Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most
common subtype of non-Hodgkin lymphoma (NHL) and accounts for
~30–40% of all newly diagnosed lymphomas among adults (1,2).
DLBCL is readily curable with combination chemotherapy, such as
R-CHOP (rituximab, cyclophosphamide, adriamycin, vincristine and
prednisone) (3,4). Although ~50% of patients with diffuse
large B-cell lymphoma can now survive for more than 10 years
(5), most patients who do not
respond to R-CHOP treatment will eventually die from the disease.
As a result, efforts have focused on the development of a more
effective and personalized treatment modality.
EBV is associated with a number of malignancies,
such as nasopharyngeal carcinoma, NHL and a subset of B-cell
lymphoma in immunosuppressed individuals (6). EBV-positive DLBCL in patients older
than 50 years is not associated with any known immunodeficiency
(7). In addition, EBV-positive
DLBCL in the elderly is characterized by activated B-cell
phenotypes via NF-κB pathway activation and high expression of
latent membrane protein 1 (LMP1) (8). EBV-LMP1 not only activates
phosphatidylinositol 3-kinase (PI3K)/Akt pathways (9,10),
but also is associated with the development of cancer stem cells
(CSC) in nasopharyngeal epithelial cell lines (30). LMP1 also increases the expression
of several markers, such as Octamer 4 (OCT4), SRY-related HMG box 2
(SOX2) and ATP-binding cassette (ABC) subfamily G member 2 (ABCG2)
(11). The three major multidrug
resistance ABC transporter proteins consist of multidrug
resistance-related protein-1 (MRP1), multidrug resistance 1 (MDR1;
P-glycoprotein) and ABCG2 (12).
These proteins specifically allow the transport of various chemical
entities, including anticancer drugs (13,14).
ABC transporters are also a major route for anthracycline (e.g.,
adriamycin) elimination (3).
Furthermore, the complete remission rates are significantly lower
in the group expressing drug resistance-related proteins than in
the group not expressing them (15). These results demonstrate that the
increased expression of ABC transporters on plasma membranes
results in increased efflux of anticancer drugs, leading to
multidrug resistance. However, detailed downstream signaling and
the role of PI3K in induction of ABC transporters and survival of
EBV-infected B cell cancer are not clear.
B-cell receptor (BCR) ligation activates
CD19-related Src family kinases (Lyn, Fyn and Blk) and Syk tyrosine
kinase, leading to Lyn- or Syk-dependent phosphoinositide 3-kinase
(PI3K) activation (16–18). Several types of B cell malignancies
appear to be dependent on the PI3K pathway for survival (19,20).
PI3K signaling, especially constitutive expression of p110α, is
associated with survival of multiple myeloma (MM) cells (21). Knockdown of p110δ, a PI3K catalytic
unit, by small interfering RNA also causes significant inhibition
of MM cell growth (22), whereas
inhibitors of the p110δ catalytic isoform (idelalisib) fail to
suppress aggressive DLBCL (23,24).
Furthermore, the catalytic subunit of PI3K association with drug
resistance in DLCBL remains unknown.
In the present study, we established EBV-infected
DLBCL and MM cell lines as in vitro models to characterize
EBV-induced drug resistance and identify molecular targets to
control refractory B cell cancer. Bortezomib, which is approved for
treatment of MM, reduced the level of p50 and p65 components of the
canonical NF-κB pathway in EBV-transformed B cells and reduced the
level of p52 in the non-canonical NF-κB pathway (25). We also investigated whether a
combination of the PI3K inhibitor and bortezomib controls
characteristics of EBV-related B cell cancer by blocking NF-κB
activity.
Materials and methods
Preparation of EBV infectious culture
supernatant and generation of EBV-infected DLBCL and MM cells
EBV supernatant stock was prepared from a B95–8 cell
line (Vircell S.L., Granada, Spain). DLBCL or MM cells were added
to EBV stock supernatant, and after 2-h incubation at 37°C,
RPMI-1640 media (Corning Inc., Corning, NY, USA) was added
(1×106 cells/ml). The cultures were incubated for 3
weeks. Analysis of surface expression of CD21 was verified by BD
Accuri™ C6 (BD Biosciences, San Jose, CA, USA) using PE-conjugated
anti-CD21 antibody (BD Biosciences).
Cell lines and reagents
HT cell lines were obtained from the American Type
Culture Collection (ATCC; Manassas, VA, USA). NCI-H929 cell lines
were obtained from the European Collection of Cell Cultures (ECACC;
Salisbury, UK). These cells were maintained in RPMI-1640 media
(Corning) supplemented with 10% fetal bovine serum (FBS; RMBIO,
Missoula, MT, USA), streptomycin and glutamine at 37°C in 5%
CO2. Bortezomib was purchased from LC Laboratories
(Woburn, MA, USA). Doxorubicin and cyclophosphamide were obtained
from Sigma-Aldrich (St. Louis, MO, USA). Bay11-7082, A66, TGX-221,
CAL-101 and LY294002 were purchased from Selleck Chemicals
(Houston, TX, USA).
Cell proliferation assay
Cells (5×104 cells/well) were cultured in
media containing 10% FBS in 96-well plates. After 24 h, cell
proliferation was measured using an AlamarBlue (Serotec Ltd.,
Kidlington, Oxford, UK) assay. AlamarBlue was added (10% by volume)
to each well, and relative fluorescence units (RFUs) were
determined 9 h later with a Wallac 1420 Victor2 multi-label plate
reader (Perkin-Elmer, Shelton, CT, USA; excitation, 530 nm;
emission, 590 nm). Experiments were performed in triplicate, and
relative fluorescence was calculated using mean fluorescence for
each culture.
RT-PCR
Total RNA was isolated using an RNeasy Mini kit
(Qiagen, Hilden, Germany) and transcribed into cDNA using oligo
(dT) primers and reverse transcriptase. PCR products were amplified
using specific primer sets for EBNA1 (upstream primer,
5′-GAGCGGGGAGATAATGTACA; downstream primer,
5′-TAAAAGATGGCCGGACAAGG), EBNA2 (upstream primer,
5′-AACCCTCTAAGACTCAAGGC; downstream primer,
5′-ACTTTCGTCTAAGTCTGCGG), LMP1 (upstream primer,
5′-CACGACCTTGAGAGGGGCCCA; downstream primer,
5′-GCCAGATGGTGGCACCAAGTC), LMP2A (upstream primer,
5′-ATGACTCATCTCAACACATA; downstream primer,
5′-CATGTTAGGCAAATTGCAAA), MDR1 (upstream primer,
5′-TTGCTGCTTACATTCAGGTTTCA; downstream primer, 5′-AGCCTATCTCCTGT
GCATTA), MRP1 (upstream primer, 5′-AAGACCAAGACGTATCAGGT; downstream
primer, 5′-CAATGGTCACGTAGACGGCAA), MRP2 (upstream primer,
5′-TCTCTCGATACTCTGTGGCAC; downstream primer,
5′-CTGGAATCCGTAGGAGATGAAGA), OCT4 (upstream primer,
5′-CGACCATCTGCCGCTTTGAG; downstream primer,
5′-CCCCCTGTCCCCCATTCCTA), and SOX2 (upstream primer,
5′-AGCAACGGCAGCTACAGCA; downstream primer,
5′-TGGGAGGAAGAGGTAACCACAG). A specific primer set for β-actin
(upstream primer, 5′-ATCCACGAAACTACCTTCAA; downstream primer,
5′-ATCCACACGGAGTACTTGC) was used as a control, and PCR was
performed using Prime Taq Premix (GeNet Bio, Chungnam, Korea). PCR
products were analyzed via agarose gel electrophoresis and
visualized with ethidium bromide under UV light using the Amersham™
Image 600 (GE Healthcare, Pittsburgh, PA, USA).
Western blot analysis
Cells were harvested and lysed in
radioimmunoprecipitation assay (RIPA) buffer (Elpis Biotech, Inc.,
Daejeon, Korea) containing a protease inhibitor cocktail
(Sigma-Aldrich) and phosphatase inhibitor (Cocktail II;
Sigma-Aldrich). Total cell lysates were subjected to SDS-PAGE.
Separated proteins were transferred to nitrocellulose membranes
(Millipore, Billerica, MA, USA), the membranes were blocked with 5%
skim milk, and conventional immunoblotting was performed using
several antibodies. Chemiluminescence was detected using an ECL kit
(Advansta Inc., Menlo Park, CA, USA) and the Amersham™ Image 600
(GE Healthcare). The following primary Abs were used: MDR1, MRP1,
MRP2, OCT4, SOX2, phospho-Src (Tyr416), Src, phospho-Syk
(Tyr323), phospho-Syk (Tyr525/526), Syk,
p110α, p110β, p110γ, p110δ, phospho-PI3K (Tyr458), PI3K,
phospho-Akt (Ser473), Akt, phospho-Akt
(Ser473), caspase-8, caspase-9, caspase-3, PARP, NF-κB
p105/p50, p100/p52, c-Rel, Rel-B and β-actin were obtained from
Cell Signaling Technology (Beverly, MA, USA); EBNA2, LMP2A and
Ref-1 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA,
USA); β-tubulin was obtained from BD Biosciences; LMP1 was obtained
from Abnova (Taipei, Taiwan); and EBNA1 was obtained from Thermo
Fisher Scientific (Rockford, IL, USA).
Small interfering RNA (siRNA)
transfection
Experimentally verified human SOX2-siRNA duplex,
OCT4-siRNA duplex, and negative control-siRNA were obtained from
Bioneer. Cells were seeded at a concentration of
1×106/well in a 6-well plate and grown overnight prior
to transfection with 300 nM siRNA using Viromer Blue reagent
(Lipocalyx GmbH, Halle, Germany) according to the manufacturer's
instructions. Cells were used for further experiments 48 h after
transfection.
Detection of NF-κB translocation by
fractionation
Nuclear and cytosol cellular fractions were prepared
using a Nuclear/Cytosol Fractionation kit (BioVision Inc., Mountain
View, CA, USA), according to the manufacturer's protocol. Briefly,
2×106 cells with or without various treatments were
harvested and suspended in 200 µl of cytosol extraction
buffer A. After incubation on ice for 10 min, cytosol extraction
buffer B was added to the cell suspension and incubated on ice for
1 min. The obtained pellets were re-suspended in 10 µl of
nuclear extraction buffer mix and designated nuclear fractions.
Measurement of NF-κB activity by NF-κB
DNA-binding ELISA
ELISA and an NF-κB p50/p65 Transcription Factor
assay kit (Abcam, Cambridge, MA, USA) were used according to the
manufacturer's protocol to quantify the DNA-binding activity of
NF-κB. Briefly, nuclear extracts were transferred to a 96-well
plate coated with a specific dsDNA sequence containing the NF-κB
response element. NF-κB proteins bound to the target sequence were
detected with a primary antibody and an HRP-conjugated secondary
antibody. Absorbance was measured at 450 nm as a relative measure
of protein-bound NF-κB. All fractions were stored at −80°C until
further use.
Statistical analysis
Data are expressed as mean ± standard deviation
(SD). Statistical analysis was conducted using one-way analysis of
the variance (ANOVA). P<0.05 were considered statistically
significant.
Results
EBV infection is capable of inducing drug
resistance in DLBCL cells
To establish EBV-transformed DLBCL and MM as a model
of EBV-related drug resistant B cell cancer, we first examined the
expression of CD21, a receptor for cellular infection of EBV, in HT
and H929 cell lines. HT and H929 had stable expression of CD21
(Fig. 1A). In addition, HT cells
showed characteristic cluster formation and rapid proliferation at
3 weeks after EBV infection (Fig.
1B). At 5 weeks, EBV-infected HT (HT/EBV) cells expressed and
maintained EBV-related mRNA and proteins (EBNA1, EBNA2, LMP1 and
LMP2A) and levels similar to the protein expression observed in
B95-8 cells (Fig. 1C).
EBV-infected HT cells also exhibited drug resistance to several
approved drugs, including cyclophosphamide, doxorubicin and
bortezomib (Fig. 1D). HT/EBV cells
upregulated the expression of MDR1, MRP1, MRP2 and stem cell
markers, including OCT4 and SOX2 (Fig.
1E). These results suggest that HT/EBV cells display relatively
high drug resistance compared with non-infected HT cells.
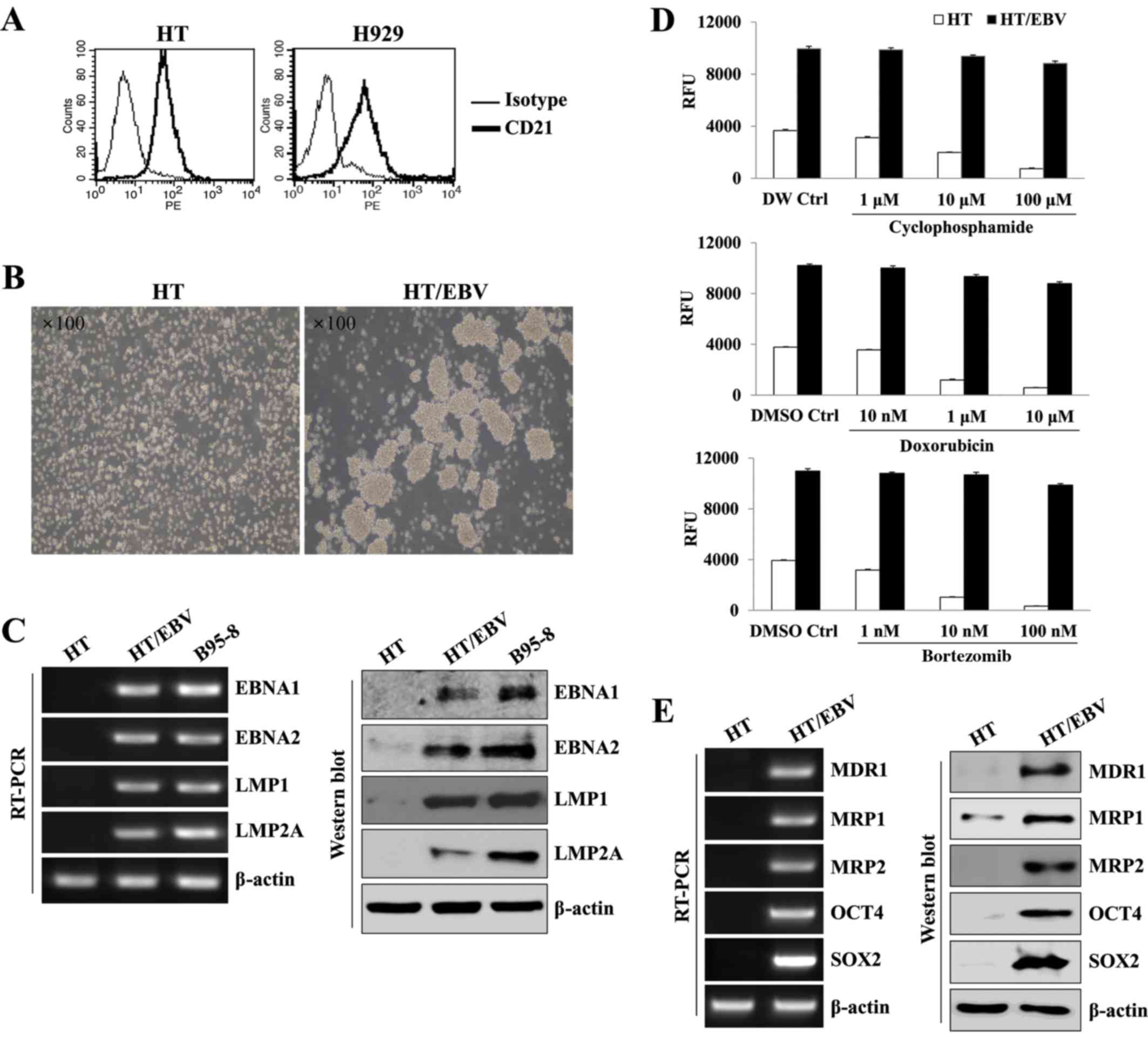 | Figure 1EBV provides DLBCL cells with
multidrug resistance. (A) CD21 surface expression on HT and H929
cell lines was analyzed by flow cytometry. The thin line represents
isotype control, and the thick line represents CD21 antigen. (B)
Phase-contrast images of HT cells with or without EBV. EBV-infected
HT cells showed rapid changes in morphology over 3 weeks, resulting
in sphere-like clumps. (C) The left and right panels show RT-PCR
and western blot analysis of EBV-related gene expression in
EBV-infected and uninfected HT cells. B95-8 cells were used as a
positive control. (D) HT or HT/EBV cells (5×104
cells/well) were cultured in 96-well plates and treated with the
indicated concentration of cyclophosphamide, doxorubicin, or
bortezomib for 24 h. Cell proliferation was determined by
AlamarBlue assays. RFU signifies relative fluorescence units. (E)
The left and right panels show increased expression of drug
resistance markers, including MDR1, MRP1, MRP2, SOX2 and OCT4, at
both the mRNA and protein levels, respectively. The data are
representative of three independent experiments. |
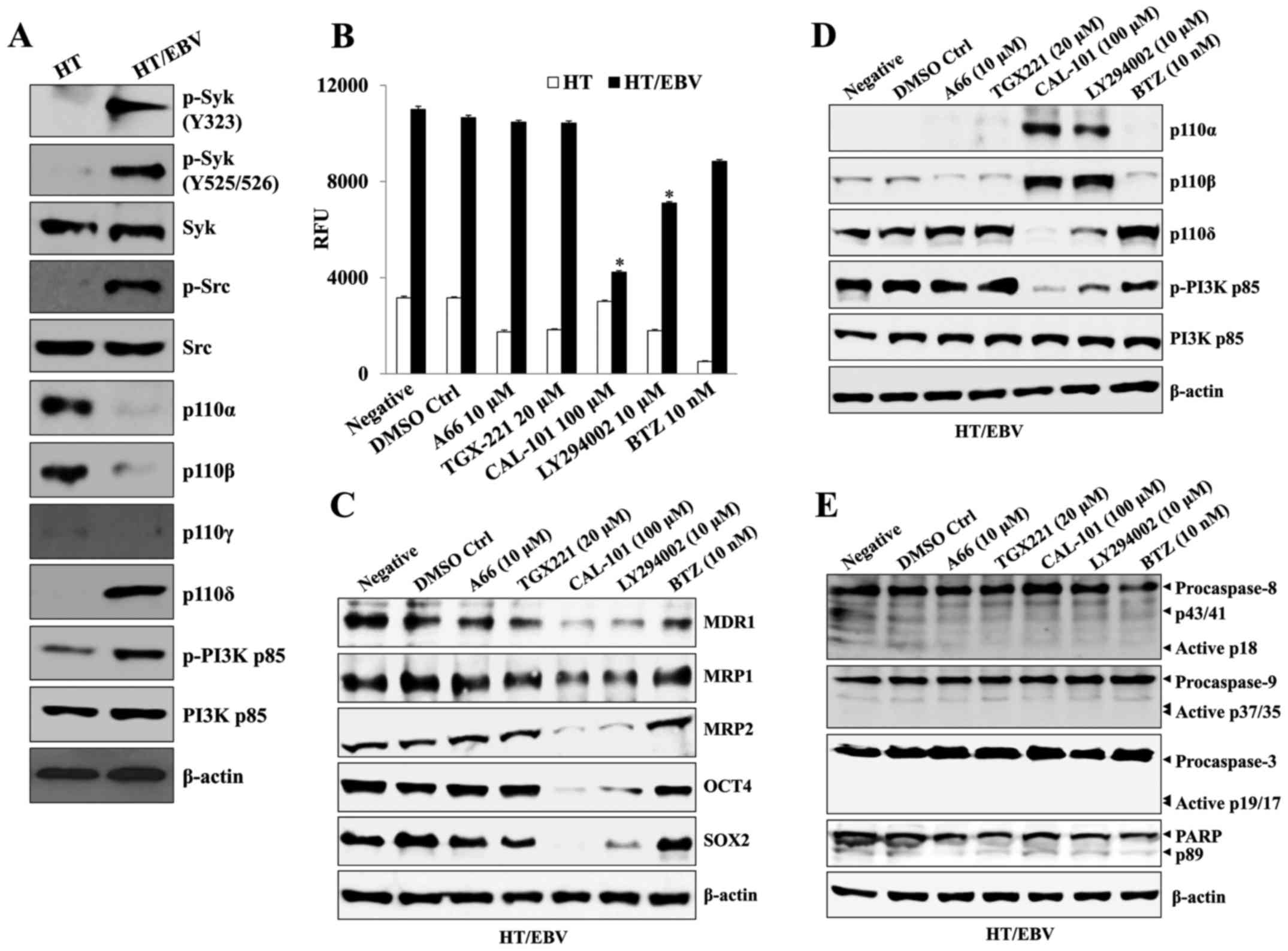
Syk/Src-mediated p110δ/Akt pathway
induces drug resistance in HT/EBV cells
Next, we investigated which p110 isoform is
associated with the generation of chemoresistant cells after EBV
infection using each inhibitor of the p110 isoform of PI3K.
EBV-infected HT cells had enhanced expression of phosphorylated
Syk/Src kinase and p110δ compared to that of non-infected HT cells,
whereas p110α and p110β activation were significantly inhibited
after EBV infection (Fig. 2A).
Although treatment with bortezomib resulted in the most effective
anti-proliferative action against non-infected HT cells (Fig. 2B), specific inhibitors of p110α and
p110β also significantly inhibited the proliferation of
non-infected HT cells (Fig. 2B).
Whereas, A66 (p110α inhibitor) and TGX-221 (p110β inhibitor) had no
effect on the proliferation rate of HT/EBV cells (Fig. 2B), but CAL-101 (a specific
inhibitor of p110δ) or LY294002 (p110α/β/δ inhibitor) significantly
reduced the growth rate of HT/EBV cells (Fig. 2B). The treatment with CAL-101 or
LY294002 of HT/EBV cells suppressed the appearance of drug
resistance-related proteins (Fig.
2C) and re-induced the expression of p110α and p110β, whereas
the level of p110δ was suppressed (Fig. 2D). However, treatment with high
doses of CAL-101 or bortezomib alone had no effect on the cleavage
of caspase-9 and -3 in H929/EBV cells (Fig. 2E). These results suggest that the
activation of the p110δ isotype of PI3K results in drug resistance
and the expression of p110δ has a critical role in the development
of drug-resistant cancer cells after EBV infection.
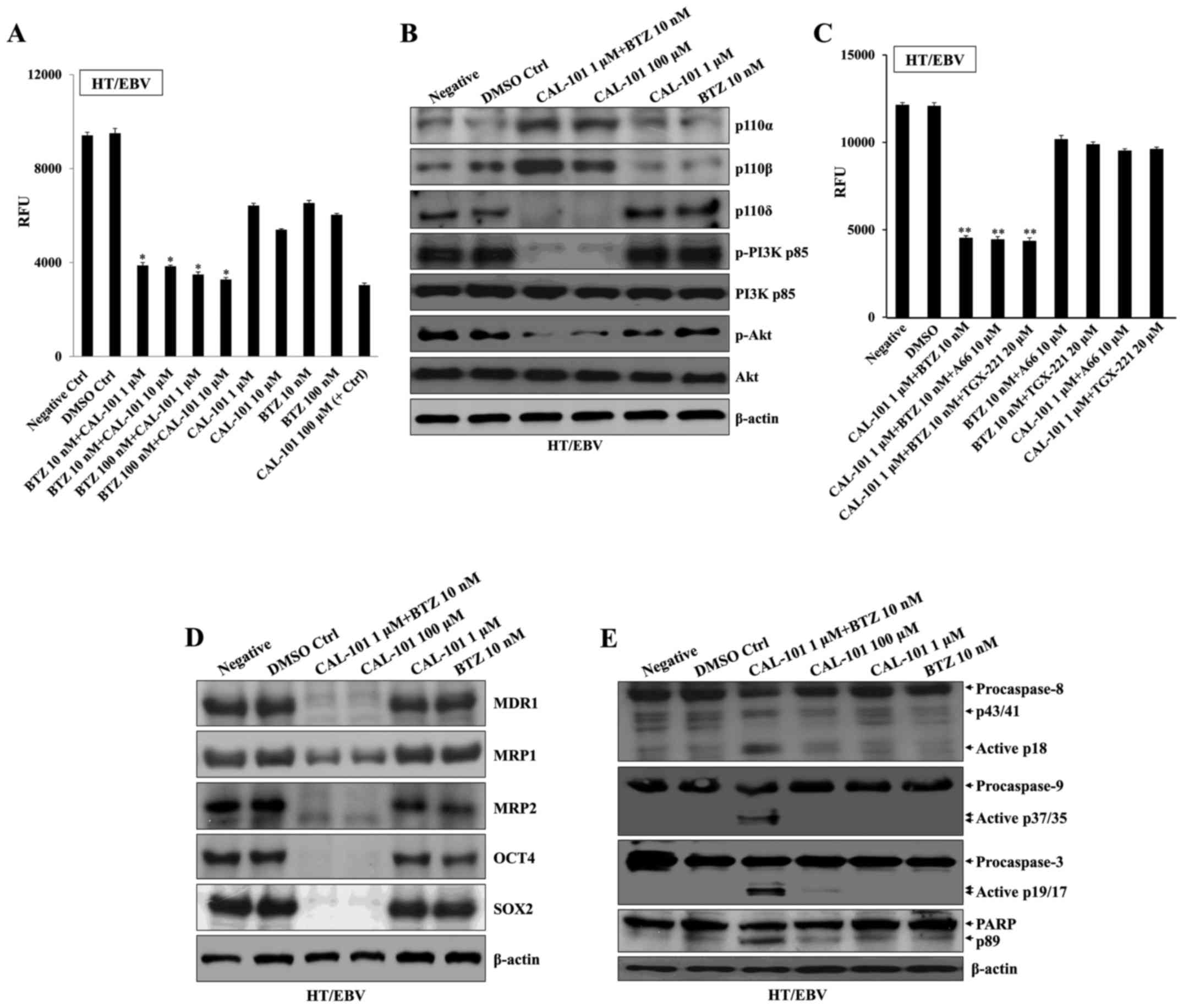
Combination of a p110δ inhibitor with
bortezomib induces cell death and reverses the characteristics of
HT/EBV cells
Next, we examined whether CAL-101 combined with
bortezomib prevents proliferation and induces cell death of HT/EBV
cells. Co-treatment of HT/EBV cells with CAL-101 (1 µM) and
bortezomib (10 nM) effectively inhibited proliferation compared
with high doses of CAL-101 (100 µM) or bortezomib (100 nM)
alone (Fig. 3A). CAL-101 combined
with bortezomib prominently restored the expression of p110α and
p110β as well as suppression of p110δ activation in HT/EBV cells
(Fig. 3B). However, additional
combination with A66 or TGX-221 failed to further inhibit the
proliferation of HT/EBV cells (Fig.
3C). The expression of MDR1, MRP1, MRP2 and stem cell markers
in H929/EBV cells was completely reversed after treatment with
CAL-101 and bortezomib (Fig. 3D).
In addition, combining CAL-101 with bortezomib activated caspase-9
and -3 synergistically and resulted in cleaved PARP, the target of
activated caspase-3 (Fig. 3E).
These results suggest that CAL-101 and bortezomib synergistically
converted the intractable DLBCL infected by EBV into treatable
DLBCL cells.
Combining CAL-101 and bortezomib reverses
drug resistance through blockage of NF-κB activation
B-cell-related cancer cells promote downstream
signaling through the PI3K and NF-κB pathways for survival and
proliferation (26,27). Inhibition of canonical NF-κB
signaling leads to apoptosis and suppression of OCT4 expression
(28). We next investigated
whether co-treatment with CAL-101 and bortezomib affects the
expression of SOX2 and OCT4 via regulation of NF-κB activity.
Treatment with an NF-κB inhibitor (Bay-11-7082) resulted in
inhibition of activation and nuclear translocation of key
components p50, p52 and Rel-B of NF-κB (Fig. 4A and B) in both the cytosol and
nuclear fractions of HT/EBV cells and reduced the expression of
SOX2 and OCT4 (Fig. 4C). To
confirm the direct involvement of OCT4 and SOX2 in drug resistance
of HT/EBV cells, OCT4 and SOX2 were silenced with siRNA (Fig. 4D), followed by treatment with
CAL-101 and bortezomib in HT/EBV cells. Gene silencing of HT/EBV
cells with OCT4 siRNA or SOX2 siRNA effectively suppressed
proliferation and increased the sensitivity to bortezomib and
co-treatment with CAL-101 and bortezomib (Fig. 4E). In addition, combinational
treatment with CAL-101 and bortezomib synergistically attenuated
NF-κB activation through inhibition of both canonical (p50) and
non-canonical (p65) pathways in H929/EBV cells (Fig. 4F). These results suggest that the
p110δ PI3K/NF-κB signaling pathway regulates drug resistance in
EBV-infected DLBCL cells.
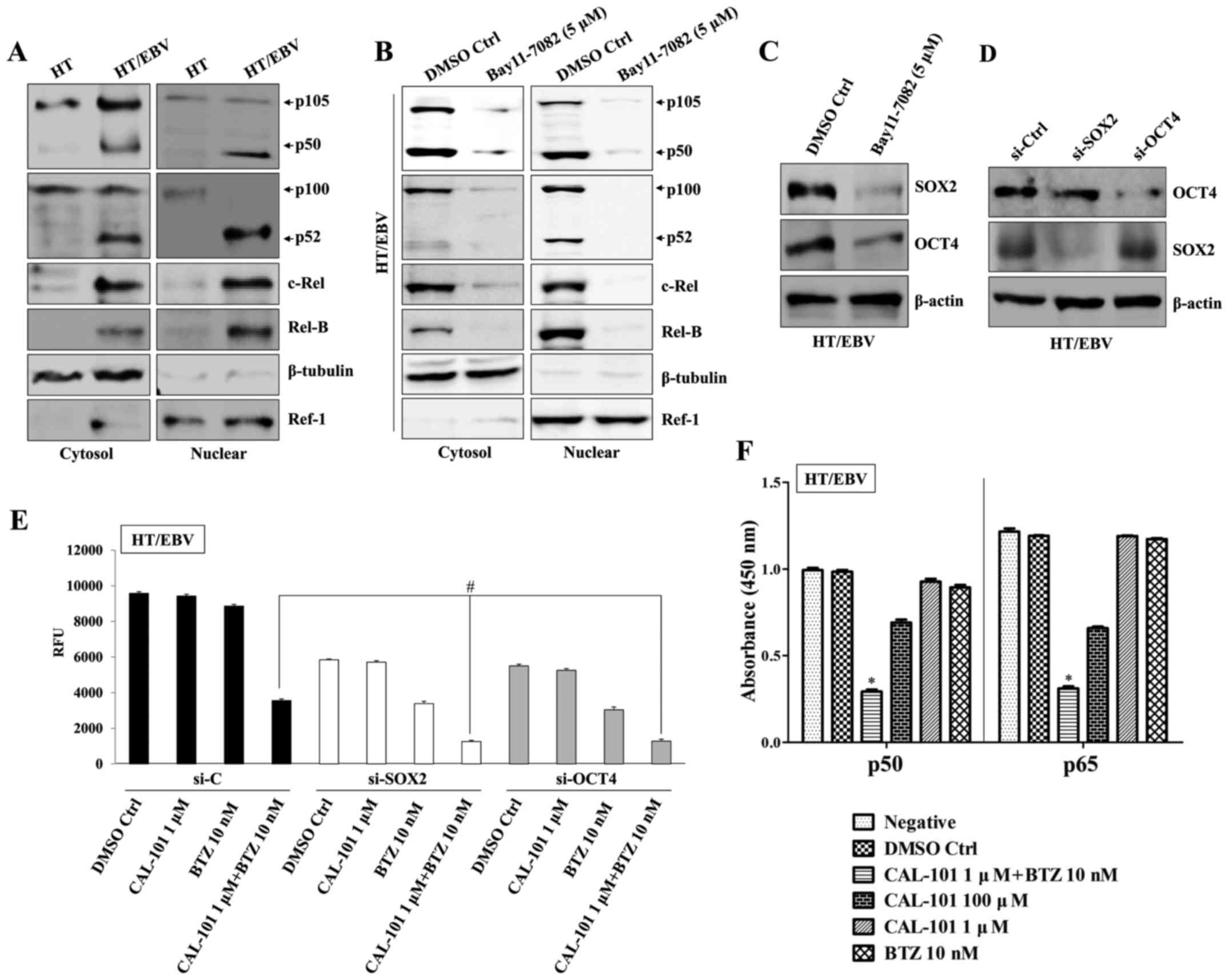 | Figure 4Combined treatment with CAL-101 and
bortezomib reverses drug resistance by blocking NF-κB activation.
(A) Cytosolic extracts (left panel) or nuclear extracts (right
panel) of HT and HT/EBV cells were analyzed by western blots using
Abs against p105/p50, p100/p52, Rel-B and c-Rel. A nuclear marker,
Ref-1 and cytosol marker, β-tubulin, were used to verify the purity
of each fraction. (B) HT/EBV cells (2.5×105 cells/ml)
were treated with the NF-κB inhibitor Bay11-7082 (5 µM) for
8 h at 37°C. (B) Cells were then washed with PBS and continuously
cultured for 24 h. Cells were harvested, and the NF-κB levels in
the cytosol and nuclear fractions were determined by western blot
analyses. (C) Western blot analysis with antibodies against SOX2
and OCT4. (D) Western blot showing specific knockdown of SOX2 or
OCT4. (E) HT/EBV cells (5×104 cells/well) were cultured
in 96-well plates, transfected with SOX2-, OCT4-, or control-siRNA
for 24 h, and then treated with the indicated drug combinations for
an additional 24 h. Cell proliferation was determined by AlamarBlue
assays. RFU signifies relative fluorescence unit.
#P<0.01. (F) HT/EBV cells (2.5×105
cells/ml) were treated with the indicated drugs for 24 h. ELISA was
used to measure NF-κB DNA-binding activity in nuclear extracts.
Transcription factors NF-κB p50 and p65 (from kits) served as
positive controls of NF-κB activity. ELISA results are expressed as
relative absorbance. *P<0.005. The data are
representative of three independent experiments. BTZ,
bortezomib. |
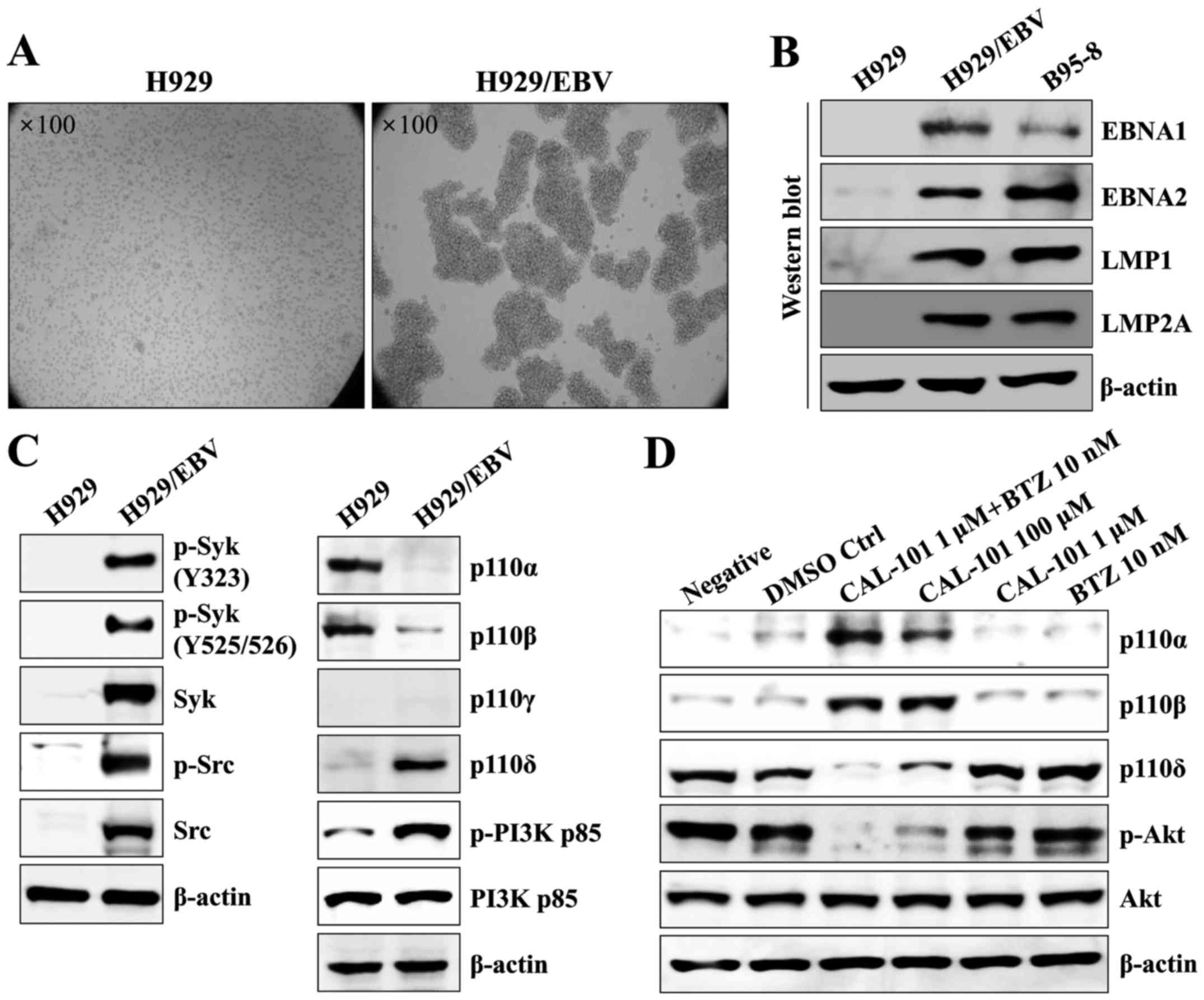
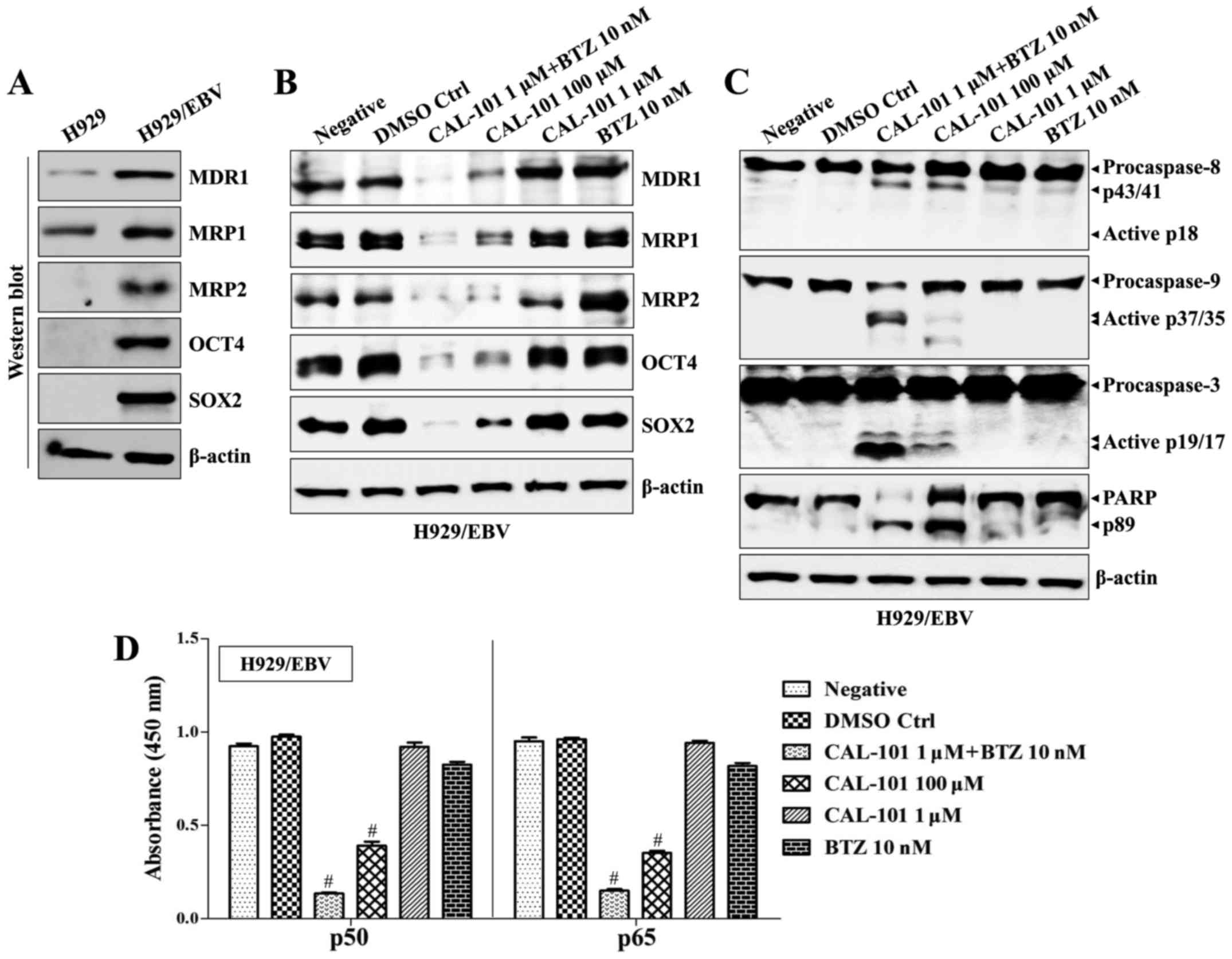
Suppression of p110δ PI3K and NF-κB
activity attenuates drug resistance of EBV-positive MM cells
The EBV genome was also detected in MM (29–31).
Furthermore, SOX2 is expressed not only in monoclonal gammopathy of
undetermined significance (MGUS), but also in symptomatic MM
(11). We next investigated
whether co-treatment with CAL-101 and bortezomib blocked the
development of drug resistance and generation of stem cell
characteristics in EBV-positive MM cells. The H929 cell line, an
EBV-negative MM cell line, was infected with EBV, and EBV-infected
H929 (H929/EBV) cells generated clumps and showed enhanced
proliferation (Fig. 5A).
EBV-related mRNAs and proteins were detected in H929/EBV cells at
similar levels to those of B95-8 cells (Fig. 5B). Although the expression of
p110α/p110β was suppressed, the p110δ isoform of PI3K increased
along with upregulation of phosphorylated Syk/Src kinase in
H929/EBV cells (Fig. 5C).
Treatment with CAL-101 and bortezomib efficiently blocked the
elevation of p110δ and restored the expression of p110α/p110β in
H929/EBV cells (Fig. 5D). Although
H929/EBV cells increased the expression of MDR1, MRP1, MRP2, OCT4
and SOX2 (Fig. 6A), a combination
of CAL-101 with bortezomib synergistically suppressed the
expression of drug resistance-related proteins and stem cell
markers (Fig. 6B) and enhanced the
expression of cleaved caspases compared with the group treated with
single drugs (Fig. 6C) in H929/EBV
cells. Furthermore, co-treatment with CAL-101 and bortezomib of
H929/EBV cells blocked the activation of canonical and
non-canonical NF-κB (Fig. 6D).
These results indicate that co-treatment with CAL-101 and
bortezomib has a suppressive effect against EBV-positive MM
cells.
Discussion
Although EBV genome-carrying cells represent a small
fraction of the total population in various EBV-related cancers
(23,25,45),
EBV infection resulted in drug resistance in DLBCL and MM cells in
the present study. Class I PI3K enzymes expressed in B cells are
heterodimeric complexes composed of regulatory (p85) and catalytic
(p110α, p110β, p110δ or p110γ) subunits (32). While the p110α and p110β catalytic
isoforms are ubiquitously expressed, the p110δ and p110γ isoforms
are largely restricted to leukocytes (33,34).
The PIK3CA gene, which encodes p110α, is mutated in many
cancers, whereas PI3K/AKT mutations are rarely found, especially in
B cell malignancies and multiple myeloma (35–37).
Although resistance to chemotherapy is acquired through a variety
of mechanisms, including activation of key pro-survival signaling
molecules, such as PI3K and NF-κB activation (38), the mechanisms underlying aberrant
expression and activity of p110δ in EBV-infected cancer cells
remain unclear. From these results, we hypothesize that
co-treatment with CAL-101 and bortezomib might reduce the
resistance to anticancer drugs by synergistic suppression of NF-κB
activity. In this study, EBV-infected cancer cells upregulated
Syk/Src-dependent p110δ PI3K activation, leading to promotion of
the expression of drug resistance-related proteins, including MDR1,
MRP1 and MRP2. A combination of CAL-101 with bortezomib attenuated
chemoresistance and the expression of stem cell markers by blocking
the nuclear translocation and accumulation of NF-κB in EBV-infected
DLBCL and MM cells. These results suggest that the regulation of
p110δ/Akt-mediated NF-κB pathway plays a critical role in the
generation of EBV-induced chemoresistant cancer cells.
EBV-positive DLBCL in the elderly accounts for 8–10%
of DLBCL among Asian patients (39). EBV-positive cells are characterized
by postgerminal center B-cell phenotypes and prominent NF-κB
activation (8). The prognosis of
EBNA2-expressing EBV-positive DLBCL patients is significantly worse
compared with EBV-negative cases (40). Most studies have also shown that
the outcome of elderly patients with EBV-positive DLBCL treated
with drug combinations (CHOP) in the presence of rituximab (R-CHOP)
is worse than that of patients who are EBV-negative under the same
treatment (41–43). MRP1 has been identified in cell
lines showing the typical multidrug resistance phenotypes, but
without elevated expression of MDR1 (P-glycoprotein) (17). Expression of MDR1 has a significant
effect on response to chemotherapy and prognosis in AIDS-related
NHL (18). Since MDR1 and MRP1 are
key efflux routes of anthracycline, which is one of the key drugs
for treatment of DLBCL, we investigated whether blocking p110δ
activation has an effect on the expression of drug
resistance-related proteins in EBV-infected cancer cells. Treatment
with high doses of CAL-101 or LY294002 significantly suppressed
proliferation and expression of MDR1, MRP1 and MRP2 in EBV-infected
B cell cancer cells. These results suggest that activation of p110δ
plays an important role in development of drug resistance induced
by EBV infection.
Bortezomib is widely used to treat both newly
diagnosed MM and relapsed or refractory MM (44,45).
However, bortezomib treatment appears to generate very
short-duration responses and results in rapid development of drug
resistance (46). Although
bortezomib, a proteasome inhibitor, has been found to induce
apoptosis in EBV lymphoblastoid cell lines through suppression of
canonical and non-canonical activity of NF-κB (25), the effects of bortezomib on the
generation of drug resistance in EBV-infected B cell cancer remain
controversial. Inhibition of NF-κB activation results in apoptosis
and suppression of OCT4 expression in human embryonic stem cells
(28). In this study, treatment
with bortezomib alone had no effect on levels of SOX2 and OCT4 in
EBV-infected DLBCL and MM cells. Notably, our results showed that
CAL-101, in combination with bortezomib, suppressed expression of
stem cell markers and induced cleavage of caspase-3 via inhibition
of NF-κB activation in EBV-infected DLBCL and MM cells. Based on
these results, we confirmed that p110δ/Akt-mediated NF-κB
activation is an essential pathway in the generation of EBV-related
cancer stem cells, and these cells might be converted into
drug-sensitive cancer cells after treatment with a combination of
CAL-101 and bortezomib.
Multiple myeloma (MM) is characterized by
uncontrolled proliferation of plasma cells (PCs) in bone marrow
(47). Despite the development of
novel targeted drugs, including proteasome inhibitors (bortezomib)
and immunomodulatory drugs (thalidomide and lenalidomide), the
incidence of MM is expected to increase due to aging populations
(48,49). In addition, MM is considered
treatable, but not curable, even after bone marrow transplantation
(50,51), because this disease eventually
results in relapse and becomes hopeless due to the development of
high malignancy and resistance to first-line anti-MM drugs
(52,53). However, the molecular
characteristics of drug-resistant refractory MM and the specific
method to convert intractable MM to a treatable condition are still
unclear and undefined. Although there is still controversy about MM
cancer stem cells and drug-resistant condition, we found that
p110δ-dependent NF-κB activation in EBV-infected MM cells is
responsible for the development of stem cell characteristics and
generation of drug resistance. Our results suggest that the
combination of a specific p110δ inhibitor and bortezomib may
convert incurable refractory hematologic disease into a manageable
condition and that this combination can be used to treat DLBCL in
elderly patients and relapsed drug-resistant MM-expressing cancer
stem cell markers.
Acknowledgments
This study was supported by the Basic Science
Research Program of Ministry of Education (no.
NRF-2015R1D1A1A01056672) and the Ministry of Science, ICT and
Future Planning (NRF-2015R1C1A2A01053732) through the National
Research Foundation (NRF) of Republic of Korea.
References
|
1
|
Lenz G and Staudt LM: Aggressive
lymphomas. N Engl J Med. 362:1417–1429. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Klein U and Dalla-Favera R: Germinal
centres: Role in B-cell physiology and malignancy. Nat Rev Immunol.
8:22–33. 2008. View
Article : Google Scholar
|
|
3
|
Andreadis C, Gimotty PA, Wahl P, Hammond
R, Houldsworth J, Schuster SJ and Rebbeck TR: Members of the
glutathione and ABC-transporter families are associated with
clinical outcome in patients with diffuse large B-cell lymphoma.
Blood. 109:3409–3416. 2007. View Article : Google Scholar
|
|
4
|
Sehn LH and Gascoyne RD: Diffuse large
B-cell lymphoma: Optimizing outcome in the context of clinical and
biologic heterogeneity. Blood. 125:22–32. 2015. View Article : Google Scholar
|
|
5
|
Maurer MJ, Ghesquières H, Jais JP, Witzig
TE, Haioun C, Thompson CA, Delarue R, Micallef IN, Peyrade F, Macon
WR, et al: Event-free survival at 24 months is a robust end point
for disease-related outcome in diffuse large B-cell lymphoma
treated with immunochemotherapy. J Clin Oncol. 32:1066–1073. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Küppers R: B cells under influence:
Transformation of B cells by Epstein-Barr virus. Nat Rev Immunol.
3:801–812. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ok CY, Papathomas TG, Medeiros LJ and
Young KH: EBV-positive diffuse large B-cell lymphoma of the
elderly. Blood. 122:328–340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Montes-Moreno S, Odqvist L, Diaz-Perez JA,
Lopez AB, de Villambrosía SG, Mazorra F, Castillo ME, Lopez M,
Pajares R, García JF, et al: EBV-positive diffuse large B-cell
lymphoma of the elderly is an aggressive post-germinal center
B-cell neoplasm characterized by prominent nuclear factor-κB
activation. Mod Pathol. 25:968–982. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roberts ML and Cooper NR: Activation of a
ras-MAPK-dependent pathway by Epstein-Barr virus latent membrane
protein 1 is essential for cellular transformation. Virology.
240:93–99. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dawson CW, Tramountanis G, Eliopoulos AG
and Young LS: Epstein-Barr virus latent membrane protein 1 (LMP1)
activates the phosphatidylinositol 3-kinase/Akt pathway to promote
cell survival and induce actin filament remodeling. J Biol Chem.
278:3694–3704. 2003. View Article : Google Scholar
|
|
11
|
Kondo S, Wakisaka N, Muramatsu M, Zen Y,
Endo K, Murono S, Sugimoto H, Yamaoka S, Pagano JS and Yoshizaki T:
Epstein-Barr virus latent membrane protein 1 induces cancer
stem/progenitor-like cells in nasopharyngeal epithelial cell lines.
J Virol. 85:11255–11264. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mo W and Zhang JT: Human ABCG2: Structure,
function, and its role in multidrug resistance. Int J Biochem Mol
Biol. 3:1–27. 2012.PubMed/NCBI
|
|
13
|
Ambudkar SV, Dey S, Hrycyna CA,
Ramachandra M, Pastan I and Gottesman MM: Biochemical, cellular,
and pharmacological aspects of the multidrug transporter. Annu Rev
Pharmacol Toxicol. 39:361–398. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ueda K: ABC proteins protect the human
body and maintain optimal health. Biosci Biotechnol Biochem.
75:401–409. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ohsawa M, Ikura Y, Fukushima H, Shirai N,
Sugama Y, Suekane T, Hirayama M, Hino M and Ueda M:
Immunohistochemical expression of multidrug resistance proteins as
a predictor of poor response to chemotherapy and prognosis in
patients with nodal diffuse large B-cell lymphoma. Oncology.
68:422–431. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kurosaki T, Takata M, Yamanashi Y, Inazu
T, Taniguchi T, Yamamoto T and Yamamura H: Syk activation by the
Src-family tyrosine kinase in the B cell receptor signaling. J Exp
Med. 179:1725–1729. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Beitz LO, Fruman DA, Kurosaki T, Cantley
LC and Scharenberg AM: SYK is upstream of phosphoinositide 3-kinase
in B cell receptor signaling. J Biol Chem. 274:32662–32666. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fujimoto M, Fujimoto Y, Poe JC, Jansen PJ,
Lowell CA, DeFranco AL and Tedder TF: CD19 regulates Src family
protein tyrosine kinase activation in B lymphocytes through
processive amplification. Immunity. 13:47–57. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Herman SE, Gordon AL, Wagner AJ, Heerema
NA, Zhao W, Flynn JM, Jones J, Andritsos L, Puri KD, Lannutti BJ,
et al: Phosphatidylinositol 3-kinase-δ inhibitor CAL-101 shows
promising preclinical activity in chronic lymphocytic leukemia by
antagonizing intrinsic and extrinsic cellular survival signals.
Blood. 116:2078–2088. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ringshausen I, Schneller F, Bogner C, Hipp
S, Duyster J, Peschel C and Decker T: Constitutively activated
phosphati-dylinositol-3 kinase (PI-3K) is involved in the defect of
apoptosis in B-CLL: Association with protein kinase Cdelta. Blood.
100:3741–3748. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hofmann C, Stühmer T, Schmiedl N, Wetzker
R, Mottok A, Rosenwald A, Langer C, Zovko J, Chatterjee M, Einsele
H, et al: PI3K-dependent multiple myeloma cell survival is mediated
by the PIK3CA isoform. Br J Haematol. 166:529–539. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ikeda H, Hideshima T, Fulciniti M, Perrone
G, Miura N, Yasui H, Okawa Y, Kiziltepe T, Santo L, Vallet S, et
al: PI3K/p110{delta} is a novel therapeutic target in multiple
myeloma. Blood. 116:1460–1468. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fruman DA and Cantley LC: Idelalisib: a
PI3Kδ inhibitor for B-cell cancers. N Engl J Med. 370:1061–1062.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Witzig TE, Reeder CB, LaPlant BR, Gupta M,
Johnston PB, Micallef IN, Porrata LF, Ansell SM, Colgan JP,
Jacobsen ED, et al: A phase II trial of the oral mTOR inhibitor
everolimus in relapsed aggressive lymphoma. Leukemia. 25:341–347.
2011. View Article : Google Scholar :
|
|
25
|
Zou P, Kawada J, Pesnicak L and Cohen JI:
Bortezomib induces apoptosis of Epstein-Barr virus
(EBV)-transformed B cells and prolongs survival of mice inoculated
with EBV-transformed B cells. J Virol. 81:10029–10036. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen L, Monti S, Juszczynski P, Daley J,
Chen W, Witzig TE, Habermann TM, Kutok JL and Shipp MA:
SYK-dependent tonic B-cell receptor signaling is a rational
treatment target in diffuse large B-cell lymphoma. Blood.
111:2230–2237. 2008. View Article : Google Scholar
|
|
27
|
Chen L, Monti S, Juszczynski P, Ouyang J,
Chapuy B, Neuberg D, Doench JG, Bogusz AM, Habermann TM, Dogan A,
et al: SYK inhibition modulates distinct PI3K/AKT-dependent
survival pathways and cholesterol biosynthesis in diffuse large B
cell lymphomas. Cancer Cell. 23:826–838. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang C, Atkinson SP, Vilella F, Lloret M,
Armstrong L, Mann DA and Lako M: Opposing putative roles for
canonical and noncanonical NF-κB signaling on the survival,
proliferation, and differentiation potential of human embryonic
stem cells. Stem Cells. 28:1970–1980. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ancín I, Sarrá J, Peris J, Romagosa V,
Domingo-Claros A and Grañena A: Demonstration of Epstein-Barr virus
in a case of multiple myeloma after renal transplantation.
Haematologica. 85:773–774. 2000.PubMed/NCBI
|
|
30
|
Tcheng WY, Said J, Hall T, Al-Akash S,
Malogolowkin M and Feig SA: Post-transplant multiple myeloma in a
pediatric renal transplant patient. Pediatr Blood Cancer.
47:218–223. 2006. View Article : Google Scholar
|
|
31
|
Voelkerding KV, Sandhaus LM, Kim HC,
Wilson J, Chittenden T, Levine AJ and Raska K Jr: Plasma cell
malignancy in the acquired immune deficiency syndrome. Association
with Epstein-Barr virus. Am J Clin Pathol. 92:222–228. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deane JA and Fruman DA: Phosphoinositide
3-kinase: Diverse roles in immune cell activation. Annu Rev
Immunol. 22:563–598. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chantry D, Vojtek A, Kashishian A,
Holtzman DA, Wood C, Gray PW, Cooper JA and Hoekstra MF: p110delta,
a novel phosphatidylinositol 3-kinase catalytic subunit that
associates with p85 and is expressed predominantly in leukocytes. J
Biol Chem. 272:19236–19241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vanhaesebroeck B, Welham MJ, Kotani K,
Stein R, Warne PH, Zvelebil MJ, Higashi K, Volinia S, Downward J
and Waterfield MD: P110delta, a novel phosphoinositide 3-kinase in
leukocytes. Proc Natl Acad Sci USA. 94:4330–4335. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ismail SI, Mahmoud IS, Msallam MM and
Sughayer MA: Hotspot mutations of PIK3CA and AKT1 genes are absent
in multiple myeloma. Leuk Res. 34:824–826. 2010. View Article : Google Scholar
|
|
36
|
Leupin N, Cenni B, Novak U, Hügli B,
Graber HU, Tobler A and Fey MF: Disparate expression of the PTEN
gene: A novel finding in B-cell chronic lymphocytic leukaemia
(B-CLL). Br J Haematol. 121:97–100. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Georgakis GV, Li Y, Rassidakis GZ,
Medeiros LJ, Mills GB and Younes A: Inhibition of the
phosphatidylinositol-3 kinase/Akt promotes G1 cell cycle arrest and
apoptosis in Hodgkin lymphoma. Br J Haematol. 132:503–511.
2006.PubMed/NCBI
|
|
38
|
Shostak K and Chariot A: NF-κB, stem cells
and breast cancer: The links get stronger. Breast Cancer Res.
13:2142011. View Article : Google Scholar
|
|
39
|
Adam P, Bonzheim I, Fend F and
Quintanilla-Martínez L: Epstein-Barr virus-positive diffuse large
B-cell lymphomas of the elderly. Adv Anat Pathol. 18:349–355. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hoeller S, Tzankov A, Pileri SA, Went P
and Dirnhofer S: Epstein-Barr virus-positive diffuse large B-cell
lymphoma in elderly patients is rare in Western populations. Hum
Pathol. 41:352–357. 2010. View Article : Google Scholar
|
|
41
|
Park S, Lee J, Ko YH, Han A, Jun HJ, Lee
SC, Hwang IG, Park YH, Ahn JS, Jung CW, et al: The impact of
Epstein-Barr virus status on clinical outcome in diffuse large
B-cell lymphoma. Blood. 110:972–978. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sato A, Nakamura N, Kojima M, Ohmachi K,
Carreras J, Kikuti YY, Numata H, Ohgiya D, Tazume K, Amaki J, et
al: Clinical outcome of Epstein-Barr virus-positive diffuse large
B-cell lymphoma of the elderly in the rituximab era. Cancer Sci.
105:1170–1175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hong JY, Yoon DH, Suh C, Huh J, Do IG,
Sohn I, Jo J, Jung SH, Hong ME, Yoon H, et al: EBV-positive diffuse
large B-cell lymphoma in young adults: Is this a distinct disease
entity? Ann Oncol. 26:548–555. 2015. View Article : Google Scholar
|
|
44
|
Richardson PG, Sonneveld P, Schuster MW,
Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D,
Lonial S, Goldschmidt H, et al Assessment of Proteasome Inhibition
for Extending Remissions (APEX) Investigators: Bortezomib or
high-dose dexamethasone for relapsed multiple myeloma. N Engl J
Med. 352:2487–2498. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
San Miguel JF, Schlag R, Khuageva NK,
Dimopoulos MA, Shpilberg O, Kropff M, Spicka I, Petrucci MT,
Palumbo A, Samoilova OS, et al VISTA Trial Investigators:
Bortezomib plus melphalan and prednisone for initial treatment of
multiple myeloma. N Engl J Med. 359:906–917. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kumar S and Rajkumar SV: Many facets of
bortezomib resistance/susceptibility. Blood. 112:2177–2178. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kuehl WM and Bergsagel PL: Multiple
myeloma: Evolving genetic events and host interactions. Nat Rev
Cancer. 2:175–187. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
48
|
Morgan GJ, Walker BA and Davies FE: The
genetic architecture of multiple myeloma. Nat Rev Cancer.
12:335–348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Palumbo A and Anderson K: Multiple
myeloma. N Engl J Med. 364:1046–1060. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Rajkumar SV and Buadi F: Multiple myeloma:
New staging systems for diagnosis, prognosis and response
evaluation. Best Pract Res Clin Haematol. 20:665–680. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kyle RA and Rajkumar SV: Treatment of
multiple myeloma: A comprehensive review. Clin Lymphoma Myeloma.
9:278–288. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Matsui W, Wang Q, Barber JP, Brennan S,
Smith BD, Borrello I, McNiece I, Lin L, Ambinder RF, Peacock C, et
al: Clonogenic multiple myeloma progenitors, stem cell properties,
and drug resistance. Cancer Res. 68:190–197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Matsui W, Huff CA, Wang Q, Malehorn MT,
Barber J, Tanhehco Y, Smith BD, Civin CI and Jones RJ:
Characterization of clonogenic multiple myeloma cells. Blood.
103:2332–2336. 2004. View Article : Google Scholar
|