Introduction
Associated with prior exposure to asbestos (1), malignant pleural mesothelioma (MPM)
is an aggressive inflammatory cancer arising from mesothelial cells
lining the pleural, peritoneal and pericardial cavities. It has a
long latency period, and because of this, the vast majority of
patients present at an advanced stage resulting in a poor prognosis
with a median survival time of 6–12 months. Current estimates
suggest that 43,000 people die from MPM each year with a mortality
rate in the order of 6.2 per million population (2,3).
Many countries have banned the use of asbestos, and data are
emerging to suggest that the incidence of mesothelioma in these
countries may be falling (4–6),
although there is some dispute regarding these projections
(7). Furthermore, the long-term
risk to younger people exposed to asbestos still present in many
buildings is not currently known but could be substantial (8). Nevertheless, it is generally accepted
that MPM mortality rates will continue to increase by 5–10% per
year in most industrialized countries for the next 2–3 decades,
despite asbestos ablation efforts (7). In addition, asbestos consumption
continues to increase in emerging countries such as such as Brazil,
and a new wave of MPM is predicted in such countries (9).
The current standard of care is a combination of
pemetrexed (or raltitrexed) and cisplatin chemotherapy (10). Unfortunately, this treatment only
has a patient response rate of between 23–40% and is non-curative
(11,12). Consequently, there is an urgent
need to identify novel therapeutic avenues in this disease to
improve patient outcomes.
Dysregulation of epigenetic transcriptional control,
particularly in the areas of promoter DNA methylation and histone
post-translational modifications, is a well-established feature of
human malignancies, including mesothelioma (13–15).
The pharmaceutical sector has devoted significant efforts to
develop agents capable of targeting the epigenetic machinery. The
potential to target the epigenetic machinery in MPM initially came
from data in a phase I trial of vorinostat a histone deacetylase
(HDAC) inhibitor, in which 4 of 13 patients with MPM (30%) who
received vorinostat had stable disease lasting more than 4 months
with two unconfirmed partial responses (16). Unfortunately, when vorinostat was
given as a second-line or third-line therapy in the phase III trial
(VANTAGE 014) of 660 pretreated advanced MPM patients it did not
improve overall survival, and the recommendation was therefore that
it was unsuitable as a therapy in MPM patients (17).
Despite this, evidence continues to emerge
suggesting that targeting the epigenetic machinery may be a viable
therapeutic option in MPM. For example, we recently demonstrated
that KAT5 (a lysine acetyltransferase) was overexpressed in MPM and
a suitable potential candidate for therapeutic intervention
(18). Furthermore, the potential
importance of targeting the epigenetic machinery in MPM has been
highlighted by data emerging regarding MPM patients who have
mutated BRCA1-associated protein 1 (BAP1). BAP1 plays critical
roles in chromatin remodeling, and loss of BAP1 in MPM cells has
now been associated with altered sensitivity to HDAC inhibitors
through regulation of a specific HDAC, HDAC2 (19).
In a recent analysis of MPM, a subset of genes was
found to be silenced by histone H3 lysine 27 trimethylation
(H3K27me3), a mark most often found at or near the promoters of
silent genes (20,21). Polycomb repressive complex 2 (PRC2)
catalyses trimethylation of Histone H3 at lysine 27 (H3K27me2/3)
(22), and contains the lysine
methyltransferase EZH2. Targeting this complex with the
methyltransferase inhibitor DZNep has been shown to be a potential
therapeutic option in MPM (23),
and in particular sensitivity to DZNep has been linked to a subset
of MPM patients that contain (BAP1) mutation (24), and a phase II clinical trial of the
EZH2 inhibitor tazmetostat (EPZ-6438) in mesothelioma is currently
in progress (clinicaltrials.gov NCT02860286).
The proteins which demethylate H3K27me3 have been
collectively identified as the Kdm6 family (20) and include Kdm6a (also known as Utx)
and Kdm6b (also known as JMJD3) both of which have been shown to
catalyze the demethylation of H3K27me3 (25,26).
The roles of these demethylases in cancer are less well defined.
There are many instances where loss of expression of these
demethylases are found in cancer through mutation (27). Likewise, there are also many
studies which have shown overexpression of these proteins in cancer
(27–29), and specific inhibitors
(GSK-J1/GSK-J4) for the Kdm6 family have now been developed
(30).
Given the pro-inflammatory nature of MPM, and the
therapeutic potential to target the PRC2 complex, it may also be
possible that the Kdm6 family is a potential candidate therapeutic
avenue. In the present study, we assessed the expression of Kdm6
family members in MPM and the effects of Kdm6 inhibition on MPM
cellular health. Our results indicate that while the Kdm6 family is
overexpressed in MPM, selective inhibition of this family may not
be suitable as a therapeutic option in MPM.
Materials and methods
Primary tumor samples
Surgical specimens were obtained as discarded tumor
samples from patients who had undergone extended
pleuropneumonectomy at Glenfield Hospital (Leicester, UK). Benign
specimens were acquired from patients never diagnosed with MPM.
Informed consent was obtained from each patient, and the study was
conducted after formal approval from the relevant Hospital Ethics
Committee (Leicestershire REC references 6742 and 6948). Samples
consisted of the following: 5 benign lesions and 17 MPM samples
(epithelioid, n=7; sarcomatoid, n=4; biphasic, n=6), and details
are provided in Table I.
 | Table IDetails of pleura/mesothelioma
samples used in the present study. |
Table I
Details of pleura/mesothelioma
samples used in the present study.
| Patients | Pathology (benign,
epithelial, biphasic, sarcomatoid) | Age (years) | Gender |
|---|
| 1 | Benign-pleural
plaque (Benign fibrous plaque and focal chronic inflammation) | 55 | Male |
| 2 | Benign-pleural
plaque (Benign hyaline plaques with chronic inflammation + reactive
changes) | 55 | Male |
| 3 | Benign-pneumothorax
(Chronic inflammatory infiltrate, mesothelial proliferation) | 30 | Male |
| 4 | Benign empyema
(Acute and chronic inflammation + fibrosis) | 68 | Male |
| 5 | Benign-pleural
plaque (Inflammatory + talc granuloma) | 55 | Male |
| 6 | Epithelial | 62 | Male |
| 7 | Epithelial | 73 | Male |
| 8 | Epithelial | 66 | Male |
| 9 | Epithelial | 56 | Female |
| 10 | Epithelial | 52 | Male |
| 11 | Epithelial | 56 | Male |
| 12 | Epithelial | 54 | Male |
| 13 | Biphasic | 54 | Male |
| 14 | Biphasic | 54 | Female |
| 15 | Biphasic | 41 | Male |
| 16 | Biphasic | 58 | Male |
| 17 | Biphasic | N/A | Male |
| 18 | Biphasic | 60 | Female |
| 19 | Sarcomatoid | 74 | Male |
| 20 | Sarcomatoid | 64 | Male |
| 21 | Sarcomatoid | 59 | Male |
| 22 | Sarcomatoid
(desmoplastic) | 64 | Male |
All research on these samples was conducted at St.
James's Hospital under the SJH/AMNCH research ethics committee
approval (041017/8704).
Cell culture
All MPM cell lines were maintained in a humidified
atmosphere containing 5% v/v CO2 in appropriate media
supplemented with 10% v/v fetal bovine serum (FBS) and penicillin
streptomycin (500 U/ml). Cell culture reagents were purchased from
Lonza (Walkersville, MD, USA). A panel of normal mesothelial cell
lines (LP9 and Met5A) and malignant MPM cell lines (NCI-H2596, MMP,
MMB, NCI-H2052, NCI-H28, Ju77, One58, RS-5, DM-3, ACC-MESO-1,
ACC-MESO-4, Y-MESO-8D, Y-MESO-9, Y-MESO-12, Y-MESO-14, NCI-H226 and
REN) were used in the present study.
ACC-MESO-1, ACC-MESO-4, Y-MESO-8D, Y-MESO-9,
Y-MESO-12 and Y-MESO-14 were generously provided by Yoshitaka
Sekido (Aichi Cancer Center Research Institute, Nagoya, Japan).
NCI-H2052, One-58 and JU77 cells were provided by
Duncan Stewart (University of Leicester, Leicester, UK). The REN
and NCI-H226 cell lines were provided by Dean Fennell (Queen's
University, Belfast, Northern Ireland). NCI-H28 and the
immortalized non-tumorigenic mesothelial cell line, Met-5A were
purchased from the ATCC (LGC Promochem, Teddington, UK). DM-3 and
RS-5 were purchased from the Leibniz-Institut DSMZ-Deutsche
Sammlung von Mikroorganismen und Zellkulturen (DMSZ).
The NCI-H226 cell line was authenticated by
STR-profiling (Source BioScience, Nottingham, UK).
Total RNA isolation and RT-PCR
amplification
Total RNA was extracted using TRI reagent according
to the manufacturer's instructions (Molecular Research Center,
Cincinnati, OH, USA). cDNA was synthesized as follows: 250 ng
(primary tumors) or 1000 ng (cell lines) 250 ng - 1 μg of
total RNA was first pre-treated by digestion with RQ1 DNase
(Promega, Madison, WI, USA). Following inactivation the RQ1 DNase
treated mRNA was converted to cDNA using RevertAid (Thermo Fisher
Scientific) and random hexamers (Roche) according to the
manufacturer's instructions. cDNA was then stored at −20°C until
use.
Expression of Kdm6a, Kdm6b and 18S rRNA was
subsequently examined by RT-PCR, using the primers outlined in
Table II. PCR cycling conditions
consisted of 95°C for 5 min followed by 35 cycles of 1 min at 95°C,
1 min at 58°C and 1 min at 72°C with a final extension at 72°C for
10 min. Products were electrophoresed on a 2% agarose gel. Product
quantification was performed using TINA 2.09c (Raytest,
Isotopenmeßgeräte GmbH, Straubenhardt, Germany) den sitometry
software. The mRNA expression was normalized to either 18S rRNA or
β-actin controls, and was expressed as a ratio of experimental gene
expression: control gene expression.
| Table IIPCR primers used in the present
study. |
Table II
PCR primers used in the present
study.
| Primer | Sequence | Expected size
(bp) |
|---|
| Kdm6a Forward |
5′-GGCACTGTTCATTGGGTTCAGG-3′ | 305 |
| Kdm6a Reverse |
5′-TTTGTCCGCCCATGCCATAT-3′ | |
| Kdm6b Forward |
5′-AGCAAACGGGATGCCTTCTCAC-3′ | 179 |
| Kdm6b Reverse |
5′-TGCACCTGGGTGCGAACTTC-3′ | |
| CXCL1 Forward |
5′-ATGGCCCGCGCTGCTCTCTC-3′ | 324 |
| CXCL1 Reverse |
5′-TCAGTTGGATTTGTCACTGTTC-3′ | |
| CXCL2 Forward |
5′-ATGGCCCGCGCCACGCTCTC-3′ | 324 |
| CXCL2 Reverse |
5′-TCAGTTGGATTTGCCATTTTTCAGC-3′ | |
| CXCL8 Forward |
5′-ATGACTTCCAAGCTGGCCGTG-3′ | 297 |
| CXCL8 Reverse |
5′-TGAATTCTCAGCCCTCTTCA-3′ | |
| 18S rRNA
Forward |
5′-GATGGGCGGCGGAAAATAG-3′ | 166 |
| 18S rRNA
Reverse |
5′-GGCGTGGATTCTGCATAATGG-3′ | |
| β-actin
Forward |
5′-TGTTTGAGACCTTCAACACCC-3′ | 529 |
| β-actin
Reverse |
5′-AGCACTGTGTTGGCGTACAG-3′ | |
Drug treatment and cellular viability
assays
GSK-J4 was purchased from Selleck (St. Louis, MO,
USA; cat. no. O7753), and dissolved in dimethyl sulfoxide (DMSO) at
a final concentration of 1 M. Cells were serum starved (0.5% v/v
FBS) for 24 h prior to addition of either drug or vehicle, and
incubated for a further 48 h. Cellular viability was assessed using
either a resazurin reduction assay as previously described
(31), or by a BrdU ELISA (Roche
Diagnostics, Ltd., Sussex, UK), according to the manufacturer's
instructions.
Cellular apoptosis (FACS)
NCI-H226 cells were plated in 6-well plates
(1×105 cells/well) and allowed to adhere overnight.
Complete media was then removed and the cells washed with 100 ml
phosphate-buffered saline (PBS). At this point, serum depleted
media (0.5% FBS) was then added and the cells incubated for a
further 24 h. Following this cells were subsequently treated with
various concentrations of GSK-J4, diluted in serum depleted media,
and incubated in the presence of drug for a further 48 h. Where
appropriate, control cells were treated with either vehicle or left
untreated with media only. Following treatment, the culture media
was transferred to labeled FACS tubes and placed on ice. Remaining
adherent cells were trypsinized and transferred to the same
corresponding FACS tubes. Cells were then centrifuged (1300 rpm for
3 min) and all supernatants were discarded. The pellet of cells was
resuspended and washed in 1 ml of 1X binding buffer (BB) diluted in
ice cold PBS, centrifuged and subsequently resuspended in 100
μl BB. A total of 2 μl Annexin V (IQ Products BV,
Groningen, the Netherlands) was added to each tube, with the
exception of the negative control and media only samples, and
incubated at 4°C for 20 min, protected from light. The cells were
washed in 1 ml 1X binding buffer and supernatant removed. Prior to
flow cytometric analysis the pellet of cells was resuspended in 400
μl BB containing 0.5 μg/ml PI (Invitrogen, Paisley,
UK), except the negative control and FMO (fluorescence minus one)
control for PI for which BB alone was used and analyzed by flow
cytometry.
Cellular apoptosis (caspase-3/-7
activation)
NCI-H226 cells were seeded at a density of
4×103 cells/well in Corning® 96 Well Flat
Clear Bottom Black Polystyrene Tissue Culture-treated 96-well
plates and allowed to adhere overnight. Subsequently, the media was
removed and the cells washed with 100 ml PBS. The cells were then
incubated in serum depleted media (0.5% FBS) for a further 24 h, at
which point they were then treated with GSK-J4 at various
concentrations for a further 48 h. Caspase-3/-7 activation was then
measured using a FluoroFire caspase-3/-7 fluorescent assay kit
according to the manufacturer's instructions (Molecutools, Dublin,
Ireland).
In silico analysis
Data-mining of available mesothelioma datasets were
conducted using Oncomine (www.oncomine.org) (32), or cBioPortal (www.cbioportal.org) (33) using the default settings. The
results shown here are in whole or part based upon data generated
by the The Cancer Genome Atlas Research Network (cancergenome.nih.gov).
Statistical analysis
Data are expressed as mean ± standard error of
multiple experiments (n=3). Statistical analysis was performed
using either Mann-Whitney or unpaired Student's t-tests, or one-way
ANOVA with Dunnett's post-test using the GraphPad Prism 5.01.
Differences were considered to be statistically significant at
P<0.05.
Results
Kdm6 family members are ubiquitously
expressed in mesothelioma cell lines
Utilizing RT-PCR, the levels of expression of Kdm6a
and Kdm6b mRNA were examined in a panel of cell lines including
those derived from normal pleura (LP9 and Met5A) and mesotheliomas.
Kdm6a and Kdm6b were readily detectable in all cell lines as shown
in Fig. 1.
Kdm6 family members are overexpressed in
MPM
To assess the expression of Kdm6 family members in
primary patient material, RT-PCR was performed on a panel of benign
pleura and MPM tumor samples isolated at surgery from patients
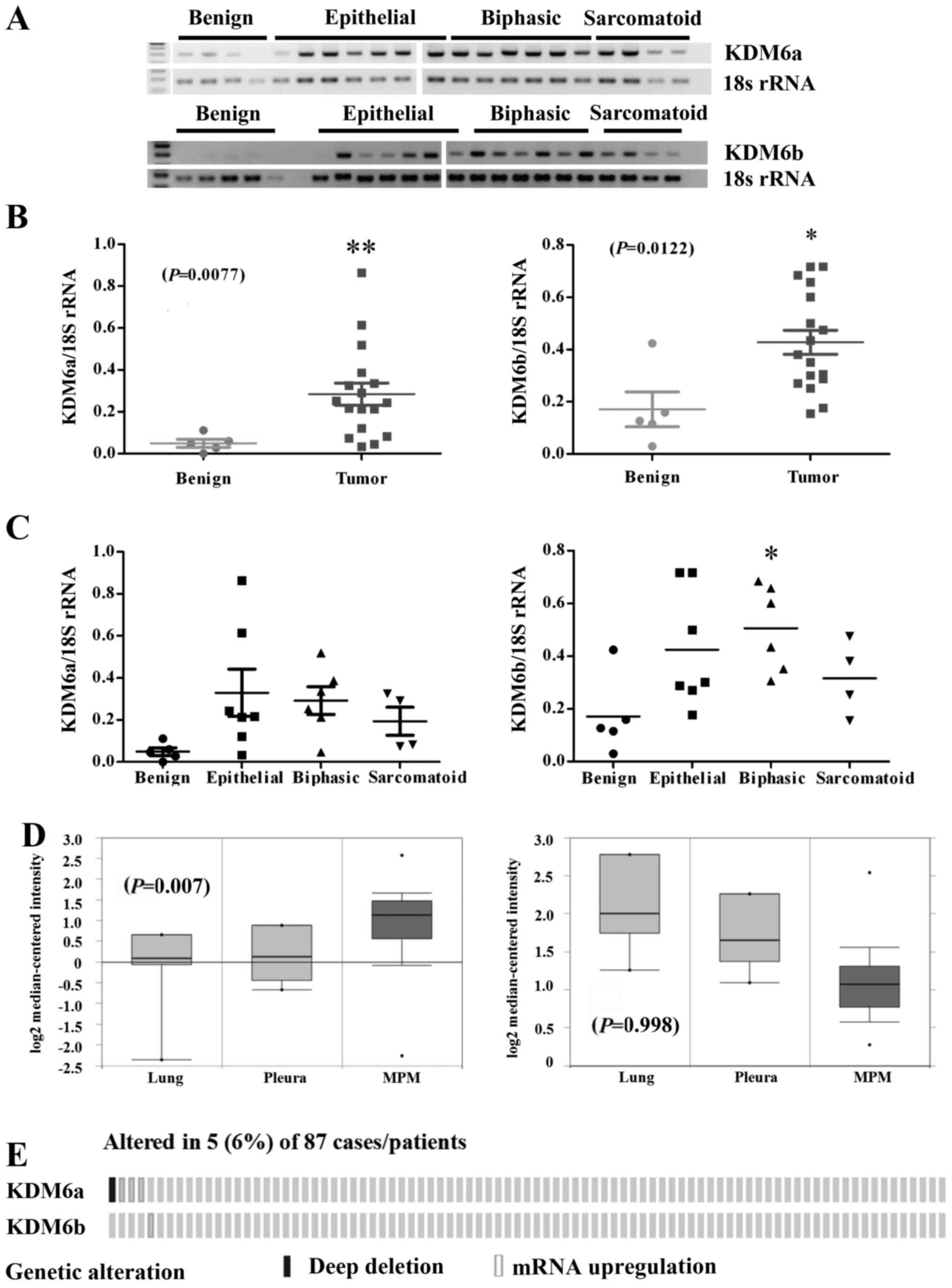
(Fig. 2A). Densitometric analysis
of the gels revealed a significant increase in the expression of
Kdm6a mRNA (P=0.0036) and Kdm6b mRNA (P=0.0122) in MPM tumor
samples compared with normal pleura (Fig. 2B). When stratified by histology,
significant overexpression of both Kdm6a mRNA was observed across
all histological subtypes, whereas only Kdm6b mRNA was
significantly altered in the biphasic subtype (Fig. 2C). Using Oncomine (32), we queried the expression of both
Kdm6 family members in the Gordon et al (34) dataset. In this dataset only Kdm6a
was shown to be significantly overexpressed in the MPM specimens
compared to benign pleura or lung (P=0.007), whereas Kdm6b levels
were not significantly altered (Fig.
2D). Using cBioPortal (33),
we also examined an available mesothelioma TCGA NGS dataset
comprising (n=87) samples (Fig.
2E). Overexpression of Kdm6a and Kdm6b were observed in a small
cohort of patients (Fig. 2E).
Overall, the results suggest that the Kdm6 family could be a
potential therapeutic target in MPM.
Inhibition of the Kdm6 family results in
decreased MPM proliferation and altered expression of
pro-inflammatory cytokines
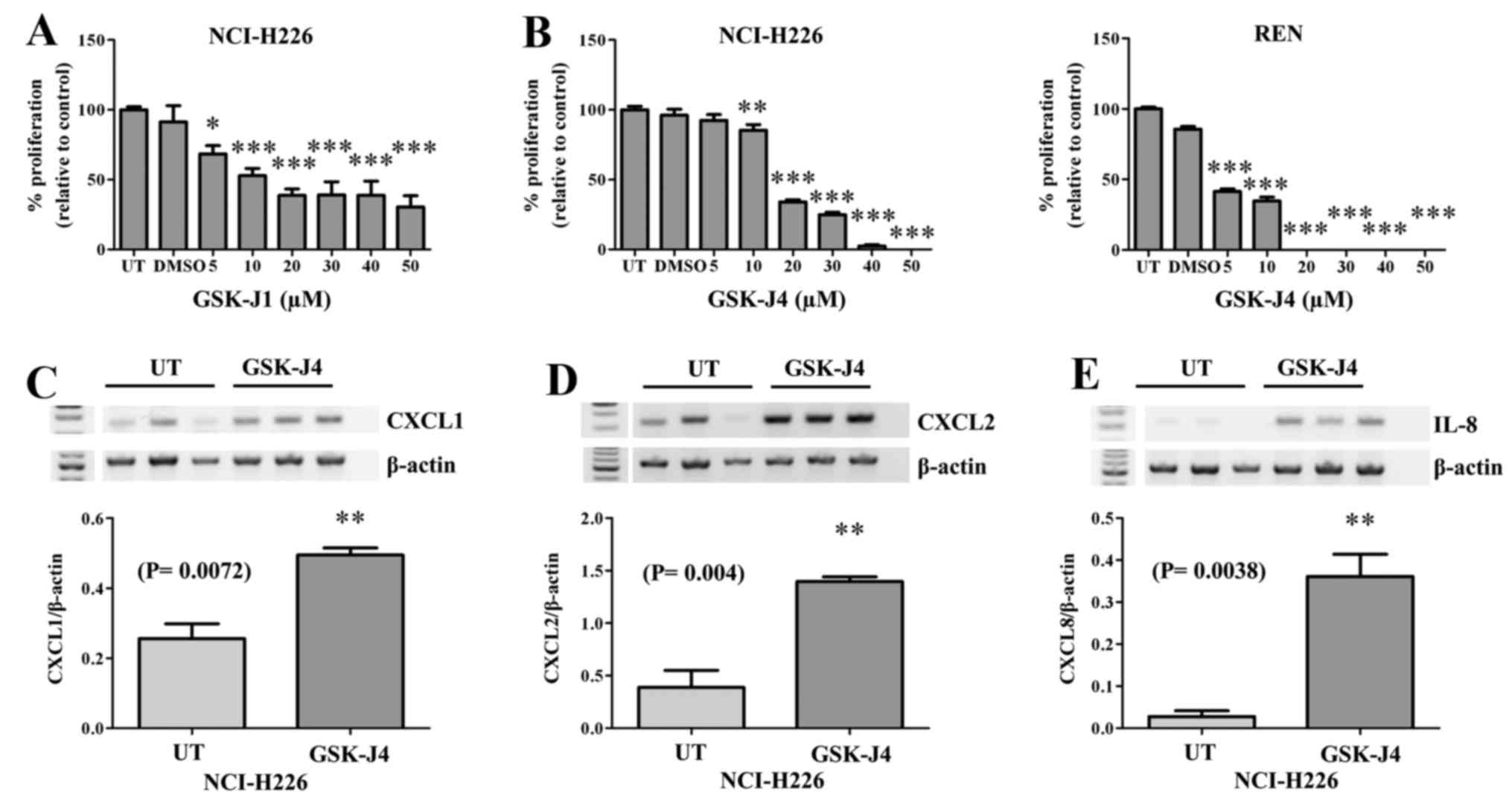
The only current selective inhibitor targeting Kdm6a
and Kdm6b is GSK-J1, with a corresponding cell-active pro-drug form
GSK-J4 (30). We initially tested
the effect of GSK-J1 on the proliferative capacity of NCI-H226
cells (Fig. 3A), the results of
which indicated that this compound could indeed inhibit MPM
cellular proliferation. We subsequently tested the ability of the
pro-drug to inhibit cellular proliferation and confirmed that
GSK-J4 inhibited MPM cellular proliferation in two MPM cell lines
tested (Fig. 3B). Given the known
role of Kdm6a as a key regulator in inflammation (27,30,35),
and the ability of GSK-J4 to block pro-inflammatory cytokine
expression (30), and the
pro-inflammatory nature of MPM, we examined the effect of GSK-J4 on
the expression of a panel of pro-inflammatory cytokines. However,
treatment of MPM cells with GSK-J4 resulted in significant
upregulation of CXCL1 (P=0.0072; Fig.
3C), CXCL2 (P= 0.004; Fig. 3D)
and CXCL8/IL-8 (P=0.0038; Fig.
3E).
Non-malignant pleural cells are more
sensitive to GSK-J4 than MPM cells
Whilst GSK-J4 has significant anti-proliferative
effects on MPM cell lines, given that expression of Kdm6a and Kdm6b
was found to be almost ubiquitous in our panel of cell lines
(Fig. 1), we subsequently examined
the sensitivity of normal mesothelial cells (LP-9) in parallel with
two malignant MPM cell lines (NCI-H226 and REN). Using either a
Resazurin based assay (Fig. 4A),
or BrdU incorporation (Fig. 4B),
we demonstrated that the normal pleural cell line is more sensitive
to GSK-J4. We re-screened the cell lines for expression of Kdm6a
and Kdm6b mRNA and confirmed that both cells were expressed
(Fig. 4C). The increased
sensitivity of the normal pleural cell line to Kdm6 inhibition was
reflected in increased levels of apoptosis in comparison with the
MPM cell lines (Fig. 5).
Discussion
MPM is an aggressive cancer with very limited
treatment options. Currently, the established first-line therapy is
a combination of cisplatin and an anti-folate. Most MPM patients do
not respond to this or other therapies, and the duration of
response is short with a rapid development of resistance (36). There is no current defined
second-line therapy, and as a consequence there is an urgent need
to identify new therapeutic options for the treatment of this
cancer. The recent demonstration that MPMs are in fact polyclonal
tumors formed by the coalescence of different independent subclones
highlights the need for new therapeutic approaches, as each clone
may carry its own distinct set of molecular alterations, and the
intra-tumoral heterogeneity arising may allow for the emergence of
drug-resistant subpopulations, and as such multi-targeted
approaches to therapy may be required to overcome the issue of
clonality (37).
EZH2 is associated with the H3K27me3 histone
post-translational modification mark common to silenced chromatin
or bivalent 'poised' promoters (20,38).
The lysine demethylases that catalyze the removal of this mark have
been identified as Kdm6a (Utx) and Kdm6b (Jmjd3). Given the
potential importance of this mark in BAP1 mutated MPM, in this
study we therefore sought to examine the expression of these lysine
demethylases in MPM. An initial screen showed that the mRNA for
both Kdm6a and Kdm6b was commonly expressed in all cell lines
derived from both normal mesothelial and malignant MPM cells
(Fig. 1).
However, when examined in patient tumors we
demonstrate that both Kdm6a and Kdm6b show significantly elevated
mRNA for these lysine demethylases in the tumors compared to benign
pleura (Fig. 2A). When separated
according to histological subtype we observed no significant
overexpression of Kdm6a across all histological subtypes (Fig. 2C), whereas elevated Kdm6b mRNA was
only significant in the biphasic subtype (Fig. 2C). In this regard the current
sample size is clearly insufficient to make any clear comparisons
across histological subtype, and that a larger sample size will be
required to address this. In silico analysis confirmed that
altered expression of these Kdms does occur in MPM, but
significantly may only be restricted to Kdm6a (Fig. 2D and E).
It could also be construed that normal pleural
tissues from patients with resected lung cancer would be a more
suitable control than the inflammatory benign pleural samples used
in the present study. In our opinion that moving forward, this may
be a useful additional control to compare non-inflammatory pleura
to inflammatory pleura. Nevertheless, given the fact that MPM is a
pro-inflammatory cancer, and that Kdm6 family members have been
shown to play key roles in the regulation of pro-inflammatory
responses (30), including the
potential role of Kdm6b as a key element in 'inflammaging' that can
contribute to tumor progression (27), our data demonstrates that
overexpression of these demethylases occurs in malignant tissue
(Fig. 2) compared to the benign
pleural plaques which contain a significant inflammatory profile
(Table I). In this regard, our
data remove a potential confounding issue; that inflammation itself
may be driving the upregulation of Kdm6a and Kdm6b in malignant
tissue.
Inhibitors specific to members of the Kdm6 family
(GSK-J1 and GSK-J4) have been developed (30), and given both the pro-inflammatory
nature of MPM, and the elevated expression of Kdm6a and Kdm6b in
our patient samples, we subsequently examined the effects of these
inhibitors on MPM cellular health and pro-inflammatory cytokine
expression. Both drugs were shown to have significant
anti-proliferative activity on MPM cell lines with the cell-active
ethyl ester pro-drug GSK-J4 showing greater anti-proliferative
effects at lower concentrations than GSK-J1 (Fig. 3A and B).
GSK-J4 was originally demonstrated to prevent
LPS-mediated induction of pro-inflammatory cytokines in human
primary macrophages (30).
However, treatment of an MPM cell line with GSK-J4 resulted in
significant upregulation of several pro-inflammatory cytokines
including IL-8 (Fig. 3C). In this
regard, induction of IL-8 by anticancer regimens has previously
been observed in an experimental model of mesothelioma (39). Furthermore, elevated levels of IL-8
have also been found to be increased in mesothelioma patients
undergoing therapy following pleurectomy or extra-pleural
pneumonectomy (EPP) (40), or in
MPM patients treated tumor necrosis factor-alpha (TNF-α) (41). It has been noted that elevated
levels of IL-8 may not be a reliable marker for predicting response
to therapy (42), and the
observation that GSK-J4 is capable of elevating the expression of
several pro-inflammatory cytokines suggests that inhibition of Kdm6
family members may have the potential to induce cytokine response
syndrome (CRS) (43), as sometimes
seen in MPM (44).
The increased production of pro-inflammatory
cytokines in MPM cells was accompanied by decreased cellular
proliferation (Fig. 4) and an
associated induction of apoptosis (Fig. 5) and necrosis (Lauren MacDonagh,
unpublished). Moreover, we were able to show that cells derived
from normal pleura were more sensitive to GSK-J4 that those derived
from MPM. These results suggest that whilst the expression of Kdm6
family members are significantly elevated in MPM, targeting the
same may not be sparing of patients normal pleura. However, our
results also indicate that in primary tissues levels of the Kdm6
family are much higher in patient tumors than in normal pleura
(Fig. 2), and this discrepancy
remains to be resolved.
In conclusion, our results suggest that the Kdm6
family may represent a novel therapeutic target for the treatment
of MPM, but given the potential for damage to the normal pleura, or
the potential for inducing a cytokine storm, further studies are
warranted. We believe that more studies will be required to further
determine whether or not targeting the Kdm6 family is possible in
MPM, or indeed even in a subset of MPM and to determine if a
multi-targeting approach combining epigenetic therapies (including
GSK-J4) is possible.
Acknowledgments
The authors are grateful to Dr Warren Thomas, Dr
Yoshitaka Sekido, Dr Dean Fennell and Dr Hannu Norppa for their
generosity in providing access to various mesothelioma cell lines.
The present study was supported in part by funding for consumables
from the Masters in Translational Oncology program (TCD) for Sian
Cregan.
References
|
1
|
Wagner JC, Sleggs CA and Marchand P:
Diffuse pleural mesothelioma and asbestos exposure in the North
Western Cape Province. Br J Ind Med. 17:260–271. 1960.PubMed/NCBI
|
|
2
|
Delgermaa V, Takahashi K, Park EK, Le GV,
Hara T and Sorahan T: Global mesothelioma deaths reported to the
World Health Organization between 1994 and 2008. Bull World Health
Organ. 89:716–724. 724A–724C. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ramazzini C: The global health dimensions
of asbestos and asbestos-related diseases. J Occup Health.
58:220–223. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Järvholm B and Burdorf A: Emerging
evidence that the ban on asbestos use is reducing the occurrence of
pleural mesothelioma in Sweden. Scand J Public Health. 43:875–881.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Price B and Ware A: Time trend of
mesothelioma incidence in the United States and projection of
future cases: An update based on SEER data for 1973 through 2005.
Crit Rev Toxicol. 39:576–588. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tan E, Warren N, Darnton AJ and Hodgson
JT: Projection of mesothelioma mortality in Britain using Bayesian
methods. Br J Cancer. 103:430–436. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Carbone M, Ly BH, Dodson RF, Pagano I,
Morris PT, Dogan UA, Gazdar AF, Pass HI and Yang H: Malignant
mesothelioma: Facts, myths, and hypotheses. J Cell Physiol.
227:44–58. 2012. View Article : Google Scholar
|
|
8
|
Gilham C, Rake C, Burdett G, Nicholson AG,
Davison L, Franchini A, Carpenter J, Hodgson J, Darnton A and Peto
J: Pleural mesothelioma and lung cancer risks in relation to
occupational history and asbestos lung burden. Occup Environ Med.
73:290–299. 2016. View Article : Google Scholar :
|
|
9
|
Algranti E, Saito CA, Carneiro AP, Moreira
B, Mendonça EM and Bussacos MA: The next mesothelioma wave:
Mortality trends and forecast to 2030 in Brazil. Cancer Epidemiol.
39:687–692. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baas P, Fennell D, Kerr KM, Van Schil PE,
Haas RL and Peters S; ESMO Guidelines Committee: Malignant pleural
mesothelioma: ESMO Clinical Practice Guidelines for diagnosis,
treatment and follow-up. Ann Oncol. 26(Suppl 5): v31–v39. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
van Meerbeeck JP, Gaafar R, Manegold C,
Van Klaveren RJ, Van Marck EA, Vincent M, Legrand C, Bottomley A,
Debruyne C and Giaccone G; European Organisation for Research and
Treatment of Cancer Lung Cancer Group; National Cancer Institute of
Canada: Randomized phase III study of cisplatin with or without
raltitrexed in patients with malignant pleural mesothelioma: An
intergroup study of the European Organisation for Research and
Treatment of Cancer Lung Cancer Group and the National Cancer
Institute of Canada. J Clin Oncol. 23:6881–6889. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vogelzang NJ, Rusthoven JJ, Symanowski J,
Denham C, Kaukel E, Ruffie P, Gatzemeier U, Boyer M, Emri S,
Manegold C, et al: Phase III study of pemetrexed in combination
with cisplatin versus cisplatin alone in patients with malignant
pleural mesothelioma. J Clin Oncol. 21:2636–2644. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baird A, Richard D, O'Byrne KJ and Gray
SG: Epigenetic Therapy in Lung Cancer and Mesothelioma. Epigenetic
Cancer Therapy. 1st. Gray SG: Academic Press; pp. 189–213. 2015,
View Article : Google Scholar
|
|
14
|
Baylin SB and Jones PA: Epigenetic
determinants of cancer. Cold Spring Harb Perspect Biol.
8:a0195052016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang X, Tang N, Rishi AK, Pass HI and
Wali A: Methylation profile landscape in mesothelioma: Possible
implications in early detection, disease progression, and
therapeutic options. Methods Mol Biol. 1238:235–247. 2015.
View Article : Google Scholar
|
|
16
|
Kelly WK, O'Connor OA, Krug LM, Chiao JH,
Heaney M, Curley T, MacGregore-Cortelli B, Tong W, Secrist JP,
Schwartz L, et al: Phase I study of an oral histone deacetylase
inhibitor, suberoylanilide hydroxamic acid, in patients with
advanced cancer. J Clin Oncol. 23:3923–3931. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Krug LM, Kindler HL, Calvert H, Manegold
C, Tsao AS, Fennell D, Öhman R, Plummer R, Eberhardt WE, Fukuoka K,
et al: Vorinostat in patients with advanced malignant pleural
mesothelioma who have progressed on previous chemotherapy
(VANTAGE-014): A phase 3, double-blind, randomised,
placebo-controlled trial. Lancet Oncol. 16:447–456. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cregan S, MacDonagh L, Gao Y, Barr MP,
O'Byrne KJ, Finn SP, Cuffe S and Gray SG: KAT5 (Tip60) is a
potential therapeutic target in malignant pleural mesothelioma. Int
J Oncol. 48:1290–1296. 2016.PubMed/NCBI
|
|
19
|
Sacco JJ, Kenyani J, Butt Z, Carter R,
Chew HY, Cheeseman LP, Darling S, Denny M, Urbé S, Clague MJ, et
al: Loss of the deubiquitylase BAP1 alters class I histone
deacetylase expression and sensitivity of mesothelioma cells to
HDAC inhibitors. Oncotarget. 6:13757–13771. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bosselut R: Pleiotropic functions of
H3K27Me3 demethylases in immune cell differentiation. Trends
Immunol. 37:102–113. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Goto Y, Shinjo K, Kondo Y, Shen L, Toyota
M, Suzuki H, Gao W, An B, Fujii M, Murakami H, et al: Epigenetic
profiles distinguish malignant pleural mesothelioma from lung
adenocarcinoma. Cancer Res. 69:9073–9082. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Conway E, Healy E and Bracken AP: PRC2
mediated H3K27 methylations in cellular identity and cancer. Curr
Opin Cell Biol. 37:42–48. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kemp CD, Rao M, Xi S, Inchauste S, Mani H,
Fetsch P, Filie A, Zhang M, Hong JA, Walker RL, et al: Polycomb
repressor complex-2 is a novel target for mesothelioma therapy.
Clin Cancer Res. 18:77–90. 2012. View Article : Google Scholar
|
|
24
|
LaFave LM, Béguelin W, Koche R, Teater M,
Spitzer B, Chramiec A, Papalexi E, Keller MD, Hricik T,
Konstantinoff K, et al: Loss of BAP1 function leads to
EZH2-dependent transformation. Nat Med. 21:1344–1349. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Agger K, Cloos PA, Christensen J, Pasini
D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE and Helin
K: UTX and JMJD3 are histone H3K27 demethylases involved in HOX
gene regulation and development. Nature. 449:731–734. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hong S, Cho YW, Yu LR, Yu H, Veenstra TD
and Ge K: Identification of JmjC domain-containing UTX and JMJD3 as
histone H3 lysine 27 demethylases. Proc Natl Acad Sci USA.
104:18439–18444. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Perrigue PM, Najbauer J and Barciszewski
J: Histone demethylase JMJD3 at the intersection of cellular
senescence and cancer. Biochim Biophys Acta. 1865:237–244.
2016.PubMed/NCBI
|
|
28
|
Paolicchi E, Crea F, Farrar WL, Green JE
and Danesi R: Histone lysine demethylases in breast cancer. Crit
Rev Oncol Hematol. 86:97–103. 2013. View Article : Google Scholar :
|
|
29
|
Shen Y, Guo X, Wang Y, Qiu W, Chang Y,
Zhang A and Duan X: Expression and significance of histone H3K27
demethylases in renal cell carcinoma. BMC Cancer. 12:4702012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kruidenier L, Chung CW, Cheng Z, Liddle J,
Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, et
al: A selective jumonji H3K27 demethylase inhibitor modulates the
proinflammatory macrophage response. Nature. 488:404–408. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Riss TL, Moravec RA, Niles AL, Duellman S,
Benink HA, Worzella TJ and Minor L: Cell viability assays. Assay
Guidance Manual (Internet). Sittampalam GS, Coussens NP, Nelson H,
Arkin M, Auld D, Austin C, Bejcek B, Glicksman M, Inglese J,
Iversen PW, et al: Eli Lilly & Company and the National Center
for Advancing Translational Sciences; Bethesda, MD: 2004
|
|
32
|
Rhodes DR, Kalyana-Sundaram S, Mahavisno
V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ,
Kincead-Beal C, Kulkarni P, et al: Oncomine 3.0: Genes, pathways,
and networks in a collection of 18,000 cancer gene expression
profiles. Neoplasia. 9:166–180. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gordon GJ, Rockwell GN, Jensen RV,
Rheinwald JG, Glickman JN, Aronson JP, Pottorf BJ, Nitz MD,
Richards WG, Sugarbaker DJ, et al: Identification of novel
candidate oncogenes and tumor suppressors in malignant pleural
mesothelioma using large-scale transcriptional profiling. Am J
Pathol. 166:1827–1840. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
De Santa F, Totaro MG, Prosperini E,
Notarbartolo S, Testa G and Natoli G: The histone H3 lysine-27
demethylase Jmjd3 links inflammation to inhibition of
polycomb-mediated gene silencing. Cell. 130:1083–1094. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bononi A, Napolitano A, Pass HI, Yang H
and Carbone M: Latest developments in our understanding of the
pathogenesis of mesothelioma and the design of targeted therapies.
Expert Rev Respir Med. 9:633–654. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Comertpay S, Pastorino S, Tanji M,
Mezzapelle R, Strianese O, Napolitano A, Baumann F, Weigel T,
Friedberg J, Sugarbaker P, et al: Evaluation of clonal origin of
malignant mesothelioma. J Transl Med. 12:3012014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Arcipowski KM, Martinez CA and
Ntziachristos P: Histone demethylases in physiology and cancer: A
tale of two enzymes, JMJD3 and UTX. Curr Opin Genet Dev. 36:59–67.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lansley SM, Varano Della Vergiliana JF,
Cleaver AL, Ren SH, Segal A, Xu MY and Lee YC: A commercially
available preparation of Staphylococcus aureus bio-products
potently inhibits tumour growth in a murine model of mesothelioma.
Respirology. 19:1025–1033. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yom SS, Busch TM, Friedberg JS, Wileyto
EP, Smith D, Glatstein E and Hahn SM: Elevated serum cytokine
levels in mesothelioma patients who have undergone pleurectomy or
extrapleural pneumonectomy and adjuvant intraoperative
photo-dynamic therapy. Photochem Photobiol. 78:75–81. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Stam TC, Swaak AJ, Kruit WH, Stoter G and
Eggermont AM: Intrapleural administration of tumour necrosis
factor-alpha (TNFalpha) in patients with mesothelioma: Cytokine
patterns and acute-phase protein response. Eur J Clin Invest.
30:336–343. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nowak AK, Millward MJ, Creaney J, Francis
RJ, Dick IM, Hasani A, van der Schaaf A, Segal A, Musk AW and Byrne
MJ: A phase II study of intermittent sunitinib malate as
second-line therapy in progressive malignant pleural mesothelioma.
J Thorac Oncol. 7:1449–1456. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lee DW, Gardner R, Porter DL, Louis CU,
Ahmed N, Jensen M, Grupp SA and Mackall CL: Current concepts in the
diagnosis and management of cytokine release syndrome. Blood.
124:188–195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nowak AK, Cook AM, McDonnell AM, Millward
MJ, Creaney J, Francis RJ, Hasani A, Segal A, Musk AW, Turlach BA,
et al: A phase 1b clinical trial of the CD40-activating antibody
CP-870,893 in combination with cisplatin and pemetrexed in
malignant pleural mesothelioma. Ann Oncol. 26:2483–2490.
2015.PubMed/NCBI
|