another inhibitor of cell autophagy,
was further applied to human U87-MG cells to confirm
CoCl2-induced cell insults (Table I). Pretreatment of human U87-MG
cells with chloroquine did not affect cell viability, cell
autophagy, caspase-3 activity, or cell apoptosis. In contrast,
chloroquine pretreatment caused significant 51, 52, 61 and 51%
decreases in CoCl2-induced alterations in cell
viability, autophagic cells, caspase-3 activation, and apoptotic
cells, respectively (Table I).
 | Table IEffects of chloroquine (CLQ) on
cobalt chloride (CoCl2)-induced insults to human glioma
cells. |
Table I
Effects of chloroquine (CLQ) on
cobalt chloride (CoCl2)-induced insults to human glioma
cells.
| Cell viability
(OD550) | Cell autophagy
(%) | Caspase-3 activity
(FI) | Apoptotic cells
(%) |
|---|
| Control | 0.412±0.089 | 6±1 | 11±3 | 5±1 |
|
CoCl2 | 0.133±0.048a | 29±7a | 41±8a | 39±7a |
| CLQ | 0.402±0.102 | 5±1 | 9±2 | 6±2 |
| CLQ+hypoxia | 0.268±0.069a,b | 14±3a,b | 16±4a,b | 19±4a,b |
RNAi was applied to human U87-MG cells to determine
the roles of p53 in CoCl2-induced autophagic death
(Fig. 6). Application of p53 siRNA
to human U87-MG cells for 12 h did not affect levels of p53
(Fig. 6A, top panel, lane 2).
However, after exposure to p53 siRNA for 24 or 48 h, amounts of p53
in human U87-MG cells were significantly decreased (lanes 3 and 4).
β-actin was analyzed as the internal control (top panel). These
protein bands were quantified and statistically analyzed (bottom
panel). Application of p53 siRNA to human U87-MG cells for 24 or 48
h caused significant 69 and 77% reductions in levels of p53,
respectively. Exposure to hypoxia induced autophagy of 28% of human
U87-MG cells (Fig. 6B).
Pretreatment with 3-MA and p53 siRNA did not affect cell autophagy
but significantly alleviated hypoxia-induced autophagy of human
U87-MG cells. Co-treatment with 3-MA and p53 siRNA synergistically
lowered hypoxia-induced cell autophagy (Fig. 6B). At the same time, pretreatment
of human U87-MG cells with a combination of 3-MA and p53 siRNA
caused noteworthy alleviations of hypoxia-induced cell apoptosis
and cell death (Fig. 6C and
D).
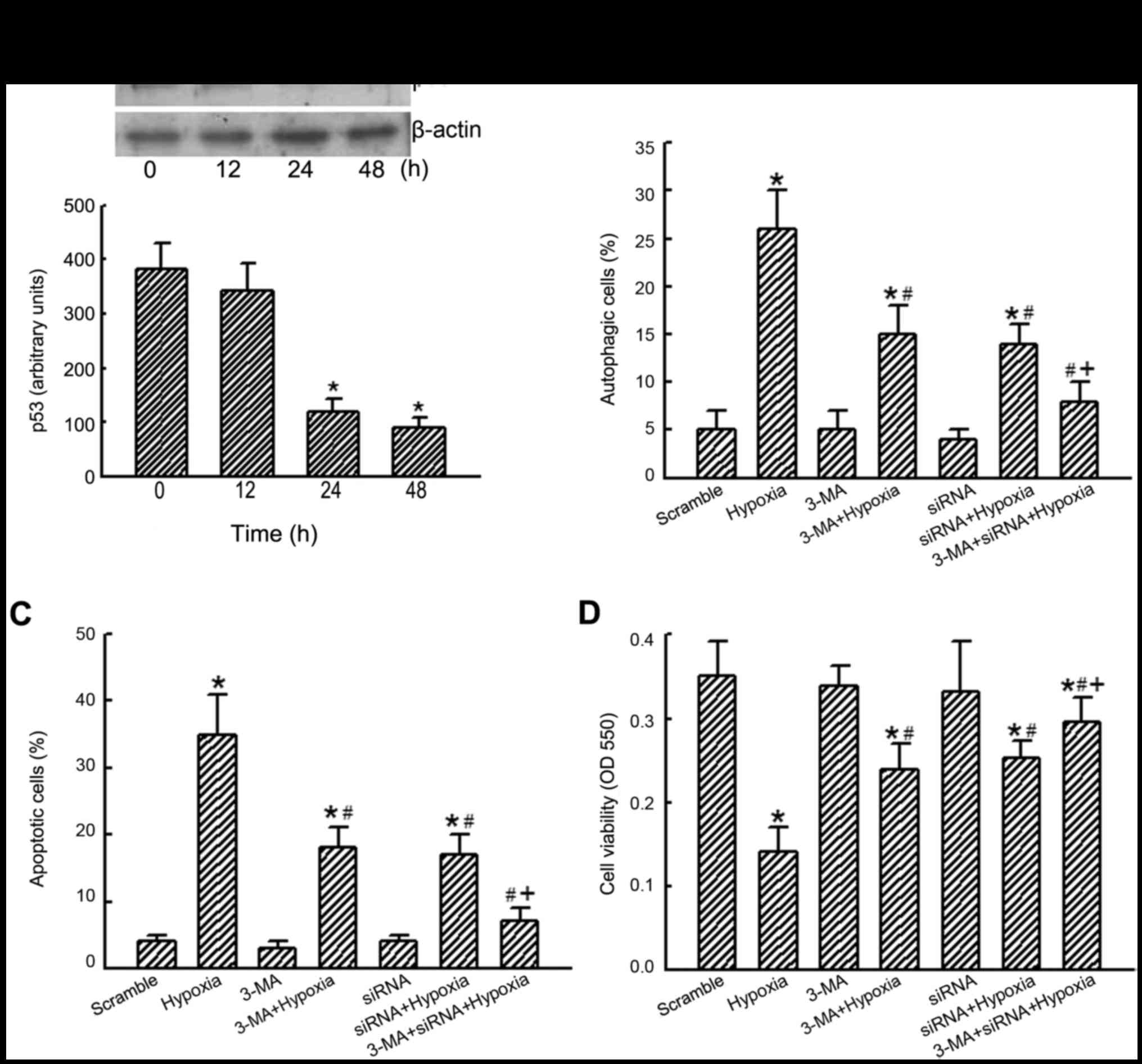 | Figure 6Roles of p53 in cobalt chloride
(CoCl2)-induced cell autophagy, apoptosis, and
viability. Human glioma U87-MG cells were exposed to p53 siRNA
(siRNA) for 6, 12 and 24 h. Scrambled siRNA was applied into
control cells as the negative control (scramble). Levels of p53
were immunodetected, and β-actin was analyzed as the internal
control (A, top panel). These protein bands were quantified and
statistically analyzed (bottom panel). U87-MG cells were pretreated
with p53 siRNA (siRNA) and 3-methyladenine (3-MA) and then exposed
to 100 µM CoCl2 for another 24 h (hypoxia). Cell
autophagy (B) and cell apoptosis (C) were determined using flow
cytometry. Cell viability was assayed using a colorimetric method
(D). Each value represents the mean ± SEM for n=6.
*,#,+p<0.05, values significantly differed from the
control, hypoxia-, and p53 siRNA-treated groups, respectively. |
To confirm the roles of p53 in
CoCl2-induced autophagic death of glioma cells, human
glioma DBTRG-05MG cells were further used as our experimental model
(Fig. 7). Exposure of DBTRG-05MG
cells to hypoxia led to a 3.6-fold induction of cell autophagy
(Fig. 7A). Pretreatment with 3-MA
did not affect autophagy of DBTRG-05MG cells but attenuated
hypoxia-induced cell autophagy by 56%. Administration of hypoxia
induced apoptosis of DBTRG-05MG cells by 20% (Fig. 7B). In contrast, pretreatment of
DBTRG-05MG cells with 3-MA did not affect cell apoptosis but caused
a significant 60% reduction in hypoxia-induced apoptosis.
Application of p53 siRNA into DBTRG-05MG cells for 24 h did not
influence autophagy and apoptosis (Fig. 7C and D). However, when knocking
down p53 translation in DBTRG-05MG cells, the hypoxia-induced
autophagy and apoptosis were simultaneously reduced by 100 and 75%,
respectively (Fig. 7C and D).
Discussion
The present study showed that CoCl2
treatment can induce autophagy and subsequent death of human
malignant glioma cells. Malignant gliomas are the most prevalent
and aggressive brain tumors (2).
In the clinic, patients suffering from malignant gliomas usually
have poor prognoses, and their median overall survival rates are
less than 1 year. Induction of hypoxia has recently been explored
as a novel strategy for treating aggressive tumors such as cancer
stem cells and chemotherapy-resistant tumor cells (19,22).
This study showed that administration of CoCl2 induced
hypoxic stress in human glioma cells. Separately, exposure of human
U87-MG cells to hypoxia triggered cell autophagy, apoptosis, and
death. Interestingly, suppressing autophagy using the inhibitor,
3-MA, concurrently lessened hypoxia-induced apoptosis and death in
human glioma U87-MG and DBTRG-05MG cells. Hence, CoCl2
treatment can induce autophagic death in human glioma cells. Brain
hypoxia is thought to have bidirectional roles in controlling cell
survival and death in human malignant glioma cells (13,14).
Hu et al reported that hypoxia-induced autophagy promotes
glioblastoma cell survival (23).
Nevertheless, our study provides in vitro data which
demonstrate that delayed hypoxia can induce autophagy and
autophagic death in human glioma cells. TMZ is an oral
chemotherapeutic drug that is used to treat brain tumors (32). A previous study further showed that
bortezomib induces cell death in GBM stem cells and TMZ-resistant
glioma cells via a hypoxia-dependent mechanism (22). As a result, discovering a novel
scheme to induce hypoxia may be beneficial for therapy of malignant
gliomas and TMZ-resistant GBM through an autophagic apoptosis
pathway.
Exposure to CoCl2 induces hypoxic insults
to human glioma cells. In a hypoxic microenvironment, HIF-1α is
highly produced and functions as a transcription factor to regulate
a large array of gene expressions in response to hypoxic conditions
(9). Characteristically, HIF-1α is
recognized as a typical marker indicating that cells are
experiencing hypoxia. Our present data revealed that
CoCl2 treatment augmented amounts of HIF-1α in human
U87-MG cells. As to the mechanism, CoCl2 can diminish
HIF-1α degradation by suppressing prolyl-4-hydroxylase activity
(33). Consequently, treatment of
human glioma cells with CoCl2 can stimulate cellular
hypoxic stress by means of enriching HIF-1α expression. At the same
time, exposure to hypoxia decreased the viability of human U87-MG
cells. Brain hypoxia often occurs in brain neoplasms and traumatic
brain injury and is recognized as a major cause of secondary injury
to neurologically critically ill patients (8). With glioblastomas, hypoxia was
reported to promote tumor survival and angiogenesis (23). In neuroblastoma cells, our previous
study demonstrated that hypoxia can induce cell death via an
apoptotic mechanism due to targeting antiapoptotic
bcl-xL gene expression (12). The present study additionally
proved the effects of hypoxia on triggering insults to human glioma
cells.
CoCl2 treatment lessens the proliferation
of human glioma U87-MG cells. Hypoxia broadly exists in diverse
types of tumor microenvironments (7,9).
During tumorigenesis, low levels of oxygen may develop to promote
tumor cell proliferation, angiogenesis, and metastasis through an
HIF-1α-involved multifaceted network (34). Our present results showed that
under extended hypoxic stress, incorporation of BrdU in human
glioma U87-MG cells was repressed in concentration- and
time-dependently. BrdU incorporation in cells can be measured to
explain cell cycle processing in order to monitor the status of
cell proliferation (35). Thus,
this study demonstrated the suppressive effects of extended hypoxia
on the proliferation of human glioma cells. In the clinic, rapid
tumor growth because of speedy cell proliferation is one of major
features and reasons illuminating the casual recurrence and poor
prognoses of human malignant gliomas (4). Li et al reported that hypoxic
conditions can activate a self-protective mechanism against glioma
proliferation (36). As a result,
CoCl2 treatment is able to prevent tumor cell
proliferation and then suppress tumor growth and angiogenesis in
human malignant gliomas.
CoCl2 treatment can induce autophagic
insults to human glioma cells. Autophagy is a self-degradative
process inside cells (15,16). Autophagosomes, acidic vesicular
organelles, and elevation in LC3-II levels are considered as
consistent markers of cell autophagy (37,38).
Our preliminary study has shown that treatment of human U87-MG
cells with CoCl2 could induce autophagosomes, acidic
vesicular organelles, and LC3-II amounts. Herein, we demonstrated
the concentration-and time-dependent effects of CoCl2
treatment on raising the proportions of human glioma cells with
acidic vesicular organelles and the levels of LC3-II protein.
Moreover, 3-MA and chloroquine are typically used as inhibitors of
cell autophagy because it can block the formation of autophagosomes
(39). Our current data showed the
repressive effects of pretreatment with 3-MA and chloroquine on
CoCl2-induced autophagy of human glioma cells.
Separately, pretreatment with Rapa, an inducer of autophagy,
synergistically amplified hypoxia-induced cell autophagy.
Therefore, we provide several lines of evidence to show that
delayed hypoxia can induce autophagy in human glioma cells.
Nevertheless, autophagic cells can reverse course and survive or
proceed to death (23,24). This study showed that hypoxia
decreased the viability of human U87-MG cells. Pretreatment with
3-MA and chloroquine caused concurrent defenses against
CoCl2-induced cell death. Consequently, CoCl2
treatment can induce autophagy and autophagic death of human glioma
cells. Bevacizumab is reported to kill high-grade gliomas via a
hypoxia-induced autophagic pathway (19). Thus, induction of prolonged hypoxia
has the potential to be a new therapeutic strategy for human
gliomas.
Autophagy-induced apoptosis is involved in
CoCl2 treatment-induced damage to human glioma cells.
Exposure of human glioma U87-MG cells to CoCl2 induced
caspase-3 activation and sequential cell cycle arrest at the
sub-G1 phase. Caspase-3 activation and cell cycle arrest
at the sub-G1 phase are two typical characteristics
indicating that cells are undergoing apoptosis (40,41).
Hence, CoCl2 treatment can induce apoptosis of human
glioma U87-MG cells. Autophagy is reported to closely cross-talk
with apoptosis (42). In general,
autophagy can block apoptosis, and certain apoptosis-associated
proteins disrupt the autophagic process. In some cases, autophagy
may induce cell apoptosis or necrosis, known as autophagic cell
death (42). This study
demonstrated that pretreatment with 3-MA alone did not influence
apoptosis of human U87-MG cells but caused significant alleviation
of CoCl2-induced apoptotic injury. Thus,
CoCl2 treatment can induce autophagic apoptosis in human
glioma cells. The key principles of cancer therapy focus on
inducing cell death and inhibiting cell proliferation (42). Our previous studies showed that
honokiol, a polyphenol, induced autophagic apoptosis in
neuroblastomas and gliomas (21,25).
The present study additionally supports hypoxia being applied to
kill glioma cells via inducing cell autophagy and consequent
autophagic apoptosis.
p53 participates in CoCl2-induced
autophagic apoptosis in human glioma cells. p53, a tumor suppressor
protein, can transcriptionally regulate expression of certain genes
and then control cell survival or death (26). Also, p53 is reported to control
cell autophagy by adjusting downstream damage-regulated autophagy
modulator (DRAM) expression, a lysosomal protein (10,40).
This study showed that knocking down p53 expression using RNAi
instantaneously attenuated CoCl2-induced autophagy and
subsequent apoptosis. Our recent study proved that activation of
p53 sequentially results in suppression of mammalian target of
rapamycin (mTOR) activity and then induction of autophagy of glioma
cells (10). Hence, p53
participates in CoCl2-induced cell autophagy in human
glioma cells. Feng et al reported that p53 can upregulate
unc-51-like kinase 1/2, which are necessary for sustained autophagy
in response to camptothecin-induced DNA damage (44). Furthermore, a previous study
demonstrated the contribution of p53 to triggering cell autophagy
and death by inducing DRAM gene expression (45). The present study indicated that
co-treatment with 3-MA and p53 siRNA synergistically protected
human glioma cells from CoCl2 treatment-induced
caspase-3 activation, cell apoptosis, and cell death. As a result,
p53 plays a crucial role in mediating hypoxia-induced cell
autophagy and subsequent cell apoptosis in human gliomas.
In conclusion, this study verified the effects of
CoCl2 in inducing hypoxic stress in human glioma U87-MG
cells. In parallel, exposure to CoCl2 led to significant
reductions in the viability and proliferation of human glioma
cells. As to the mechanism, treatment of human U87-MG cells with
CoCl2 meaningfully augmented levels of acidic vesicular
autophagosomes and the cellular LC3-II protein. The gain-and
loss-of-function strategies further showed induction of autophagy
in human glioma cells by CoCl2. Pretreatment with 3-MA
and chloroquine decreased the proportion of human U87-MG cells
undergoing autophagy and concurrently attenuated
CoCl2-triggered caspase-3 activation and cell apoptosis.
Consecutively, knocking down p53 synthesis by RNAi could weaken
hypoxia-induced cell autophagy, apoptosis, and death. In contrast,
co-treatment with 3-MA and p53 siRNA synergistically protected
human glioma cells against CoCl2- induced autophagic
death. The CoCl2-induced autophagic apoptosis were
supplementarily confirmed in human glioma DBTRG-05MG cells. Taken
together, CoCl2 treatment can induce autophagic
apoptosis of human glioma cells via a p53-dependent pathway.
Induction of delayed hypoxia may have the potential to be
clinically applied for treating human malignant gliomas. An in
vivo intracranial model is being investigated to confirm our
in vitro findings on the suppressive effects of hypoxia on
glioma growth through an autophagic apoptosis pathway.
Acknowledgments
This study was supported by grants from the Chi-Mei
Medical Center (102CM-TMU-14-1), Wan-Fang Hospital (105-swf-04),
and the Health and Welfare Surcharge of Tobacco Products
(MOHW105-TDU-B-212-134001), Taiwan. The authors express their
gratitude to Ms. Yi-Ling Lin for her technical support and data
collection during the experiments.
References
|
1
|
Wacker MR, Hoshino T, Ahn DK, Davis RL and
Prados MD: The prognostic implications of histologic classification
and bromodeoxyuridine labeling index of mixed gliomas. J
Neurooncol. 19:113–122. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jordan JT, Gerstner ER, Batchelor TT,
Cahill DP and Plotkin SR: Glioblastoma care in the elderly. Cancer.
122:189–197. 2016. View Article : Google Scholar
|
|
3
|
Kalkan R: Hypoxia is the driving force
behind GBM and could be a new tool in GBM treatment. Crit Rev
Eukaryot Gene Expr. 25:363–369. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Preusser M, Lim M, Hafler DA, Reardon DA
and Sampson JH: Prospects of immune checkpoint modulators in the
treatment of glioblastoma. Nat Rev Neurol. 11:504–514. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Curtin JF, Liu N, Candolfi M, Xiong W,
Assi H, Yagiz K, Edwards MR, Michelsen KS, Kroeger KM, Liu C, et
al: HMGB1 mediates endogenous TLR2 activation and brain tumor
regression. PLoS Med. 6:e102009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gerstner ER, Duda DG, di Tomaso E, Ryg PA,
Loeffler JS, Sorensen AG, Ivy P, Jain RK and Batchelor TT: VEGF
inhibitors in the treatment of cerebral edema in patients with
brain cancer. Nat Rev Clin Oncol. 6:229–236. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Freese KE, Kokai L, Edwards RP, Philips
BJ, Sheikh MA, Kelley J, Comerci J, Marra KG, Rubin JP and Linkov
F: Adipose-derived stems cells and their role in human cancer
development, growth, progression, and metastasis: A systematic
review. Cancer Res. 75:1161–1168. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lingsma HF, Roozenbeek B, Steyerberg EW,
Murray GD and Maas AI: Early prognosis in traumatic brain injury:
From prophecies to predictions. Lancet Neurol. 9:543–554. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Semenza GL: Surviving ischemia: Adaptive
responses mediated by hypoxia-inducible factor 1. J Clin Invest.
106:809–812. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin CJ, Lin YL, Luh F, Yen Y and Chen RM:
Preclinical effects of CRLX101, an investigational
camptothecin-containing nanoparticle drug conjugate, on treating
glioblastoma multiforme via apoptosis and antiangiogenesis.
Oncotarget. 7:42408–42421. 2016.PubMed/NCBI
|
|
11
|
Chan DA, Krieg AJ, Turcotte S and Giaccia
AJ: HIF gene expression in cancer therapy. Methods Enzymol.
435:323–345. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chio CC, Lin JW, Cheng HA, Chiu WT, Wang
YH, Wang JJ, Hsing CH and Chen RM: MicroRNA-210 targets
antiapoptotic Bcl-2 expression and mediates hypoxia-induced
apoptosis of neuroblastoma cells. Arch Toxicol. 87:459–468. 2013.
View Article : Google Scholar
|
|
13
|
Joseph JV, Conroy S, Pavlov K, Sontakke P,
Tomar T, Eggens-Meijer E, Balasubramaniyan V, Wagemakers M, den
Dunnen WF and Kruyt FA: Hypoxia enhances migration and invasion in
glioblastoma by promoting a mesenchymal shift mediated by the
HIF1α-ZEB1 axis. Cancer Lett. 359:107–116. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun W, Jelkmann W and Depping R:
Prolyl-4-hydroxylase 2 enhances hypoxia-induced glioblastoma cell
death by regulating the gene expression of hypoxia-inducible
factor-α. Cell Death Dis. 5:e13222014. View Article : Google Scholar
|
|
15
|
Codogno P, Mehrpour M and Proikas-Cezanne
T: Canonical and non-canonical autophagy: Variations on a common
theme of self-eating? Nat Rev Mol Cell Biol. 13:7–12.
2011.PubMed/NCBI
|
|
16
|
Kaur J and Debnath J: Autophagy at the
crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol.
16:461–472. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yoshimori T: Autophagy: A regulated bulk
degradation process inside cells. Biochem Biophys Res Commun.
313:453–458. 2004. View Article : Google Scholar
|
|
18
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nanegrungsunk D, Onchan W, Chattipakorn N
and Chattipakorn SC: Current evidence of temozolomide and
bevacizumab in treatment of gliomas. Neurol Res. 37:167–183. 2015.
View Article : Google Scholar
|
|
20
|
Dolma S, Selvadurai HJ, Lan X, Lee L,
Kushida M, Voisin V, Whetstone H, So M, Aviv T, Park N, et al:
Inhibition of dopamine receptor D4 impedes autophagic flux,
proliferation, and survival of glioblastoma stem cells. Cancer
Cell. 29:859–873. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin CJ, Chen TL, Tseng YY, Wu GJ, Hsieh
MH, Lin YW and Chen RM: Honokiol induces autophagic cell death in
malignant glioma through reactive oxygen species-mediated
regulation of the p53/PI3K/Akt/mTOR signaling pathway. Toxicol Appl
Pharmacol. 304:59–69. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bota DA, Alexandru D, Keir ST, Bigner D,
Vredenburgh J and Friedman HS: Proteasome inhibition with
bortezomib induces cell death in GBM stem-like cells and
temozolomide-resistant glioma cell lines, but stimulates GBM
stem-like cells' VEGF production and angiogenesis. J Neurosurg.
119:1415–1423. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu YL, DeLay M, Jahangiri A, Molinaro AM,
Rose SD, Carbonell WS and Aghi MK: Hypoxia-induced autophagy
promotes tumor cell survival and adaptation to antiangiogenic
treatment in glioblastoma. Cancer Res. 72:1773–1783. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xue H, Yuan G, Guo X, Liu Q, Zhang J, Gao
X, Guo X, Xu S, Li T, Shao Q, et al: A novel tumor-promoting
mechanism of IL6 and the therapeutic efficacy of tocilizumab:
Hypoxia-induced IL6 is a potent autophagy initiator in glioblastoma
via the p-STAT3-MIR155-3p-CREBRF pathway. Autophagy. 12:1129–1152.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yeh PS, Wang W, Chang YA, Lin CJ, Wang JJ
and Chen RM: Honokiol induces autophagy of neuroblastoma cells
through activating the PI3K/Akt/mTOR and endoplasmic reticular
stress/ERK1/2 signaling pathways and suppressing cell migration.
Cancer Lett. 370:66–77. 2016. View Article : Google Scholar
|
|
26
|
Vousden KH and Lu X: Live or let die: The
cell's response to p53. Nat Rev Cancer. 2:594–604. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ho MH, Liao MH, Lin YL, Lai CH, Lin PI and
Chen RM: Improving effects of chitosan nanofiber scaffolds on
osteoblast proliferation and maturation. Int J Nanomed.
9:4293–4304. 2014.
|
|
28
|
Chang CY, Lui TN, Lin JW, Lin YL, Hsing
CH, Wang JJ and Chen RM: Roles of microRNA-1 in hypoxia-induced
apoptotic insults to neuronal cells. Arch Toxicol. 90:191–202.
2016. View Article : Google Scholar
|
|
29
|
Lee YE, Hong CY, Lin YL and Chen RM:
MicroRNA-1 participates in nitric oxide-induced apoptotic insults
to MC3T3-E1 cells by targeting heat-shock protein-70. Int J Biol
Sci. 11:246–255. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu GJ, Wang W, Lin YL, Liu SH and Chen RM:
Oxidative stress-induced apoptotic insults to rat osteoblasts are
attenuated by nitric oxide pretreatment via GATA-5-involved
regulation of Bcl-X L gene expression and protein translocation.
Arch Toxicol. 90:905–916. 2016. View Article : Google Scholar
|
|
31
|
Wu TT, Tai YT, Cherng YG, Chen TG, Lin CJ,
Chen TL, Chang HC and Chen RM: GATA-2 transduces LPS-induced il-1β
gene expression in macrophages via a toll-like receptor
4/MD88/MAPK-dependent mechanism. PLoS One. 8:e724042013. View Article : Google Scholar
|
|
32
|
Messaoudi K, Clavreul A and Lagarce F:
Toward an effective strategy in glioblastoma treatment. Part II:
RNA interference as a promising way to sensitize glioblastomas to
temozolomide. Drug Discov Today. 20:772–779. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kaelin WG Jr and Ratcliffe PJ: Oxygen
sensing by metazoans: The central role of the HIF hydroxylase
pathway. Mol Cell. 30:393–402. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mamlouk S and Wielockx B:
Hypoxia-inducible factors as key regulators of tumor inflammation.
Int J Cancer. 132:2721–2729. 2013. View Article : Google Scholar
|
|
35
|
Kajita K, Mori I, Kitada Y, Taguchi K,
Kajita T, Hanamoto T, Ikeda T, Fujioka K, Yamauchi M, Okada H, et
al: Small proliferative adipocytes: Identification of proliferative
cells expressing adipocyte markers. Endocr J. 60:931–939. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li W, Winters A, Poteet E, Ryou MG, Lin S,
Hao S, Wu Z, Yuan F, Hatanpaa KJ, Simpkins JW, et al: Involvement
of estrogen receptor β5 in the progression of glioma. Brain Res.
1503:97–107. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu YT, Tan HL, Shui G, Bauvy C, Huang Q,
Wenk MR, Ong CN, Codogno P and Shen HM: Dual role of
3-methyladenine in modulation of autophagy via different temporal
patterns of inhibition on class I and III phosphoinositide
3-kinase. J Biol Chem. 285:10850–10861. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen N and Karantza-Wadsworth V: Role and
regulation of autophagy in cancer. Biochim Biophys Acta.
1793:1516–1523. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Galluzzi L, Kepp O and Kroemer G:
Caspase-3 and prostaglandins signal for tumor regrowth in cancer
therapy. Oncogene. 31:2805–2808. 2012. View Article : Google Scholar
|
|
42
|
Mariño G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sui X, Chen R, Wang Z, Huang Z, Kong N,
Zhang M, Han W, Lou F, Yang J, Zhang Q, et al: Autophagy and
chemotherapy resistance: A promising therapeutic target for cancer
treatment. Cell Death Dis. 4:e8382013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Feng Z, Zhang H, Levine AJ and Jin S: The
coordinate regulation of the p53 and mTOR pathways in cells. Proc
Natl Acad Sci USA. 102:8204–8209. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Crighton D, Wilkinson S, O'Prey J, Syed N,
Smith P, Harrison PR, Gasco M, Garrone O, Crook T and Ryan KM:
DRAM, a p53-induced modulator of autophagy, is critical for
apoptosis. Cell. 126:121–134. 2006. View Article : Google Scholar : PubMed/NCBI
|