Introduction
Head and neck squamous cell carcinomas (HNSCCs)
arise from epithelia of the oral cavity, pharynx, larynx, nasal
cavity or paranasal sinuses, and they frequently invade the
surrounding bones that comprise the facial skeleton because of the
anatomical proximity (1). The
clinical importance of bone invasion of HNSCC includes a high
morbidity rate and a worse prognosis. It is thus essential to
control bone invasion in the treatment of HNSCC (1).
The progression of HNSCC cells from epithelia to
bone induces the production of cancer-associated factors
synthesized by either HNSCC cells or stromal cells activated by
HNSCC cells (2). These factors
induce the expression of receptor activator of nuclear factor κβ
ligand (RANKL) in the osteoblasts, which results in osteoclast
activation and bone destruction. Bone destruction leads to the
release of growth factors preserved in bone, including insulin-like
growth factor-I (IGF-I) and transforming growth factor-β (TGF-β)
(3,4). These growth factors promote HNSCC
bone invasion and further increase the production of
cancer-associated factors, which triggers a vicious circle
(2).
Semaphorins are a large family of molecules
containing a cysteine-rich sema domain. They were originally shown
to regulate axonal growth cone guidance in the developing central
nervous system (5–7). To date, >30 semaphorins have been
identified and classified into eight subgroups based on their
species of origin and sequence similarity. Semaphorin receptors,
i.e., the plexins, also contain a sema domain. Nine known plexins,
i.e., Plexin-A1 to -A4, -B1 to -B3, -C1, and -D1 are grouped
according to their structure similarity. Semaphorin-plexin
interactions were initially thought to be limited to the central
nervous system, but these interactions are now implicated widely in
the non-nervous systems, i.e., the cardiovascular (8), endocrine (9), gastrointestinal (10), immune (11) and respiratory (12) systems.
Semaphorin 4D (Sema4D) is an axon guidance molecule
produced by oligodendrocytes (13). Sema4D is also expressed by
T-lymphocytes and eosinophils and functions in the immune system
(13). Sema4D was identified in
the bone microenvironment, and shown to be expressed by osteoclasts
that act through Plexin-B1 on osteoblasts to inhibit their
differentiation through inhibiting IGF-I signaling (14). Sema4D is also highly expressed in
many cancer tissues including cancer of the prostate, colon,
breast, lung and HNSCC (15).
Moreover, Sema4D was recently reported to promote
osteoclastogenesis and bone metastasis in breast cancer (7).
These findings prompted us to explore whether HNSCC
cells could use Sema4D to invade bone. The results of our present
study demonstrate that 1) a high amount of Sema4D was present in
the tissue samples of bone lesions of HNSCC, and 2) the expression
of Sema4D was correlated with IGF-I expression. We also observed
that 3) HNSCC cell lines express high levels of Sema4D driven by
IGF-I, and 4) Sema4D increases the proliferation, migration and
invasion of HNSCC cells. Our results demonstrate that 5)
osteoblasts produce RANKL in response to Sema4D, which activates
osteoclasts, further stimulating bone resorption. Finally, we
observed 6) the inhibition of tumor growth and decreased numbers of
osteoclasts in a mouse model of HSC-2 cells with knocked down
Sema4D compared to controls.
Materials and methods
Clinical samples
All of the HNSCC patients whose cases were
retrospectively examined herein were treated at the Department of
Oral and Maxillofacial Surgery, Okayama University Hospital
(Okayama, Japan) between 2000 and 2010, and the diagnosis was
pathologically confirmed HNSCC (six cases of soft tissue HNSCC,
include three cases of T4N0M0, two cases of T4N1M0 and a case of
T4N2M0. Nine cases of HNSCC with jaw bone invasion include four
cases of T4N0M0, two cases of T4N1M0 and three cases of T4N2M0). No
patient had received chemotherapy or radiation therapy before
surgery. The study was approved by the ethics committee of the
Okayama University Graduate School of Medicine, Dentistry, and
Pharmaceutical Sciences (protocol no. 1949), including the use of
an advertisement poster in place of patient consent. Written
consent was not acquired for this retrospective study.
We had access to the patient records prior to the
data anonymization. The surgically resected tissues were collected
as part of the patients' routine care by the authors. All patient
information was anonymized and de-identified prior to the analysis.
The sections were deparaffinized and then autoclaved in 0.2%
citrate buffer for 15 min for antigen retrieval. Sections were
incubated with 3% hydrogen peroxide for 30 min to block endogenous
peroxidase activity.
Primary rabbit anti-human Sema4D polyclonal IgG,
rabbit anti-human Plexin-B1 polyclonal IgG, rabbit anti-human IGF-I
polyclonal IgG and rabbit anti-human IGFI-R polyclonal IgG
antibodies (Abcam, Cambridge, MA, USA) were used for
immunohistochemisty analyses. The specimens were incubated with a
1:100 dilution of the antibody overnight at 4°C, followed by three
washes with Tris-buffered saline (TBS). The slides were then
treated with a streptoavidin-biotin complex (Envision System
Labeled Polymer, horseradish peroxidase (HRP); Dako, Carpinteria,
CA, USA) for 60 min at a dilution of 1:100. The immunoreaction was
visualized using a 3,3′-diaminobenzidine (DAB) substrate-chromogen
solution (Dako Cytomation Liquid DAB Substrate Chromogen System,
Dako). Finally, the sections were immersed in an ethanol and xylene
bath and mounted for examination. Sections were photographed and
the DAB density was detected using ImageJ (NIH, Bethesda, MD, USA).
Thereafter, the threshold of density was set automatically and the
α value (0–255) was calculated also by ImageJ software.
Cell culture and reagents
Human HNSCC cell lines HSC-2 and SAS and bone
marrow-derived stroma cell line ST-2 cells were obtained from the
Cell Engineering Division of RIKEN BioResource Center (Tsukuba,
Ibaraki, Japan). Both cancer cell lines were cultured in Dulbecco's
modified Eagle's medium/Ham's F-12 nutrient mixture (DMEM/F-12)
supplemented with 10% fetal bovine serum (FBS). The bone marrow
cells were isolated from 5-week-old male C57BL/6 mice obtained from
Charles River (Yokoyama, Japan).
Bone marrow cells was cultured in the α-modification
of minimum essential medium (αMEM; Sigma, St. Louis, MO, USA)
supplemented with 10% fetal calf serum (FCS) (Hyclone Laboratories,
Logan, UT, USA) and penicillin/streptomycin solution in an
atmosphere of 5% CO2/air at 37°C. Linsitinib (an IGF-I
receptor inhibitor), SCH772984 (an ERK inhibitor) and MK2206 (an
AKT inhibitor) were purchased from MedChem Express (Princeton, NJ,
USA). Primary mouse osteoblasts (i.e., osteoblasts from a cranial
bone culture kit F-2) were purchased from Cosmo Bio (Tokyo, Japan)
and cultured in αMEM supplemented with 10% FCS.
Immunoblot analysis
Cell monolayer cultures were rinsed with ice-cold
phosphate-buffered saline (PBS) and lysed in an ice-cold lysis
buffer (50 mM Tris-HCl, pH 7.4, containing 150 mM NaCl, 1% Triton
X-100, 1% NP-40, 10 mM NaF, 100 mM leupeptin, 2 mg/ml aprotinin and
1 mM phenylmethyl sulfonyl fluoride). Protein quantification was
performed by Protein Assay System (Thermo Fisher Scientific,
Waltham, MA, USA). Cell lysates containing 10 µg of total
protein in the lysis buffer were electrophoresed in 12% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels,
and the proteins were then transferred to nylon membranes
(Immobilon-P; Millipore, Bedford, MA, USA). The membranes were
blocked with 2% non-fat dry milk in TBS overnight at 4°C and then
incubated with rabbit anti-human Sema4D polyclonal IgG, rabbit
anti-human Plexin-B1 polyclonal IgG (Abcam) or rabbit anti-mouse
RANKL polyclonal IgG (Santa Cruz Biotechnology, Santa Cruz, CA,
USA) antibodies used at a 1:200 dilution. HRP-conjugate of goat
anti-rabbit polyclonal IgG was used as the secondary antibody at a
1:1000 dilution (Amersham Biosciences, Buckinghamshire, UK). Bands
were visualized by the ECL chemiluminescence detection method
(RPN2109; Amersham Biosciences).
Cell proliferation
Cells were seeded at a density of 3×103
cells per well in a 96-well plate and cultured for 24 h. They were
then cultured in the presence of recombinant human Sema4D (R&D
Systems, Minneapolis, MN, USA). An MTS assay was performed to
obtain a relative cell number after 48 h of incubation under the
experimental procedure specified by the manufacturer (Cell Titer 96
AQueous One Solution Cell Proliferation Assay; Promega, Madison,
WI, USA).
Invasion and migration assay
We studied the invasion and migration of cells by
using Boyden chambers with and without Matrigel® (BD
Biosciences, Franklin Lakes, NJ, USA). Cells in the logarithmic
growth phase were detached by trypsin-EDTA, and 3×104
cells in medium without FBS were added to polycarbonate membranes
(pore size 8.0 µm). Sema4D was added to the lower chamber,
and the system was incubated at 37°C for 24 h in 5% CO2.
After incubation and fixation, the cells on the upper side of the
membrane were removed with a cotton swab and the remaining cells
were stained with 2% Crystal Violet (Sigma). The number of stained
cells on the lower side of the membrane in four microscopic fields
was counted, and the average of cells in three wells was
determined.
TRAP staining and osteoclast activity
assay
Bone marrow macrophages (BMMs) were prepared as
previously described (16). Bone
marrow cells were isolated from 5-week-old male C57BL/6 mice
obtained from Charles River and cultured with 10 ng/ml of
macrophage colony-stimulating factor (M-CSF; R&D Systems) for
24 h, floating cells were collected and cultured with 30 ng/ml
M-CSF for 3 days. Thereafter, cells that were attached to the
plates were collected as BMMs.
BMMs (4×104) were seeded on 48-well
plates and cultured with 30 ng/ml M-CSF in the presence or absence
of Sema4D (R&D Systems) and/or 50 ng/ml RANKL (Peprotech,
London, UK) for 6 days. The medium was completely changed every 2
days. The cells were then fixed and stained for TRAP (Sigma), and
the number of TRAP-positive multinucleated cells (nuclear number
>3) in each well was counted.
For the osteoclast activity assay, BMMs
(1×105) were seeded on hydroxyapatite-coated plates
(Osteo Assay Surface 24-well plate, Corning, NY, USA) and cultured
as above. The area of pits on the plates was quantified by ImageJ.
For RANKL inhibitory assay, BMMs (1×105) were seeded on
24-well plates and cultured without RANKL with 30 ng/ml M-CSF in
the presence or absence of Sema4D and osteoprotegerin (OPG;
Peprotech) for 6 days. The number of TRAP-positive multi-nucleated
cells (nuclear number >3) in each well was counted.
Knockdown of Sema4D in HNSCC cells
We purchased Sema4D short hairpin RNAs (shRNAs)
cloned into a pLKO.1-puro plasmid under the control of human
phosphoglycerate kinase eukaryotic promoter for stable expression
(Mission® shRNA Plasmid DNA, SHCLND-NM_006378, Sigma)
and scrambled sequence shRNA constructs (Mission Non-Target shRNA
Control Vector, SHC002, Sigma). HNSCC cells are transfected with
shRNA-pLKO.1-puro using Lipofectamine 3000® (Thermo
Fisher Scientific) according to the manufacturer's
instructions.
After 2 weeks in medium containing the selection
antibiotic puromycin, cells stably expressing the shRNA were
selected from transfected cultures. Sema4D knockdown was confirmed
by performing an immunoblot analysis and evaluated by using ImageJ.
The five types of shRNA used were: shSema4D#1,
CCGGGCCTGTGACTTGGTTTCTCTTCTCGAGAAGAGAAACCA AGTCACAGGCTTTTTG;
shSema4D#2,
CCGGCGAACCAAAGATCGTCATCAACTCGAGTTGATGACGATCTTTGGTTCGTTTTTG;
shSema4D#3,
CCGGGCCTGTGTTCTATGCACTCTTCTCGAGAAGAGTGCATAGAACACAGGCTTTTTG;
shSema4D#4,
CCGGCCTGAACTTAACATCCTTTAACTCGAGTTAAAGGATGTTAAGTTCAGGTTTTTG; and
shSema4D#5,
CCGGGTACGGTCTTATGGGCAGAAACTCGAGTTTCTGCCCATAAGACCGTACTTTTTG.
Bone xenograft model
All animal experiments were approved by the
Institutional Animal Care and Use Committee of Okayama University.
The bone xenograft model was prepared as previously described
(17). Five-week-old male athymic
mice (nu/nu) were obtained from CLEA Japan (Tokyo). HSC-2
cells, 5×106 per mouse, were inoculated into the
paraperiosteal tissue of the tibial metaphysis (17). Six mice per group were used.
Tumor sizes and body weights were measured weekly,
and the former were recorded in cubic millimeters (length ×
width2/2). The animals were sacrificed at day 28, and
the tumor tissues were removed and weighed.
Immunohistochemistry for xenograft tumor
specimens
Paraffin blocks of specimens were cut at 4 µm
thickness. Immunohistochemistry (IHC) was performed with rabbit
anti-human Sema4D polyclonal IgG, rabbit anti-human Plexin-B1
polyclonal IgG, rabbit anti-human IGF-I polyclonal IgG, rabbit
anti-human IGFI-R polyclonal IgG, rabbit anti-human Ki67 polyclonal
IgG and rabbit anti-mouse CD31 polyclonal IgG antibodies used at a
1:100 dilution (Abcam). Sections were incubated with the primary
antibodies at 4°C for 16 h, and visualized with the Envision system
(Dako). The number of TRAP-positive multinucleated osteoclasts in
three visual fields under a microscope (×200) were counted, and the
osteoclast number per bone perimeter (per 100 mm) was calculated as
previously described (18).
Statistical analysis
We analyzed the data (except those from the clinical
samples) by using the unpaired Student's t-test for comparisons of
two groups, and Fisher's protected least significant difference
(Fisher's PLSD) for multiple group comparisons. Results are
expressed as the mean ± SD. Values of p<0.05 were considered
significant. The data from clinical samples were analyzed using the
Mann-Whitney U test because of the non-parametric distribution of
data. The relationships between pairs of groups were analyzed by
using the calculated correlation coefficient.
Results
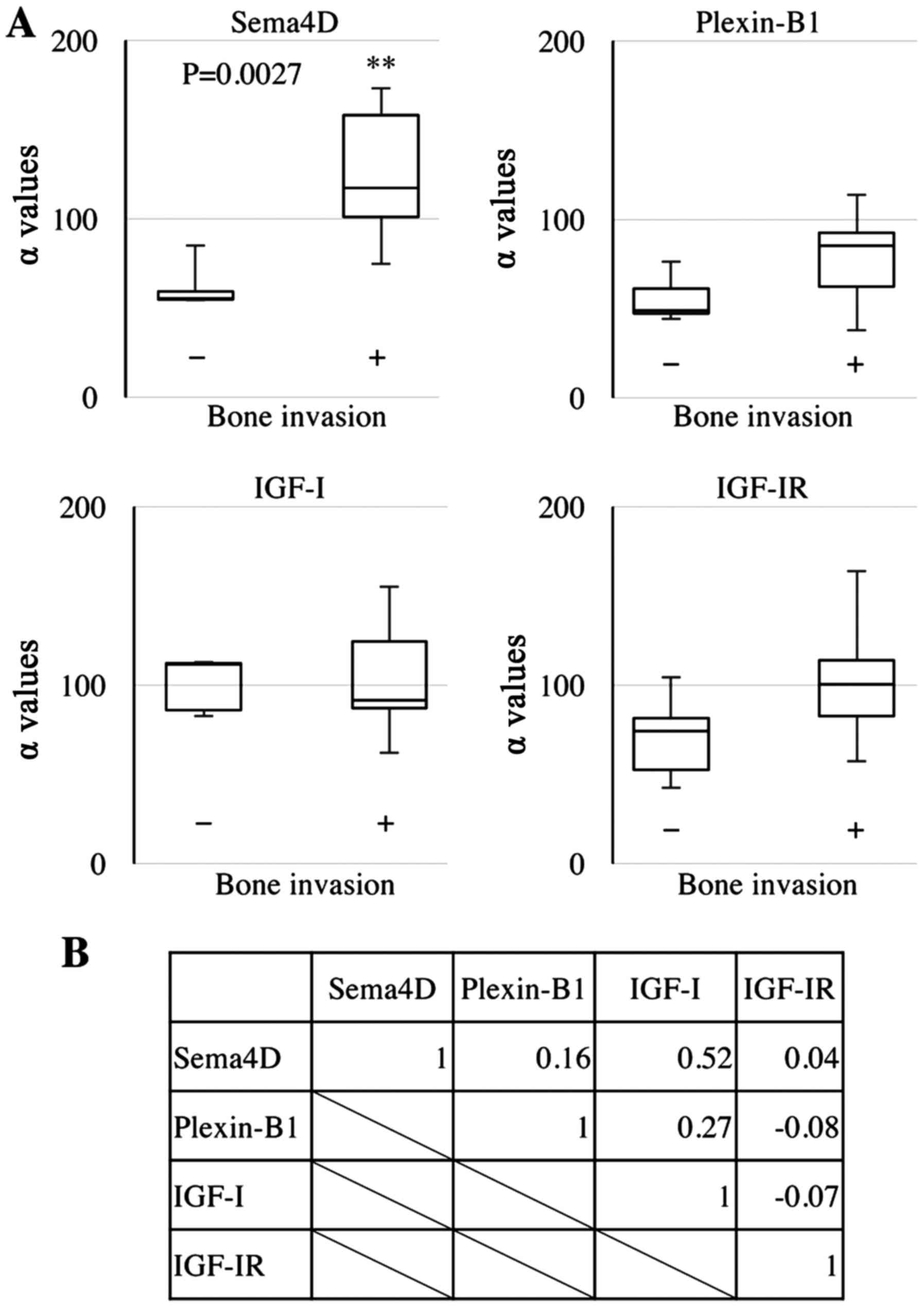
Sema4D is highly expressed in the HNSCC
tumors with bone invasion and correlates with IGF-I expression
We examined the expression of Sema4D in different
HNSCC tumors, and found that its level was 2.1-fold higher in
HNSCCs with bone invasion compared to HNSCCs without bone invasion
(p=0.0027) (Fig. 1A). Of note,
Sema4D expression was significantly correlated with IGF-I
expression (correlation coefficient: 0.52) (Fig. 1B).
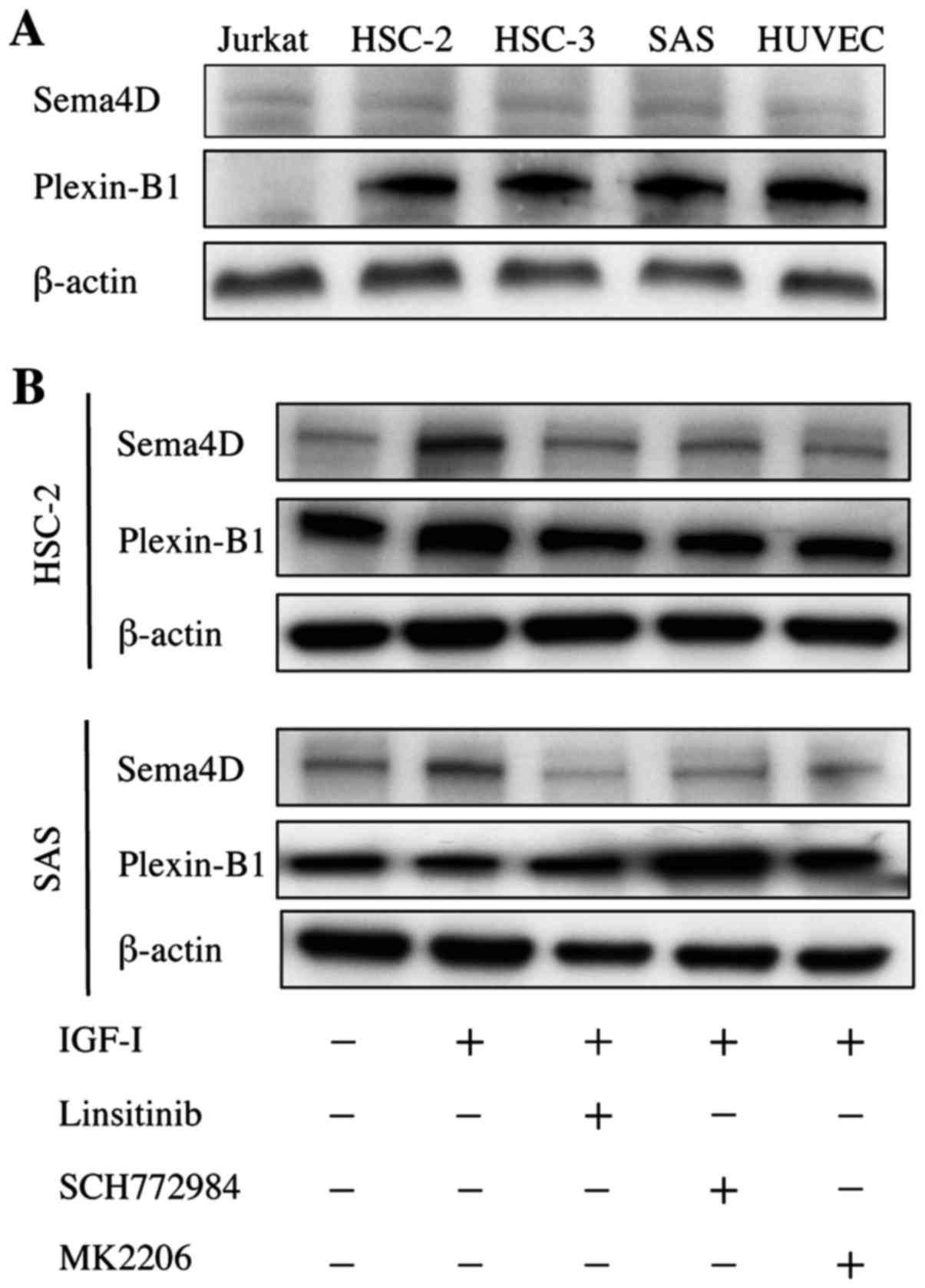
Sema4D and Plexin-B1 are expressed in
HNSCC cells in vitro
Since Sema4D was highly expressed in HNSCC tumors
with bone invasion, we examined whether Sema4D was expressed in
HNSCC cell lines HSC-2, HSC-3 and SAS in vitro. All three
HNSCC cell lines exhibited weak Sema4D expression, but this
expression was comparable to that of the Jurkat T cells used as the
positive control for Sema4D (Fig.
2A).
Plexin-B1 was strongly expressed in all three HNSCC
cell lines similarly to HUVEC cells, which were the positive
control for Plexin-B1 (Fig. 2A).
Noteworthy, IGF-I increased the expression of Sema4D by 2.1- and
1.5-fold in HSC-2 and SAS cells, respectively (Fig. 2B). Moreover, Linsitinib (an IGF-IR
inhibitor), SCH772984 (an ERK inhibitor) and MK2206 (an AKT
inhibitor) suppressed IGF-I-induced Sema4D expression. Taken
together, our findings demonstrate that Sema4D and Plexin-B1 were
expressed in HNSCC cells and that Sema4D expression was regulated
by IGF-IR downstream signaling including ERK and AKT. TGF-β did not
increase Sema4D expression in HSC-2 or SAS cells (data not
shown).
Sema4D upregulates HNSCC cellular
activity
To examine the effect of Sema4D in vitro, we
treated HNSCC cells with different concentrations of Sema4D and
measured cell proliferation by MTS assay. With 100 ng/ml Sema4D,
the proliferation was stimulated 1.5-fold and 1.3-fold (Fig. 3A and B) and the cell migration was
enhanced 1.8-fold and 2.7-fold in HSC-2 and SAS cells, respectively
(Fig. 3C and D). With 100 ng/ml
Sema4D, invasion was enhanced 2.6-fold and 2.4-fold in HSC-2 and
SAS cells, respectively (Fig. 3E and
F).
Sema4D enhances osteoclastogenesis and
bone resorptive activity
Since Sema4D increased proliferation of HNSCC cell
lines, we next studied the effect of Sema4D on osteoclasts. BMMs
were treated with Sema4D in the presence of M-CSF and RANKL for 6
days. At the concentration of 100 ng/ml, Sema4D increased the
osteoclastogenesis by 1.4-fold and the osteolytic activity by
3.8-fold (Fig. 4A and B). Since
Plexin-B1 is known to be expressed in osteoblasts (14), we postulated that Sema4D enhances
osteoclastogenesis through Plexin-B1 on osteoblasts. To test this
hypothesis, we treated ST-2 cells and primary mouse osteoblasts
with Sema4D and then evaluated the expression of RANKL by
immunoblotting. As shown in Fig.
4C, RANKL expression was upregulated by 100 ng/ml Sema4D
1.5-fold in ST-2 cells, and was induced in primary mouse
osteoblasts. To elucidate the role of RANKL in Sema4D-stimulated
osteoclastogenesis, we treated BMMs with Sema4D without exogenous
soluble RANKL with different concentrations of OPG. Fig. 4D shows that Sema4D promoted
osteoclastogenesis without exogenous soluble RANKL and that OPG
counteracted Sema4D-induced osteoclastogenesis, supporting the
finding that Sema4D increased the RANKL expression in
osteoblasts.
Knockdown of Sema4D inhibits bone
invasion of xenograft HNSCC in athymic mice
To further investigate whether Sema4D production by
HNSCC cells was responsible for bone invasion, we used shRNA to
manipulate Sema4D expression in HNSCC cells. Sema4D shRNAs were
purchased and transfected into HSC-2 cells. Immunoblot analysis
showed that the Sema4D expression in HSC-2 cells was knocked down
by 66.7% by shSema4D#4, which were thus used in the subsequent
experiments (Fig. 5A).
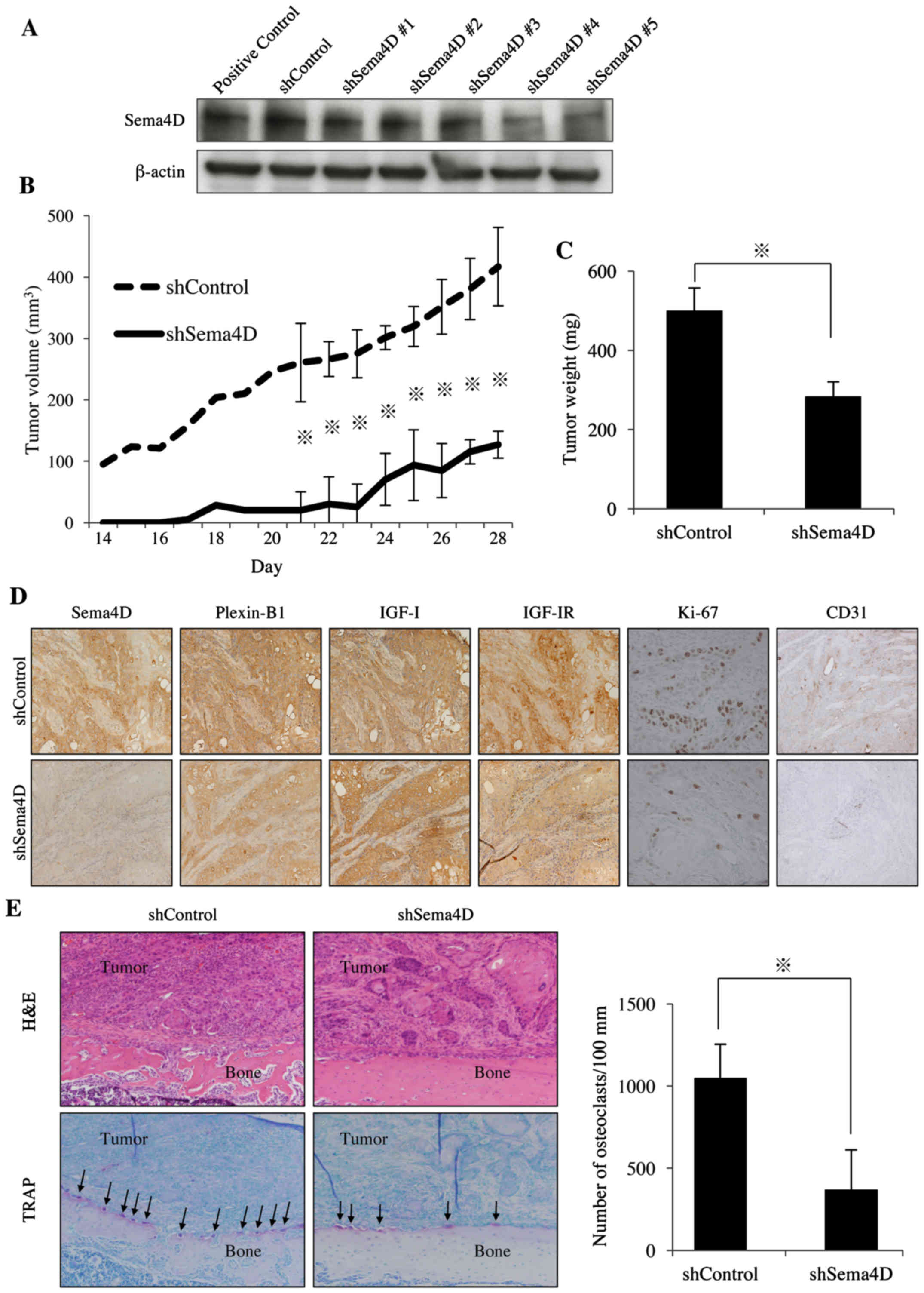 | Figure 5Effect of knockdown of Sema4D on the
growth of HSC-2 xenografts and osteoclastogenesis in athymic mice.
(A) Knockdown of Sema4D with shRNA was confirmed by immunoblot
analysis. (B and C) HSC-2 cells expressing the shControl or
shSema4D were inoculated into the paraperiosteal tissue of the
tibial metaphysis. Tumor volume and weight were recorded.
*p<0.05. (D) IHC staining for Sema4D, Plexin-B1,
IGF-I, IGF-IR, Ki-67 and CD31 in tumors from the HSC-2 expressing
shControl or shSema4D. Sema4D, Plexin-B1, IGF-I, IGF-IR, CD31: ×200
magnification. Ki67: ×400 magnification. Ki67-positive and total
numbers of cells were counted in 5 randomly selected areas at a
magnification of ×400. CD31-positive neovessels in each tumor were
counted in the five most vascularized areas at a magnification of
×200, and the numbers were averaged. Images shown are from a
representative animal of each group. (E) Hematoxylin and eosin
(H&E) and TRAP staining of tumor specimens from the HSC-2
expressing shControl or shSema4D. Magnification, ×20. The number of
TRAP-positive osteoclasts (arrow) in each group was counted.
*p<0.05. |
In the animal experiment, the growth rate of
xenograft tumors was decreased in Sema4D knockdown group compared
to that of the control group (Fig.
5B). The tumor volumes at the end of the experiment (day 28)
for the groups treated with shControl and shSema4D were 416.9±63.8
and 127.2±21.9 mm3, respectively, indicating an
approximately 69.4% decrease in tumor growth rate in
shSema4D-treated group. The end tumor weights at day 28 for the
groups treated with shControl and shSema4D were 500.0±57.7 and
283.3±37.3 mg, respectively, indicating an approximately 43.4%
decrease in the tumor growth rate for the shSema4D-treated group
(Fig. 5C). These results suggest
that knockdown of Sema4D effectively inhibited xenograft tumor
growth of HNSCC cells in athymic mice. Our IHC analysis of the
tumor specimens showed lower level of Sema4D in the knockdown
group, which is consistent with the immunoblot results (Fig. 5A and D). IHC staining with an
anti-Ki67 antibody showed that the percentage of Ki67-positive
cancer cells in the groups treated with shControl and shSema4D were
significantly decreased from 55.8±3.0 to 15.2±5.6, respectively,
representing a 72.3% decrease in cell proliferation (Fig. 5D). Vessel density in the groups
treated with shControl and shSema4D, as shown by CD31-positive
vessels, was 59.0±16.7 and 20.8±2.5, respectively, representing a
64.7% decrease in tumor angiogenesis (Fig. 5D). Knockdown of Sema4D exerted
antitumor effects in HNSCC in in vivo experiments via the
suppression of cancer cell proliferation and angiogenesis.
To determine whether Sema4D in HNSCC cells
influences osteoclastogenesis in vivo, we evaluated the
number of TRAP-positive multinucleated osteoclasts in tumor
specimens. As shown in Fig. 5E,
the numbers of osteoclasts were greatly reduced in the
shSema4D-treated group compared to the control group. The
quantitative analysis demonstrated a 65.2% decrease in osteoclast
formation following the Sema4D knockdown. Taken together, these
results indicate that the expression of Sema4D in HNSCC is
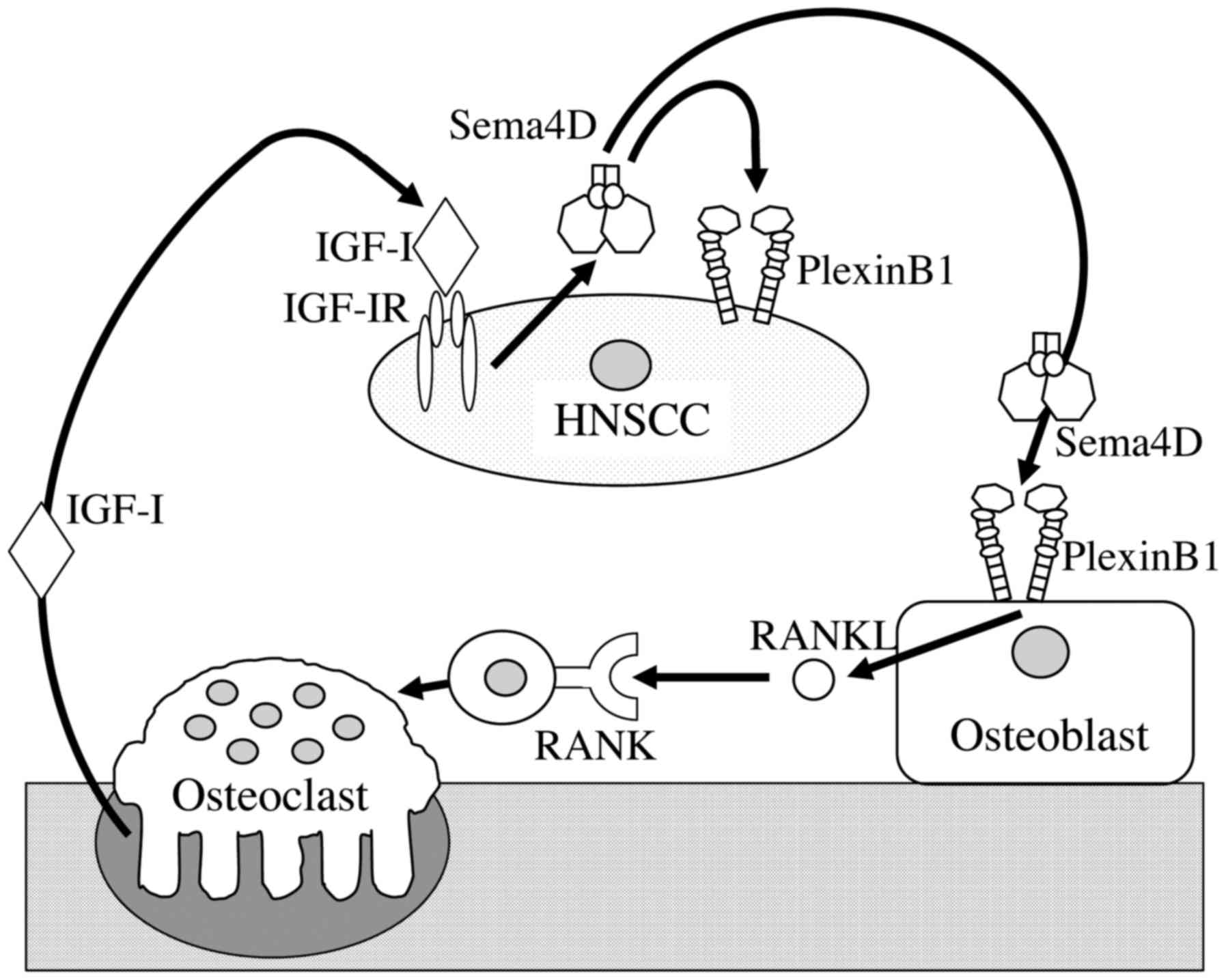
important for osteoclastogenesis and for bone invasion (Fig. 6).
Discussion
Previous research indicated that the axon guidance
molecule Sema4D has an important role in cancer progression
(19). Sema4D shed by HNSCC cells
acts through Plexin-B1 on vascular endothelial cells to promote
cell migration and tube formation, which promotes tumor
angiogenesis (15). An
immunohistochemical analysis of Sema4D in a large tumor sample
collection revealed that Sema4D overexpression was present in 80%
of the HNSCCs (15). Sema4D has
been shown to act directly on breast (19–21)
and ovarian (22) cancer cells,
inducing invasion and metastasis.
The results of our present study demonstrate that
both Sema4D and Plexin-B1 were expressed in HNSCC cell lines and
that Sema4D increased cell proliferation, migration and invasion,
suggesting that Sema4D shed by HNSCC cells acts through Plexin-B1
on HNSCC cells themselves in an autocrine manner. A prior study
showed that Sema4D activates the small GTPase RhoA, which is
related to cytoskeleton reorganization and focal adhesion formation
(23). Moreover, Sema4D has also
been shown to regulate PYK2 [a non-receptor tyrosine kinase of the
focal adhesion kinase (FAK) family] and modulate cell motility
(24). Sema4D causes the AKT and
MAPK phosphorylation (24,25), the signaling of which suppresses
apoptosis and promotes cell multiplication, respectively. These
findings suggest that Sema4D plays a direct role in disease
progression of HNSCC, possibly through the signaling pathways
mentioned above.
In the bone microenvironment, Sema4D expression in
osteoclasts and Plexin-B1 expression in osteoblasts has been
observed (14). To date, however,
there has been no report describing the expression levels of Sema4D
and Plexin-B1 in bone lesions of HNSCC. Our present findings
demonstrate that a high expression of Sema4D is present in HNSCCs
with bone invasion compared to those without bone invasion.
Moreover, Sema4D expression was correlated with IGF-I expression in
tumor tissue samples. These findings prompted us to investigate
whether IGF-I regulates Sema4D expression.
Of note, IGF-I greatly increased Sema4D expression
in HNSCC cells, which is consistent with the results we obtained
from the clinical samples. These data support our hypothesis that
IGF-I released from bone regulates Sema4D expression and promotes
bone invasion of HNSCC. In physiological conditions, IGF-I released
from bone is resorbed by osteoclasts, although it was originally
stored by osteoblasts, and it induces osteoblast differentiation.
In contrast, Sema4D produced by osteoclasts inhibits osteoblast
differentiation by inhibiting IGF-I signaling (14). In other words, osteoclasts control
bone formation by osteoblasts based on a balance between Sema4D and
IGF-I signaling. In osteolytic lesions of HNSCC, a high production
of Sema4D by HNSCC cells 'tips the scale' of bone homeostasis in
favor of resorption.
Mechanisms underlying the semaphorin enhancement of
osteoclastogenesis have been reported (7,26,27).
Mice with knock-out of Sema6D receptor plexin-A1
(plexin-A1−/−) have been phenotyped as having
osteopetrosis due to the inhibition of osteoclastogenesis. In the
presence of Sema6D, plexin-A1 forms a complex with TREM2
(triggering receptor expressed on myeloid cells 2) and DAP12
(DNAX-activation protein 12), which promote osteoclastogenesis
(26). Sema4D plays a role in
functions of osteoclasts including spreading, adhesion, migration
and resorption through β3 integrin subunit downstream signaling
(27). Sema4D also promotes IL-8
expression in osteoblasts, which promotes osteoclastogenesis and
bone metastasis of breast cancer (7).
In the present study, we observed that the RANKL
expression in osteoblasts was upregulated by Sema4D. Strengthening
this result, the OPG that was used to counteract RANKL successfully
inhibited Sema4D-induced osteoclastogenesis. We also demonstrated
that Sema4D shed by HNSCC cells was important for
osteoclastogenesis in vivo and for bone invasion, using an
animal model. However, we note here that activation of
osteoclastogenesis by Sema4D might also be through the
above-mentioned factor that has been implicated in a different type
of cancer.
Bisphosphonates (28) and an anti-RANKL antibody
(denosumab) (29,30) are widely used to manage
hypercalcemia of malignancy and skeletal metastases. These drugs
target osteoclasts and do not have antitumor activity for cancer
cells. The results presented here suggest that targeting Sema4D
could be a novel treatment strategy for anti-osteoclastic and
antitumor action in HNSCC therapy.
Acknowledgments
This work was supported by a Grant-in-Aid for
Scientific Research (B) (JSPS KAKENHI grant no. JP26293429) to A.S.
and a Grant-in-Aid for Scientific Research (C) (JSPS KAKENHI grant
no. JP26463004 and JP17K11836) to S.I. from the Ministry of
Education, Culture, Sports, Science, and Technology of Japan. The
authors would like to thank Ms. Kazuko Funakoshi for the expert
technical assistance in histological preparations.
References
|
1
|
Ampil FL, Ghali GE, Caldito G and Hardin
JC Jr: Treatment of head and neck cancer with bone or cartilage
invasion by surgery and postoperative radiotherapy. J Oral
Maxillofac Surg. 62:408–411. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shimo T and Sasaki A: Mechanism of
cancer-induced bone destruction: An association of connective
tissue growth factor (CTGF/CCN2) in the bone metastasis. Jpn Dent
Sci Rev. 47:13–22. 2011. View Article : Google Scholar
|
|
3
|
Weilbaecher KN, Guise TA and McCauley LK:
Cancer to bone: A fatal attraction. Nat Rev Cancer. 11:411–425.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shimo T, Kubota S, Yoshioka N, Ibaragi S,
Isowa S, Eguchi T, Sasaki A and Takigawa M: Pathogenic role of
connective tissue growth factor (CTGF/CCN2) in osteolytic
metastasis of breast cancer. J Bone Miner Res. 21:1045–1059. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luo Y, Raible D and Raper JA: Collapsin: A
protein in brain that induces the collapse and paralysis of
neuronal growth cones. Cell. 75:217–227. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kolodkin AL, Matthes DJ and Goodman CS:
The semaphorin genes encode a family of transmembrane and secreted
growth cone guidance molecules. Cell. 75:1389–1399. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang YH, Buhamrah A, Schneider A, Lin YL,
Zhou H, Bugshan A and Basile JR: Semaphorin 4D promotes skeletal
metastasis in breast cancer. PLoS One. 11:e01501512016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Epstein JA, Aghajanian H and Singh MK:
Semaphorin signaling in cardiovascular development. Cell Metab.
21:163–173. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Messina A and Giacobini P: Semaphorin
signaling in the development and function of the gonadotropin
hormone-releasing hormone system. Front Endocrinol (Lausanne).
4:1332013.
|
|
10
|
Kang S, Okuno T, Takegahara N, Takamatsu
H, Nojima S, Kimura T, Yoshida Y, Ito D, Ohmae S, You DJ, et al:
Intestinal epithelial cell-derived semaphorin 7A negatively
regulates development of colitis via αvβ1 integrin. J Immunol.
188:1108–1116. 2012. View Article : Google Scholar
|
|
11
|
Mizui M, Kumanogoh A and Kikutani H:
Immune semaphorins: Novel features of neural guidance molecules. J
Clin Immunol. 29:1–11. 2009. View Article : Google Scholar
|
|
12
|
Shanks K, Nkyimbeng-Takwi EH, Smith E,
Lipsky MM, DeTolla LJ, Scott DW, Keegan AD and Chapoval SP:
Neuroimmune semaphorin 4D is necessary for optimal lung allergic
inflammation. Mol Immunol. 56:480–487. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Moreau-Fauvarque C, Kumanogoh A, Camand E,
Jaillard C, Barbin G, Boquet I, Love C, Jones EY, Kikutani H,
Lubetzki C, et al: The transmembrane semaphorin Sema4D/CD100, an
inhibitor of axonal growth, is expressed on oligodendrocytes and
upregulated after CNS lesion. J Neurosci. 23:9229–9239.
2003.PubMed/NCBI
|
|
14
|
Negishi-Koga T, Shinohara M, Komatsu N,
Bito H, Kodama T, Friedel RH and Takayanagi H: Suppression of bone
formation by osteoclastic expression of semaphorin 4D. Nat Med.
17:1473–1480. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Basile JR, Castilho RM, Williams VP and
Gutkind JS: Semaphorin 4D provides a link between axon guidance
processes and tumor-induced angiogenesis. Proc Natl Acad Sci USA.
103:9017–9022. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shimo T, Matsumoto K, Takabatake K, Aoyama
E, Takebe Y, Ibaragi S, Okui T, Kurio N, Takada H, Obata K, et al:
The role of sonic hedgehog signaling in osteoclastogenesis and jaw
bone destruction. PLoS One. 11:e01517312016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Goda T, Shimo T, Yoshihama Y, Hassan NM,
Ibaragi S, Kurio N, Okui T, Honami T, Kishimoto K and Sasaki A:
Bone destruction by invading oral squamous carcinoma cells mediated
by the transforming growth factor-beta signalling pathway.
Anticancer Res. 30:2615–2623. 2010.PubMed/NCBI
|
|
18
|
Kuroda Y, Hisatsune C, Nakamura T, Matsuo
K and Mikoshiba K: Osteoblasts induce Ca2+
oscillation-independent NFATc1 activation during
osteoclastogenesis. Proc Natl Acad Sci USA. 105:8643–8648. 2008.
View Article : Google Scholar
|
|
19
|
Sakurai A, Doçi CL and Gutkind JS:
Semaphorin signaling in angiogenesis, lymphangiogenesis and cancer.
Cell Res. 22:23–32. 2012. View Article : Google Scholar :
|
|
20
|
Swiercz JM, Worzfeld T and Offermanns S:
ErbB-2 and met reciprocally regulate cellular signaling via
plexin-B1. J Biol Chem. 283:1893–1901. 2008. View Article : Google Scholar
|
|
21
|
Worzfeld T, Swiercz JM, Looso M, Straub
BK, Sivaraj KK and Offermanns S: ErbB-2 signals through Plexin-B1
to promote breast cancer metastasis. J Clin Invest. 122:1296–1305.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ye S, Hao X, Zhou T, Wu M, Wei J, Wang Y,
Zhou L, Jiang X, Ji L, Chen Y, et al: Plexin-B1 silencing inhibits
ovarian cancer cell migration and invasion. BMC Cancer. 10:6112010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Capparuccia L and Tamagnone L: Semaphorin
signaling in cancer cells and in cells of the tumor
microenvironment - two sides of a coin. J Cell Sci. 122:1723–1736.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Basile JR, Afkhami T and Gutkind JS:
Semaphorin 4D/plexin-B1 induces endothelial cell migration through
the activation of PYK2, Src, and the phosphatidylinositol
3-kinase-Akt pathway. Mol Cell Biol. 25:6889–6898. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aurandt J, Li W and Guan K-L: Semaphorin
4D activates the MAPK pathway downstream of plexin-B1. Biochem J.
394:459–464. 2006. View Article : Google Scholar :
|
|
26
|
Kumanogoh A and Kikutani H: Immunological
functions of the neuropilins and plexins as receptors for
semaphorins. Nat Rev Immunol. 13:802–814. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dacquin R, Domenget C, Kumanogoh A,
Kikutani H, Jurdic P and Machuca-Gayet I: Control of bone
resorption by semaphorin 4D is dependent on ovarian function. PLoS
One. 6:e266272011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ressler S, Mlineritsch B and Greil R:
Zoledronic acid for adjuvant use in patients with breast cancer.
Expert Rev Anticancer Ther. 11:333–349. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stopeck AT, Lipton A, Body J-J, Steger GG,
Tonkin K, de Boer RH, Lichinitser M, Fujiwara Y, Yardley DA,
Viniegra M, et al: Denosumab compared with zoledronic acid for the
treatment of bone metastases in patients with advanced breast
cancer: A randomized, double-blind study. J Clin Oncol.
28:5132–5139. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fizazi K, Carducci M, Smith M, Damião R,
Brown J, Karsh L, Milecki P, Shore N, Rader M, Wang H, et al:
Denosumab versus zoledronic acid for treatment of bone metastases
in men with castration-resistant prostate cancer: A randomised,
double-blind study. Lancet. 377:813–822. 2011. View Article : Google Scholar : PubMed/NCBI
|