Introduction
Malignant melanoma (MM) and osteosarcoma (OS) are
the representatives of aggressive tumors that are highly resistant
to multidisciplinary treatment including chemo-, radio-, and
immunotherapy (1,2). Apo2 ligand/tumor necrosis
factor-related apoptosis-inducing ligand (Apo2L/TRAIL) is a member
of the tumor necrosis factor superfamily. It has emerged as a
promising cancer-selective anticancer drug since it exhibits potent
cytotoxicity toward various cancer cell types with minimal
cytotoxicity toward normal cells (3–5).
Binding of TRAIL to two death receptors (DRs), TRAIL receptor
(TRAIL-R)1/DR4 and TRAIL-R2/DR5 triggers the extrinsic and
intrinsic apoptotic pathways (6,7). It
also triggers pathways leading to other modes of cell death such as
autophagy (8,9) and necroptosis (10,11).
However, MM and OS are resistant to TRAIL-induced cytotoxicity,
despite expressing DRs. In addition to their inherent resistance,
the acquired resistance of MM and OS cells to the drug dampens
TRAIL treatment (12).
Consequently, the combined application of medicines that enable to
reduce the TRAIL resistance is necessary for effective TRAIL
therapy of these cancers.
Ca2+ regulates many complicated cellular
processes such as cell activation, proliferation, and death.
Recently, Ca2+ is emerging as a new target for cancer
treatment. Various cancer cell types exhibit tumor-specific traits
in Ca2+ dynamics, which contribute to tumorigenesis,
malignant phenotypes, drug resistance, increased proliferation, and
survival (13–15). Growing body of evidence suggests
that a variety of Ca2+-permeable channels regulate
Ca2+ remodeling and survival in cancer cells (16). However, Ca2+ promotes
not only survival but also different modalities of cell death
including apoptosis, necrosis, autophagy, and anoikis in cancer
cells (17). Intracellular
Ca2+ overload was early thought to be a critical
mediator of necrotic cell death by leading to the increase in the
permeability of the mitochondrial membrane (mitochondrial
permeability transition) and the resulting dysfunction.
Ca2+/calpain, an intracellular Ca2+-dependent
cysteine protease, is activated by the rise in the cytosolic
Ca2+ concentration ([Ca2+]cyt) and
critically involved in cancer cell apoptosis through the processing
of the mitochondria-localized pro-apoptotic molecule,
apoptosis-inducing factor (18,19).
An excess, persistent rise in mitochondrial Ca2+
concentration ([Ca2+]mit) increases the
permeability of the inner membrane, thereby leading to release of
pro-apoptotic proteins, the collapse of mitochondrial integrity,
and activation of the intrinsic apoptotic pathway. Although the
dual effect of Ca2+ remodeling is thought to be due to
the differences in the magnitude, timing, duration, and the space
of the Ca2+ surge generated (13), at present, no model can depict the
dual role for Ca2+ remodeling. Thus, drugs targeting
overall Ca2+ signals may modulate both pro-death and
pro-survival pathways non-specifically, thereby compromising the
antitumor effect. Therefore, it is necessary to characterize the
cellular parameters and machinery that decide the two types of
Ca2+ signal and minimize the onset of the pro-survival
Ca2+ pathway by the therapy. To date, Ca2+
remodeling in melanoma and osteosarcoma is poorly characterized,
and the role for Ca2+ in their malignant phenotypes and
survival remains unclear.
In this study, we analyzed the impact of TRAIL on
Ca2+ remodeling in MM and OS cells and the possible role
of Ca2+ in their survival and TRAIL resistance. The
results showed that acute TRAIL treatment modulates Ca2+
dynamics and that Ca2+ protects these tumor cells to
TRAIL-induced apoptotic and non-apoptotic cell death. We also found
that Ca2+ remodeling in the mitochondria through
mitochondrial uniporter (MCU), mitochondrial permeability
transition pore (MPTP), and a Ca2+ transport pathway
sensitive to capsazepine and AMG9810 play a vital role in the
protection. The findings suggest that mitochondrial Ca2+
removal facilitates non-apoptotic cell death induction by TRAIL and
may have therapeutic potential in the treatment of these
TRAIL-resistant cancers.
Materials and methods
Materials
Soluble recombinant human TRAIL was obtained from
Enzo Life Sciences (San Diego, CA, USA). AMG9810, capsazepine,
CGP-37157, atractyloside, thapsigargin (Tg), necrostatin-1, and the
pan-caspase-inhibitor z-VAD-fluorometheylketone (z-VAD-FMK) were
obtained from Sigma-Aldrich (St. Louis, MO, USA). All insoluble
reagents were dissolved in dimethylsulfoxide and diluted with high
glucose-containing Dulbecco's modified Eagle's medium
(Sigma-Aldrich) supplemented with 10% fetal bovine serum
(Sigma-Aldrich; FBS/DMEM) or Hank's balanced salt solution (HBSS)
(pH 7.4) to a final concentration of <0.1% before use.
Cell culture
Human MM (A375, A2058) and OS (MG63, SAOS-2, HOS)
cell lines were obtained from Health Science Research Resource Bank
(Osaka, Japan) and cultured in FBS/DMEM in a 5% CO2
incubator. Cells were harvested by incubating with 0.25%
trypsin-EDTA (Thermo Fisher Scientific, Rochester, NY, USA) for 5
min at 37°C.
Cell growth and apoptosis
measurements
Cell growth was measured by WST-8 assay using the
Cell Counting Kit-8 (Dojindo, Kumamoto, Japan), a colorimetric
assay based on the formation of a water-soluble formazan product as
previously described (20) with
minor modifications. Briefly, cells (8×103/well) were
seeded in 96-well plates and cultured with the agents to be tested
for 72 h at 37°C in a 5% CO2 incubator. Then 1/10 volume
of WST-8 reagent was added, incubated for 1 h at 37°C and
absorbance at 450 nm was measured using a microplate reader (ARVO
MX, Perkin-Elmer Japan, Tokyo, Japan). Apoptotic cell death was
quantitatively assessed by double-staining with fluorescein
isothiocyanate (FITC)-conjugated Annexin V and propidium iodide
(PI) as previously described (21). Briefly, cells
(2×105/well) in 24-well plates were incubated with the
agents to be tested for 24 h in 10% FBS-containing medium at 37°C.
The cells were then stained with FITC-conjugated Annexin V and PI
using a commercially available kit (Annexin V FITC Apoptosis
Detection kit I; BD Biosciences, Tokyo, Japan). The stained cells
were evaluated in the FACSCalibur and analyzed using CellQuest
software (BD Biosciences). Four cellular subpopulations were
assessed: viable cells (Annexin V−/PI−);
early apoptotic cells (Annexin V+/PI-); late apoptotic cells
(Annexin V+/PI+); and necrotic/damaged cells
(Annexin V−/PI+). Annexin V+ cells
were considered to be apoptotic cells.
Ca2+ measurements
Changes in [Ca2+]cyt and
[Ca2+]mit were measured using the cytosol
Ca2+-reactive fluorescence probe Fluo 4-AM and
mitochondrial Ca2+-reactive fluorescence probe rhod 2-AM
(both were obtained from Dojindo), respectively as previously
described (22). To improve its
mitochondrial localization, rhod 2-AM was reduced to the colorless,
nonfluorescent dihydrorhod 2-AM by sodium borohydride, according to
the manufacturer's protocol. Cells were loaded with 4 μM
each of Fluo 4-AM or dihydrorhod 2-AM for 40 min at 37°C and washed
with HBSS. Then, the cells (1×106/ml) were resuspended
in HBSS in 96-well plates. The cells were manually added with the
agents to be tested. Then, the cells were measured for fluorescence
at 5 sec intervals up to 3 min in a microplate reader (Fluoroskan
Ascent, Thermo Fisher Scientific) with excitation and emission at
485 and 538 nm (for Fluo 4-AM), respectively and 542 and 592 nm
(for rhod 2-AM), respectively. For Ca2+-independent
experiments, cells were suspended in HBSS supplemented with 1 mM
EGTA in place of 1 mM CaCl2.
Caspase-3/7 activation, membrane
integrity, and cell death assay
Caspase-3/7 activation, membrane integrity, and cell
death were simultaneously measured by Muse™ Cell Analyzer (Merck
Millipore, Darmstadt, Germany) using Muse Caspase-3/7 kit. Briefly,
cells (1×105/ml) in 24-well plates were treated with the
agents to be tested for 24 h in 10% FBS/DMEM at 37°C and then
stained with a novel Caspase-3/7 reagent NucView™ and
7-amino-actinomycin D (7-AAD), a dead cell marker in the kit. 7-AAD
is excluded from healthy and early apoptotic cells, while permeates
late apoptotic and dead cells. Consequently, four populations of
cells can be distinguished by the kit; Live cells:
Caspase−/7-AAD−; early apoptotic cells:
Caspase+/7-AAD−; late apoptotic/dead cells:
Caspase+/7-AAD+; necrotic cells:
Caspase−/7-AAD+.
Statistical analysis
Data were analyzed by one-way analysis of variance
(ANOVA) followed by the Tukey's post-hoc test using an add-in
software for Excel 2016 for Windows (SSRI, Tokyo, Japan). All
values are expressed as means ± SD, and P<0.05 was considered to
be significant.
Results
Analyses of Ca2+ dynamics in
MM and OS cells stimulated with TRAIL
To determine the impact of TRAIL on Ca2+
dynamics in tumor cells, we measured the effect of TRAIL on
[Ca2+]cyt and [Ca2+]mit
in osteosarcoma cells in parallel. Treatment with soluble
recombinant human TRAIL resulted in a robust increase in
[Ca2+]cyt in HOS cells in a dose-dependent
manner (Fig. 1A and B). The
increase occurred rapidly (within minutes) and persistently (lasted
at least for 10 min). TRAIL at concentrations of ≥50 ng/ml had a
significant effect in parallel with the cytotoxic effect. In
parallel, [Ca2+]mit was increased in a
dose-dependent manner (Fig. 1C and
D). Depending on the cellular conditions,
[Ca2+]mit was elevated maximally at 50 ng/ml,
and higher concentrations of TRAIL had a smaller effect. We
observed similar results in an array of MM and OS cells including
SAOS-2, MG63, A375 and A2058 cells (not shown).
 | Figure 1TRAIL modulates Ca2+
dynamics in malignant cells. HOS cells were loaded with 4 μM
Fluo 4-AM (A and B) and dihydrorhod 2-AM (C and D), respectively,
for 40 min at 37°C, washed with HBSS. The dye-loaded cells
(1×106/ml) were resuspended in the
Ca2+-containing medium in 96-well plates. After addition
of 25, 50, or 100 ng/ml TRAIL to the cells, fluorescence was
immediately measured in triplicate in a microplate reader at 0,
1,2,3,5,10 min with excitation and emission at 485 and 538 nm (for
Fluo 4-AM), respectively and 542 and 592 nm (for dihydrorhod 2-AM),
respectively. The data show means ± SD in a representative
experiment (N=3). (B and D) Data were analyzed by ANOVA followed by
the Tukey's post-hoc test. *P<0.05;
**P<0.01; ***P<0.001 vs. control
(CTRL). The number of parentheses represents the data analyzed. |
The MCU inhibitor Ruthenium 360 (Ru360)
suppresses mitochondrial Ca2+ load in MM and OS
cells
MCU is a major molecular machinery responsible for
the physiological Ca2+ load into the mitochondrial
matrix (23). The role of MCU in
mitochondrial Ca2+ remodeling has been studied in few
tumor cells including breast cancer cells (24) and neuroblastoma cells (25). Since the role of MCU in
mitochondrial Ca2+ remodeling in MM and OS cells is
unknown, we examined the impact of MCU-specific agents on their
mitochondrial Ca2+ dynamics. The MCU inhibitor Ru360
caused a significant decrease in [Ca2+]mit,
while the mitochondrial Na+-Ca2+ exchanger
(NCLX) inhibitor CGP-37157 increased
[Ca2+]mit in HOS and SAOS-2 cells (Fig. 2A and B). EGTA and the mitochondrial
permeability transition pore (MPTP) inhibitor cyclosporine A (CysA)
significantly decreased [Ca2+]mit in HOS
cells, but not in SAOS-2 cells (Fig.
2A and B). On the other hand, atractyloside, an MPTP opener,
significantly reduced [Ca2+]mit in both MM
and OS cells (Fig. 2C and D),
indicating that Ca2+ extrusion through the MPTP
participates in regulating [Ca2+]mit.
Capsazepine and AMG9810 reduce
[Ca2+]mit cooperatively with TRAIL in MM and
OS cells
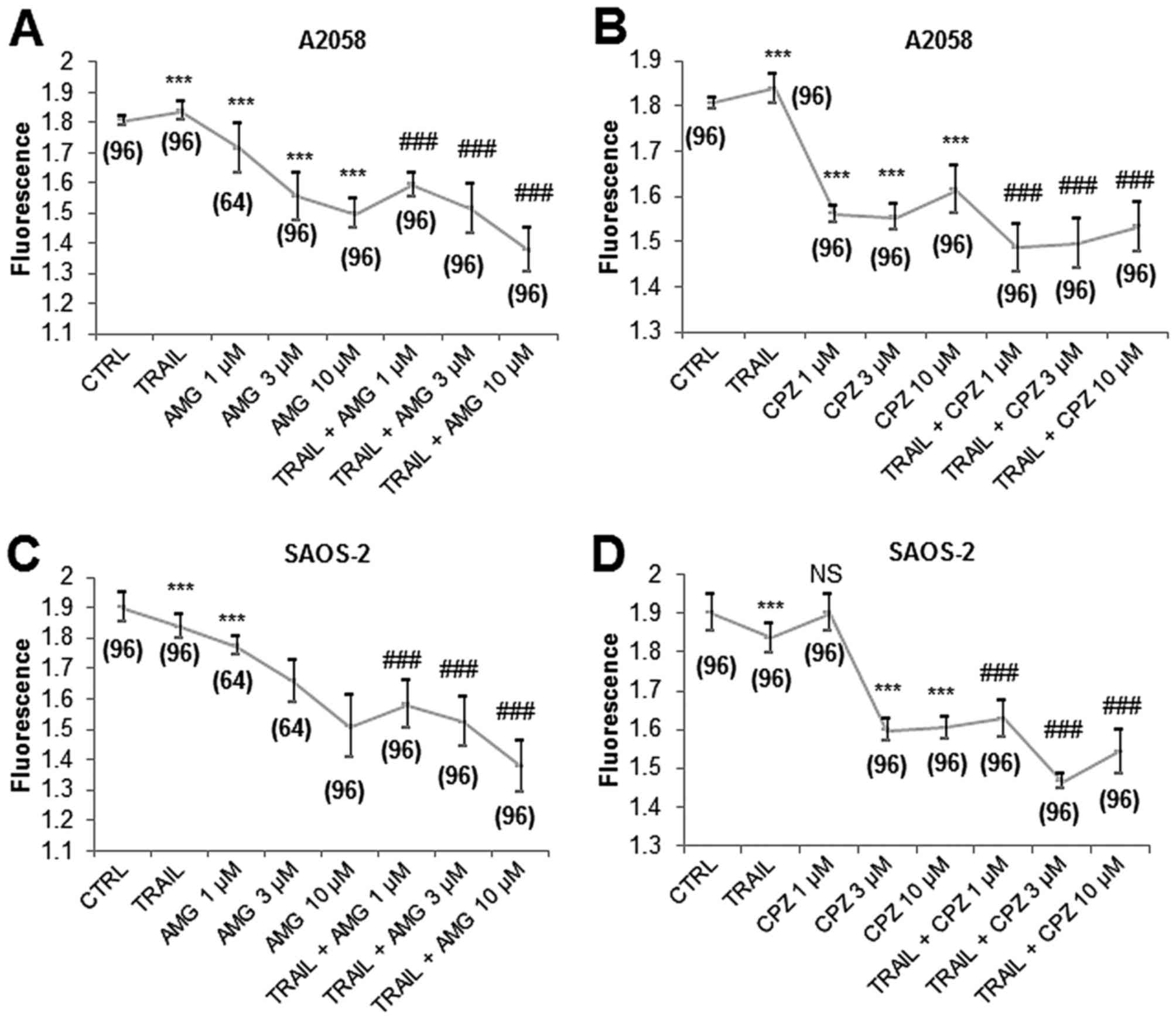
We found that capsazepine and AMG9810 modulated
mitochondrial Ca2+ dynamics in MM and OS cells. AMG9810
alone at concentrations ranging from 1 to 10 μM decreased
[Ca2+]mit in A2058 cells in a dose-dependent
manner (Fig. 3A). Capsazepine
alone reduced [Ca2+]mit maximally at 1–3
μM (Fig. 3B). When used
with TRAIL and AMG9810 together, [Ca2+]mit
was dropped to the level lower than that observed with each agent
alone (Fig. 3A). Meanwhile,
capsazepine enhanced the effects of TRAIL on
[Ca2+]mit with a maximal effect at 1–3
μM (Fig. 3B). Essentially
similar results were obtained for SAOS-2 cells (Fig. 3C and D). These results show that
capsazepine and AMG9810 reduce [Ca2+]mit
cooperatively with TRAIL in MM and OS cells.
Ca2+ removal decreases MM and
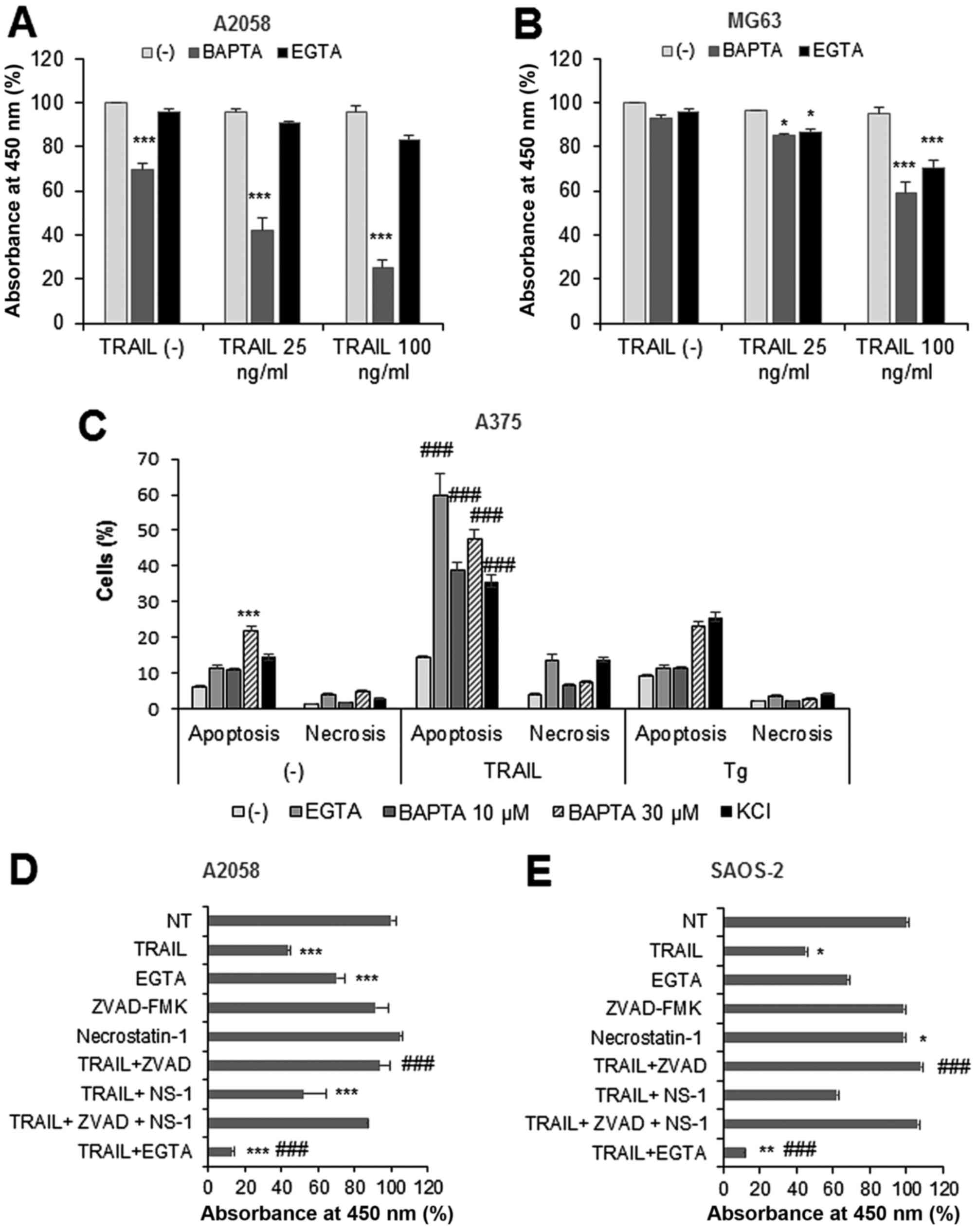
OS cell viability and potentiates TRAIL cytotoxicity
To determine the role of Ca2+ remodeling
in TRAIL cytotoxicity toward MM and OS cells, we examined the
effect of Ca2+ removal on the cytotoxicity. Treatment
with TRAIL up to 100 ng/ml for 24 h minimally decreased (4–6%
decrease) the viability of A2058 and MG63 cells. Treatment with the
intracellular Ca2+-chelator BAPTA (30 μM)
moderately decreased the viability of A2058 cells (maximum of 30%
reduction), while the extracellular Ca2+-chelator EGTA
(0.2–0.5 mM) had minimal effect (Fig.
4A), and both Ca2+-chelators decreased the viability
of MG63 cells only modestly (<10%) (Fig. 4B). BAPTA and EGTA sensitized both
cells to TRAIL, and this effect became pronounced as the
concentration was increased, although their effects varied
depending on the cell lines tested (Fig. 4A and B).
To determine the cell death modality, we performed
flow cytometric analyses using Annexin V/PI double staining.
Likewise, KCl, a potent TRAIL-sensitizer (21), BAPTA or EGTA remarkably increased
apoptotic (Annexin V+) cells compared with TRAIL or
either agent alone at 24 h (Fig.
4C). Small but significantly higher levels of necrotic (Annexin
V−/PI+) cells were observed in TRAIL +
EGTA-treated cells compared with TRAIL or EGTA alone (Fig. 4C). BAPTA and EGTA enhanced
Tg-induced apoptosis, while had no significant effect on Tg-induced
necrotic cell death. TRAIL toxicity, as well as TRAIL sensitization
by the Ca2+-chelators, became pronounced as the
incubation period was prolonged. As a result, TRAIL treatment for
72 h substantially decreased the viability of A2058 and SAOS-2
cells (56.4 and 54.8% reduction, respectively), while EGTA
treatment alone reduced them moderately (30 and 32.2% reduction).
When used together, TRAIL and EGTA considerably decreased cell
viability (maximum of 90%) (Fig. 4D
and E). The TRAIL cytotoxicity was entirely blocked by the
pan-caspase-inhibitor z-VAD-FMK, while necrostatin-1, a specific
inhibitor of necroptosis, had only a modest inhibitory effect,
indicating that the TRAIL primarily induces apoptosis in these
cells. Collectively, these results show that Ca2+
removal sensitizes MM and OS cells to TRAIL-induced apoptosis,
although the effect varied considerably depending on the cell line
tested.
Ca2+ removal sensitizes MM and
OS cells to TRAIL-induced non-apoptotic cell death
The ability of TRAIL to kill MM and OS cells varied
considerably in different experiments. Under certain conditions,
TRAIL had the minimal cytotoxic effect toward SAOS-2 and HOS cells
(Fig. 5A and B). These cells were
resistant to the cytotoxic and TRAIL-sensitizing effects of the
Ca2+ chelators. As a result, TRAIL and chelator alone or
in combination had the minimal cytotoxic effect except for that
TRAIL + EGTA significantly reduced the viability of SAOS-2 cells
(43.6% reduction). Also, z-VAD-FMK did not inhibit the effect of
TRAIL + EGTA. Treatment with Ru360 (5–30 μM) for 24 h had
the minimal cytotoxic and TRAIL-sensitizing effect (not shown).
However, during another 48 h, Ru360 alone significantly decreased
the viability of SAOS-2 and HOS cells (Fig. 5C and D). When Ru360 and TRAIL
applied together, only a small increase of cell killing was
observed compared with that induced by Ru360 alone. The cell death
induced by Ru360 (not shown) or TRAIL + Ru360 was enhanced rather
than inhibited by z-VAD-FMK (Fig. 5C
and D). Although 5 μM CysA substantially decreased the
viability of SAOS-2 cells, but not HOS cells, this cytotoxic effect
was entirely counteracted by TRAIL. Moreover, atractyloside also
enhanced TRAIL cytotoxicity in these apoptosis-resistant cells
(Fig. 5E). On the other hand,
consistent with our previous study with A375 cells (26), H2O2 markedly
sensitized the cells to TRAIL cytotoxicity, and this effect was
completely blocked by z-VAD-FMK, indicating that
H2O2 amplifies TRAIL-induced apoptosis. These
results show that agents that reduce
[Ca2+]mit with different mechanisms of action
sensitize these cells to TRAIL-induced non-apoptotic cell
death.
Capsazepine and AMG9810 kill or sensitize
MM and OS cells in a caspase-independent manner
To further explore the possible relationship between
mitochondrial Ca2+ removal and TRAIL sensitization, we
assessed the impact of capsazepine and AMG9810 (3–30 μM)
alone or in combination with TRAIL on tumor cell survival. Both
AMG9810 and capsazepine had a minimal cytotoxic effect for 24 h
(not shown). Treatment with AMG9810 at concentrations of ≥3
μM for 72 h reduced the viability of A2058 cells and at
concentrations of ≥10 μM potentiated TRAIL cytotoxicity
toward them in a dose-dependent manner (Fig. 6A). SAOS-2 cells were more resistant
to AMG9810 treatment so that only the highest concentration of the
agent exhibited substantial cytotoxic effect and enhanced TRAIL
cytotoxicity (Fig. 6C). The effect
of capsazepine seemed to be complicated and dependent on the cell
lines tested. Capsazepine (10 μM) was more cytotoxic and
more efficient in potentiating TRAIL cytotoxicity than 30 μM
capsazepine in A2058 cells (Fig.
6B) while exhibiting no significant cytotoxicity nor
TRAIL-sensitizing effect in SAOS-2 cells (Fig. 6D). Usually, HOS cells were highly
resistant to TRAIL and AMG9810 alone or in combination, and the
cytotoxic effect of TRAIL + AMG9810 was significantly augmented by
z-VAD-FMK (Fig. 6E). On the other
hand, capsazepine alone decreased their viability remarkably (82.9%
reduction), and the effect was comparable to that of TRAIL +
capsazepine. z-VAD-FMK also enhanced the cytotoxic effect of TRAIL
+ AMG9810 in SAOS-2 cells (Fig.
6F).
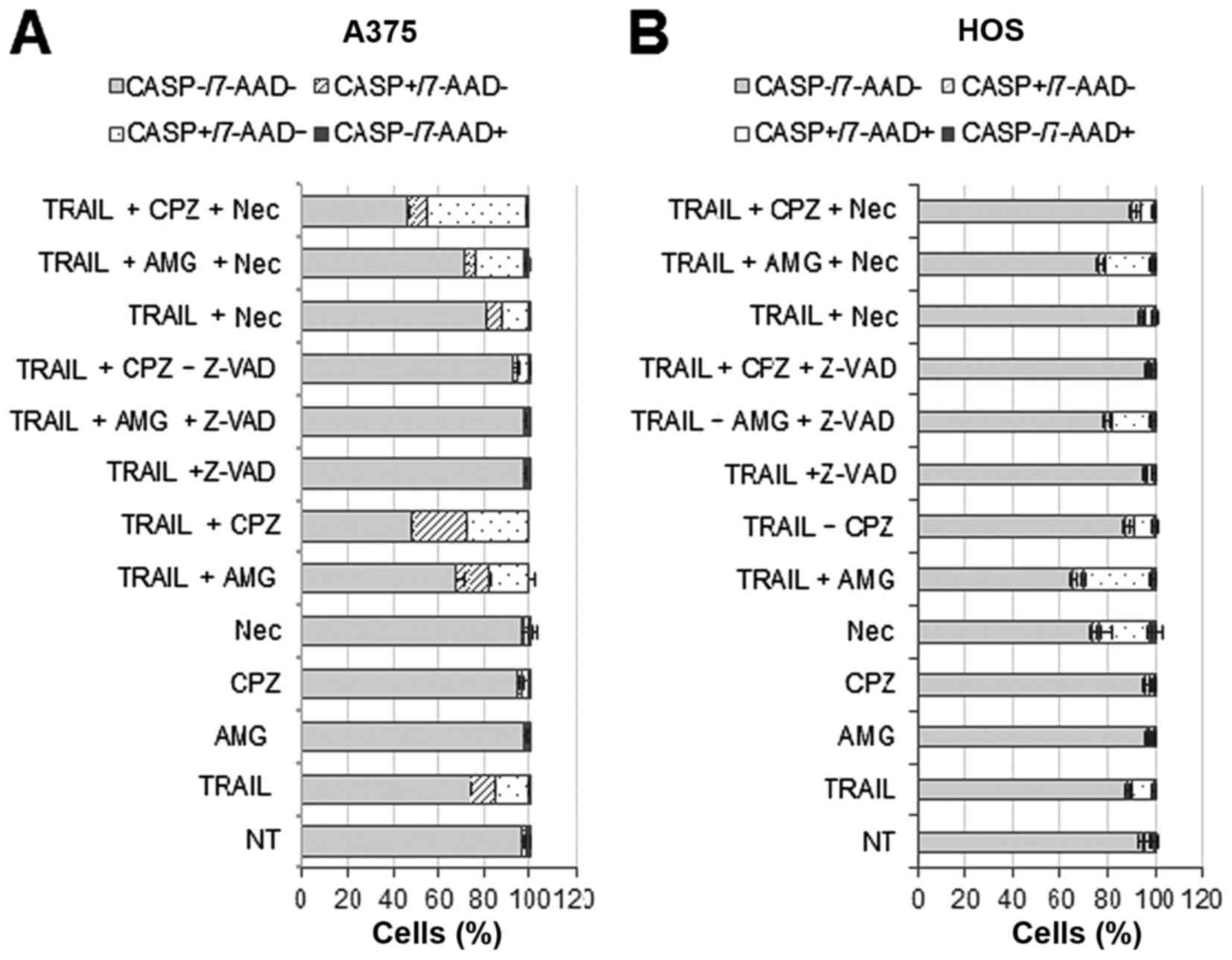
Capsazepine and AMG9810 initially amplify
TRAIL-induced caspase-3/7 activation and cell membrane damage in MM
and OS cells
The results presented so far suggested that
capsazepine and AMG9810 potentiate TRAIL cytotoxicity in a
caspase-independent manner. To determine whether these two agents
indeed modulate cell death independently of apoptosis, we examined
their effect on caspase-3/7 activation. We simultaneously assessed
caspase-3/7 activation and cell membrane damage/death by using a
caspase-3/7-specific substrate and 7-AAD, respectively. The latter
is a nucleus-staining dye, which is excluded by healthy cells,
while it can penetrate cell membranes of dying or dead cells.
Results showed that in A375 cells, capsazepine and AMG9810 alone
minimally increased caspase-3/7 activated (caspase+)
cells and damaged (7-AAD+) cells at 24 h (Fig. 7A). TRAIL treatment modestly
increased both caspase+/7-AAD− and
caspase+/7-AAD+ cells in A375 cells, and
z-VAD-FMK blocked this effect. Capsazepine, and to a lesser extent,
AMG9810 potentiated the effect of TRAIL, and z-VAD-FMK also
abrogated the amplification (Fig.
7A). Necrostatin-1 inhibited the effect of TRAIL only modestly,
while reducing the increase in
caspase+/7-AAD− cells, but not
caspase+/7-AAD+ cells by TRAIL + AMG9810.
Strikingly, necrostain-1 enhanced the increase in
caspase+/7-AAD+ cells by TRAIL + capsazepine
(Fig. 7A). On the other hand, in
HOS cells, AMG9810 was more potent than capsazepine in potentiating
the effect of TRAIL, and z-VAD-FMK blocked the effect of
capsazepine and AMG9810 (Fig. 7B).
Unlike A375 cells, necrostatin-1 alone moderately increased
caspase+/7-AAD+ cells while blunting the rise
in such cell population by TRAIL, TRAIL + AMG9810, or TRAIL +
capsazepine (Fig. 7B). These
results indicate that capsazepine and AMG9810 initially amplify
TRAIL-induced caspase-3/7 activation, cell membrane damage, and
caspase-dependent cell death depending on the cell types.
Discussion
In the present study, we analyzed the effect of
TRAIL on Ca2+ dynamics in MM and OS cells and the
possible role of Ca2+ in the control of their survival
and TRAIL sensitivity. Our results revealed that acute TRAIL
treatment modulates Ca2+ remodeling in an array of MM
and OS cell lines, as indicated by a rapid and persistent increase
in [Ca2+]cyt and
[Ca2+]mit (Fig.
1A–D). In parallel with its cytotoxicity, TRAIL increased
Ca2+ levels at the two intracellular sites in a
dose-dependent manner. The mitochondria take up or release
Ca2+ depending on [Ca2+]cyt,
thereby serving as a critical intracellular Ca2+
reservoir that maintains [Ca2+]cyt. According
to this paradigm, the increases in [Ca2+]cyt
and [Ca2+]mit may occur in parallel.
Strikingly, however, the rise in [Ca2+]cyt
was usually dose-dependent while depending on the cellular
conditions, the elevation in [Ca2+]mit was
maximum at 50 ng/ml, and higher concentrations of TRAIL had a
smaller effect. These findings indicate that the mitochondrial
Ca2+ responses involve both
[Ca2+]cyt-dependent and
[Ca2+]cyt-independent processes.
Mitochondrial Ca2+ homeostasis is maintained by a
well-balanced mitochondrial Ca2+ uptake and efflux. The
MCU complex consists of the channel-forming subunit of the
uniporter MCU and multiple components such as MICU1/2, MCUb, MCUR1,
and EMRE. These complex molecules are proven to be an essential
mitochondrial Ca2+ uptake machinery in different cell
types including cancer cells (26–28).
The mitochondrion releases Ca2+ through
several different pathways including NCLX,
Ca2+/H+ antiporter (29) and MPTP (30–33).
However, the role of MPTP is still a matter of debate, because
other observations suggest its minimal contribution to
mitochondrial Ca2+ extrusion (34). In this study, we showed that the
MCU inhibitor Ru360 decreased [Ca2+]mit,
while the NCLX antagonist CGP-37157 increased it in MM and OS cells
(Fig. 2A and B). The results
indicate that Ca2+ uptake through MCU and
Ca2+ extrusion through NCLX are key regulators of
[Ca2+]mit in our cell systems. Cyclosporine
A, which targets cyclophilin D, a critical component of the MPTP
opening (32,33), affected
[Ca2+]mit in some but not all cell types.
Whereas, atractyloside, which opens MPTP by modulating adenine
nucleotide translocator (34),
reduced [Ca2+]mit in different cell types
(Fig. 2C and D). These findings
suggest that Ca2+ extrusion through MPTP is also
necessary for the control of [Ca2+]mit, yet
cyclophilin D plays a dispensable role in the MPTP opening in our
cell systems as previously reported by other groups (35,36).
It is noteworthy that capsazepine and AMG9810
markedly reduce [Ca2+]mit and potentiate
TRAIL-induced drop in [Ca2+]mit in MM and OS
cells (Fig. 3A–D). These findings
indicate that a Ca2+ transport pathway sensitive to
these agents plays a pivotal role in regulating
[Ca2+]mit in them. The two agents are known
to act as potent antagonists of TRPV1 (37,38),
a molecule which localizes to the plasma membrane and serves as a
non-selective cation channel. Recently, TRPV1 was shown to also
exist in the ER and mitochondria in non-transformed cells and
cancer cells. The intracellular TRPV1 contributes to
Ca2+ release and ER stress (39–41).
These facts suggest a close functional relationship among this
channel, ER, and mitochondria in the regulation of Ca2+
signaling and survival of cancer cells. It is now widely accepted
that ROS and Ca2+ mutually regulate one another and
cooperatively control cell survival and death (13). Several groups, including us, have
previously demonstrated that ROS plays a critical role in TRAIL
cytotoxicity toward different malignant cell types (42–44).
Moreover, TRPV1 is one of TRP channels that are
activated by ROS (45).
Collectively, TRPV1 might play a role in the regulation of
Ca2+ dynamics, survival, and death of MM and OS cells.
However, to date, the role of TRPV1 in the control of
Ca2+ in MM and OS is poorly documented. Mergler et
al (46) reported the
expression of TRPV1 in human uveal melanoma cells and
Ca2+ regulation by it. The TRPV1 agonist capsaicin is
shown to induce an increase in [Ca2+]mit in
G292 human OS cells independently of the extracellular
Ca2+ and depletion of intracellular Ca2+
(47). Since the extracellular
Ca2+ entry appears to be dispensable for increasing
[Ca2+]mit (Fig.
2A and B), an intracellular TRPV1 might play a role in the
regulation of [Ca2+]mit homeostasis. However,
at present, we failed to detect any TRPV1 in the intracellular
sites in these cells (data not shown). Thus, the occurrence and the
role of TRPV1 remain to be studied.
Another significant finding in this study was that
depletion of Ca2+ potentiated TRAIL cytotoxicity toward
MM and OS cells (Figs. 4 and
5). The finding strongly suggests
that Ca2+ protects them from cell death. Both
intracellular and extracellular Ca2+ seemed to play a
role in this pro-survival function while the position of the two
Ca2+ varied depending on cell lines. Depletion of
Ca2+ enhanced apoptotic, but not necrotic cell death
induced by TRAIL and Tg (Fig. 4C),
indicating that Ca2+ primarily prevents apoptosis. It is
noteworthy that the effect of Ca2+ removal was more
pronounced in TRAIL-sensitive cells than in TRAIL-resistant cells
(compare Fig. 4 with Fig. 5), and that z-VAD-FMK blocked the
effect in the TRAIL-sensitive cells, but not in TRAIL-resistant
cells. Collectively, MM and OS cells may each have distinct
cellular statuse with different TRAIL sensitivity. One is
relatively TRAIL-sensitive status, where they readily undergo
apoptosis in response to TRAIL. The other is TRAIL-resistant status
where another non-apoptotic cell death is necessary for efficient
cell killing because only a small cell population undergo
apoptosis. Ca2+ removal also potentiated TRAIL-induced
non-apoptotic cell death (Fig. 5),
indicating that Ca2+ also prevents this cell death
modality.
The data presented in this study revealed the
critical role of mitochondrial Ca2+ in the prevention of
cell death. We found that the reduction in
[Ca2+]mit by inhibiting Ca2+
uptake through MCU sensitized MM and OS cells to TRAIL-induced
non-apoptotic cell death (Fig. 5).
The finding is consistent with several recent studies in other
cancer cell types. Curry and colleagues (27) reported that MCU silencing
potentiates caspase-independent cell death in MDA-MB-231 breast
cancer cells. The authors demonstrated that caspase-independent
cell death induced by the Ca2+-ionophore ionomycin is
potentiated by MCU silencing whereas caspase-dependent cell death
caused by Bcl-2 inhibition is unaffected. Moreover, the
potentiation of caspase-independent cell death occurs independently
of overall [Ca2+]cyt changes.
Marchi et al (48) reported that the in silico
microRNA miR-25 downregulates MCU expression and reduces
mitochondrial Ca2+ uptake in HeLa cells and colon cancer
cells and that this downregulation correlates with resistance to
apoptotic stimuli. In this case, this MCU manipulation does not
affect [Ca2+]cyt. Moreover, we found that the
inhibition of a Ca2+ transport pathway by capsazepine
and AMG9810 led to the decrease in [Ca2+]mit
(Fig. 3) and sensitized MM and OS
cells to TRAIL-induced non-apoptotic cell death. Strikingly,
capsazepine and AMG9810 eventually amplified cell killing in a
caspase-independent manner (Fig.
6) while initially (within 24 h) potentiated TRAIL-induced
caspase-3/7 activation and apoptosis (Fig. 8). The reduction in
[Ca2+]mit by promoting Ca2+
extrusion through MPTP also amplified TRAIL cytotoxicity (Fig. 5), providing further support for the
pro-survival role of mitochondrial Ca2+. TRAIL was
recently shown to induce necroptosis, the programmed necrotic cell
death (10,11). However, the Ca2+
modulation had the minimal effect on necrosis (Fig. 4) and necrostatin-1, a specific
inhibitor of necroptosis, had no or only a modest inhibitory effect
on the cell death induced by TRAIL (Fig. 4), TRAIL + capsazepine, and TRAIL +
AMG9810 (Fig. 7). These findings
suggest that necroptosis plays a minor role in the cell killing yet
the silencing of an essential molecular component in this cell
death modality such as receptor-interacting protein 1/3 may be
necessary to verify this view.
In conclusion, we demonstrate in this study that
mitochondrial Ca2+ acts as a pro-survival factor in MM
and OS cells by preventing apoptosis and non-apoptotic cell death.
The findings suggest that mitochondrial Ca2+ may serve
as a promising target for overcoming the resistance of these
cancers to TRAIL.
Acknowledgments
The authors thank Drs T. Ito, T. Tokunaga, and A.
Onoe for their technical assistance. This work was supported in
part by JSPS KAKENHI grant no. 15K09750 to Y.S-K.).
References
|
1
|
Ivanov VN, Bhoumik A and Ronai Z: Death
receptors and melanoma resistance to apoptosis. Oncogene.
22:3152–3161. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Guiho R, Biteau K, Heymann D and Redini F:
TRAIL-based therapy in pediatric bone tumors: How to overcome
resistance. Future Oncol. 11:535–542. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ashkenazi A: Targeting the extrinsic
apoptosis pathway in cancer. Cytokine Growth Factor Rev.
19:325–331. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Amarante-Mendes GP and Griffith TS:
Therapeutic applications of TRAIL receptor agonists in cancer and
beyond. Pharmacol Ther. 155:117–131. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
de Miguel D, Lemke J, Anel A, Walczak H
and Martinez-Lostao L: Onto better TRAILs for cancer treatment.
Cell Death Differ. 23:733–747. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang S: The promise of cancer therapeutics
targeting the TNF-related apoptosis-inducing ligand and TRAIL
receptor pathway. Oncogene. 27:6207–6215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sayers TJ: Targeting the extrinsic
apoptosis signaling pathway for cancer therapy. Cancer Immunol
Immunother. 60:1173–1180. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Herrero-Martín G, Høyer-Hansen M,
García-García C, Fumarola C, Farkas T, López-Rivas A and Jäättelä
M: TAK1 activates AMPK-dependent cytoprotective autophagy in
TRAIL-treated epithelial cells. EMBO J. 28:677–685. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He W, Wang Q, Xu J, Xu X, Padilla MT, Ren
G, Gou X and Lin Y: Attenuation of TNFSF10/TRAIL-induced apoptosis
by an autophagic survival pathway involving TRAF2- and
RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy. 8:1811–1821.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jouan-Lanhouet S, Arshad MI,
Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van
Herreweghe F, Takahashi N, Sergent O, Lagadic-Gossmann D,
Vandenabeele P, et al: TRAIL induces necroptosis involving
RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ.
19:2003–2014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sosna J, Philipp S, Fuchslocher Chico J,
Saggau C, Fritsch J, Föll A, Plenge J, Arenz C, Pinkert T, Kalthoff
H, et al: Differences and similarities in TRAIL- and tumor necrosis
factor-mediated necroptotic signaling in cancer cells. Mol Cell
Biol. 36:2626–2644. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dimberg LY, Anderson CK, Camidge R,
Behbakht K, Thorburn A and Ford HL: On the TRAIL to successful
cancer therapy? Predicting and counteracting resistance against
TRAIL-based therapeutics. Oncogene. 32:1341–1350. 2013. View Article : Google Scholar
|
|
13
|
Hempel N and Trebak M: Crosstalk between
calcium and reactive oxygen species signaling in cancer. Cell
Calcium. Jan 18–2017.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Villalobos C, Sobradillo D,
Hernández-Morales M and Núñez L: Calcium remodeling in colorectal
cancer. Biochim Biophys Acta. 1864:843–849. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Danese A, Patergnani S, Bonora M,
Wieckowski MR, Previati M, Giorgi C and Pinton P: Calcium regulates
cell death in cancer: Roles of the mitochondria and
mitochondria-associated membranes (MAMs). Biochim Biophys Acta. Jan
10–2017.Epub ahead of print. View Article : Google Scholar
|
|
16
|
Nilius B and Szallasi A: Transient
receptor potential channels as drug targets: From the science of
basic research to the art of medicine. Pharmacol Rev. 66:676–814.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Orrenius S, Gogvadze V and Zhivotovsky B:
Calcium and mitochondria in the regulation of cell death. Biochem
Biophys Res Commun. 460:72–81. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Storr SJ, Carragher NO, Frame MC, Parr T
and Martin SG: The calpain system and cancer. Nat Rev Cancer.
11:364–374. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Moretti D, Del Bello B, Allavena G and
Maellaro E: Calpains and cancer: Friends or enemies? Arch Biochem
Biophys. 564:26–36. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Akita M, Suzuki-Karasaki M, Fujiwara K,
Nakagawa C, Soma M, Yoshida Y, Ochiai T, Tokuhashi Y and
Suzuki-Karasaki Y: Mitochondrial division inhibitor-1 induces
mitochondrial hyperfusion and sensitizes human cancer cells to
TRAIL-induced apoptosis. Int J Oncol. 45:1901–1912. 2014.PubMed/NCBI
|
|
21
|
Suzuki Y, Inoue T, Murai M,
Suzuki-Karasaki M, Ochiai T and Ra C: Depolarization potentiates
TRAIL-induced apoptosis in human melanoma cells: Role for
ATP-sensitive K+ channels and endoplasmic reticulum
stress. Int J Oncol. 41:465–475. 2012.PubMed/NCBI
|
|
22
|
Suzuki Y, Yoshimaru T, Inoue T and Ra C:
Mitochondrial Ca2+ flux is a critical determinant of the
Ca2+ dependence of mast cell degranulation. J Leukoc
Biol. 79:508–518. 2006. View Article : Google Scholar
|
|
23
|
Marchi S and Pinton P: The mitochondrial
calcium uniporter complex: Molecular components, structure and
physiopathological implications. J Physiol. 592:829–839. 2014.
View Article : Google Scholar :
|
|
24
|
Tosatto A, Sommaggio R, Kummerow C,
Bentham RB, Blacker TS, Berecz T, Duchen MR, Rosato A, Bogeski I,
Szabadkai G, et al: The mitochondrial calcium uniporter regulates
breast cancer progression via HIF-1α. EMBO Mol Med. 8:569–585.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang X, Wang B, Zeng H, Cai C, Hu Q, Cai
S, Xu L, Meng X and Zou F: Role of the mitochondrial
Ca2+ uniporter in
Pb2+-induced oxidative stress in human
neuroblastoma cells. Brain Res. 1575:12–21. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tochigi M, Inoue T, Suzuki-Karasaki M,
Ochiai T, Ra C and Suzuki-Karasaki Y: Hydrogen peroxide induces
cell death in human TRAIL-resistant melanoma through intracellular
superoxide generation. Int J Oncol. 42:863–872. 2013.PubMed/NCBI
|
|
27
|
Curry MC, Peters AA, Kenny PA,
Roberts-Thomson SJ and Monteith GR: Mitochondrial calcium uniporter
silencing potentiates caspase-independent cell death in MDA-MB-231
breast cancer cells. Biochem Biophys Res Commun. 434:695–700. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
De Stefani D, Patron M and Rizzuto R:
Structure and function of the mitochondrial calcium uniporter
complex. Biochim Biophys Acta. 1853:2006–2011. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brand MD: Electroneutral efflux of
Ca2+ from liver mitochondria. Biochem J. 225:413–419.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Altschuld RA, Hohl CM, Castillo LC, Garleb
AA, Starling RC and Brierley GP: Cyclosporin inhibits mitochondrial
calcium efflux in isolated adult rat ventricular cardiomyocytes. Am
J Physiol. 262:H1699–H1704. 1992.PubMed/NCBI
|
|
31
|
Bernardi P and von Stockum S: The
permeability transition pore as a Ca2+ release channel:
New answers to an old question. Cell Calcium. 52:22–27. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Di Lisa F, Carpi A, Giorgio V and Bernardi
P: The mitochondrial permeability transition pore and cyclophilin D
in cardioprotection. Biochim Biophys Acta. 1813:1316–1322. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gutiérrez-Aguilar M and Baines CP:
Structural mechanisms of cyclophilin D-dependent control of the
mitochondrial permeability transition pore. Biochim Biophys Acta.
1850:2041–2047. 2015. View Article : Google Scholar :
|
|
34
|
Klingenberg M: The ADP and ATP transport
in mitochondria and its carrier. Biochim Biophys Acta.
1778:1978–2021. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zamorano S, Rojas-Rivera D, Lisbona F,
Parra V, Court FA, Villegas R, Cheng EH, Korsmeyer SJ, Lavandero S
and Hetz C: A BAX/BAK and cyclophilin D-independent intrinsic
apoptosis pathway. PLoS One. 7:e377822012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Briston T, Lewis S, Koglin M, Mistry K,
Shen Y, Hartopp N, Katsumata R, Fukumoto H, Duchen MR, Szabadkai G,
et al: Identification of ER-000444793, a Cyclophilin D-independent
inhibitor of mitochondrial permeability transition, using a
high-throughput screen in cryopreserved mitochondria. Sci Rep.
6:377982016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bevan S, Hothi S, Hughes G, James IF, Rang
HP, Shah K, Walpole CS and Yeats JC: Capsazepine: A competitive
antagonist of the sensory neurone excitant capsaicin. Br J
Pharmacol. 107:544–552. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gavva NR, Tamir R, Qu Y, Klionsky L, Zhang
TJ, Immke D, Wang J, Zhu D, Vanderah TW, Porreca F, et al: AMG 9810
[(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4]
dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1)
antagonist with antihyperalgesic properties. J Pharmacol Exp Ther.
313:474–484. 2005. View Article : Google Scholar
|
|
39
|
Zhao R and Tsang SY: Versatile roles of
intracellularly located TRPV1 channel. J Cell Physiol.
232:1957–1965. 2017. View Article : Google Scholar
|
|
40
|
Thomas KC, Roberts JK, Deering-Rice CE,
Romero EG, Dull RO, Lee J, Yost GS and Reilly CA: Contributions of
TRPV1, endovanilloids, and endoplasmic reticulum stress in lung
cell death in vitro and lung injury. Am J Physiol Lung Cell Mol
Physiol. 302:L111–L119. 2012. View Article : Google Scholar :
|
|
41
|
Stock K, Kumar J, Synowitz M, Petrosino S,
Imperatore R, Smith ES, Wend P, Purfürst B, Nuber UA, Gurok U, et
al: Neural precursor cells induce cell death of high-grade
astrocytomas through stimulation of TRPV1. Nat Med. 18:1232–1238.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mellier G and Pervaiz S: The three Rs
along the TRAIL: Resistance, re-sensitization and reactive oxygen
species (ROS). Free Radic Res. 46:996–1003. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Suzuki-Karasaki M, Ochiai T and
Suzuki-Karasaki Y: Crosstalk between mitochondrial ROS and
depolarization in the potentiation of TRAIL-induced apoptosis in
human tumor cells. Int J Oncol. 44:616–628. 2014.
|
|
44
|
Voltan R, Secchiero P, Casciano F, Milani
D, Zauli G and Tisato V: Redox signaling and oxidative stress:
Cross talk with TNF-related apoptosis inducing ligand activity. Int
J Biochem Cell Biol. 81:364–374. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kozai D, Ogawa N and Mori Y: Redox
regulation of transient receptor potential channels. Antioxid Redox
Signal. 21:971–986. 2014. View Article : Google Scholar
|
|
46
|
Mergler S, Derckx R, Reinach PS, Garreis
F, Böhm A, Schmelzer L, Skosyrski S, Ramesh N, Abdelmessih S, Polat
OK, et al: Calcium regulation by temperature-sensitive transient
receptor potential channels in human uveal melanoma cells. Cell
Signal. 26:56–69. 2014. View Article : Google Scholar
|
|
47
|
Chien CS, Ma KH, Lee HS, Liu PS, Li YH,
Huang YS and Chueh SH: Dual effect of capsaicin on cell death in
human osteosarcoma G292 cells. Eur J Pharmacol. 718:350–360. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Marchi S, Lupini L, Patergnani S, Rimessi
A, Missiroli S, Bonora M, Bononi A, Corrà F, Giorgi C, De Marchi E,
et al: Downregulation of the mitochondrial calcium uniporter by
cancer-related miR-25. Curr Biol. 23:58–63. 2013. View Article : Google Scholar :
|