Introduction
Glioblastoma (GBM) is the most common and aggressive
primary brain tumor, accounting for more than 50% of all brain
tumors (1). Even with the current
standard treatment composed of maximal surgical removal,
radiotherapy and chemotherapy with temozolomide (TMZ), the median
survival period of GBM patients is approximately 14 months
(2,3). Multimodal treatment with radiotherapy
and TMZ increases the survival rate compared with radiotherapy
alone. Despite the current treatment regime, the prognosis for GBM
patients is still very poor because of tumor recurrence. There are
several causes of recurrence including unclear tumor margins
following surgical removal, a fast growth rate and resistance to
chemotherapy and radiotherapy (4).
Particularly for the GBM stem cell (GSC) subpopulation, possessing
a high carcinogenic potential and self-renewing ability has been
identified and is believed to contribute to GBM propagation and
resistance to conventional therapies (5). Thus, the development of new
anticancer agents targeting both cancer cells and cancer stem cells
in GBM could be a promising therapeutic strategy to more
effectively eradicate malignant tumors.
Ginseng has been consumed as traditional medicine
for thousands of years in Asia (6). There are extensive reports regarding
ginseng's many pharmacological effects on the endocrine, immune,
cardiovascular and the central nervous systems (7). There are also many reports that
ginseng has antiaging and antioxidant properties (8). The various pharmacological and
biological effects of ginseng are mainly mediated by compounds from
the saponin group, called ginsenosides (9). Accumulating evidence has revealed
that the major functional components of ginsenoside have
antiinflammatory, antidiabetic and antitumor effects (10,11).
While >40 ginsenosides have been identified, their chemical
structures have difficulties with absorption when orally
administered (12). More than 80%
of the known ginsenosides can be classified into two structural
groups: the protopanaxadiol (PPD)-type (e.g., Rb1, Rb2, Rb3, Rc,
Rd, Rg3, Rh2 and Rs1) that have sugar moieties attached to the β-OH
at C3 and/or C20 and the protopanaxatriol (PPT)-type (e.g., Re, Rf,
Rg1, Rg2 and Rh1) that have sugar moieties attached to the α-OH at
C6 and/or the β-OH at C20. Compound K
[20-O-β-d-glucopyranosyl-20(S)-protopanaxadiol, CK] is the major
metabolite of ginsenosides Rb1, Rb2, Rc and Rd that are synthesized
by intestinal bacteria after oral administration of ginseng
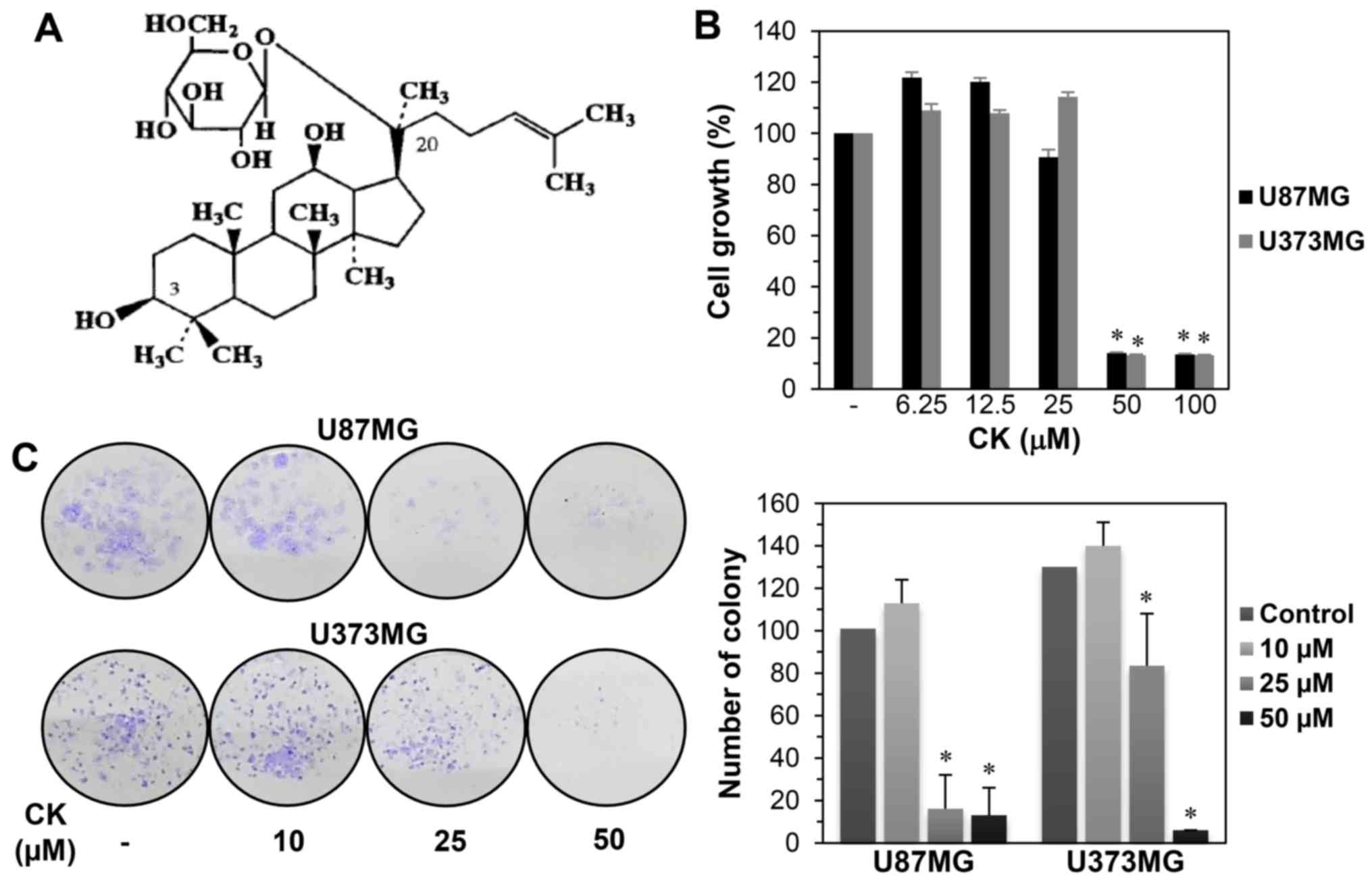
(Fig. 1A) (13). CK is absorbed in the
gastrointestinal tract before being slowly metabolized (14,15).
Several studies have indicated that CK possesses anticancer
activity against a variety of tumor cell types by inhibiting cell
proliferation and inducing apoptosis (16,17).
CK also has antimetastatic potential in various tumor cells
(18,19). Despite the long-term study of the
CK anticancer efficacy, there has been no research on GBM cells,
including GSCs.
In the present study, we investigated the anticancer
effect of CK on GBM and the underlying mechanisms involved. Our
results showed that CK inhibited the growth and metastatic ability
of GBM cells, inducing cell cycle arrest and apoptosis.
Furthermore, we identified a therapeutic effect of CK against the
cancer stem cell-like phenotypes of GBM cells, GSCs. Our findings
provide evidence that CK may be a potential therapeutic drug
against GBM.
Materials and methods
Materials
Compound K was obtained from Ilhwa Co., Ltd., (Guri,
Korea) and dissolved in dimethyl sulfoxide (DMSO) at a
concentration of 50 mM as a stock solution. Gelatin and Matrigel
were purchased from Sigma-Aldrich (St. Louis, MO, USA) and BD
Biosciences (San Jose, CA, USA), respectively. Anti-MMP-2 (64 kDa),
anti-MMP-9 (84 kDa), anti-cyclin D1 (36 kDa), anti-cyclin D3 (31
kDa), anti-PARP (89, 116 kDa), anti-cleaved caspase-3 (Asp175, 17,
19 kDa), anti-cleaved caspase-9 (Asp330, 37 kDa), anti-phospho-PI3K
[p85 (Tyr458)/p55 (Tyr199), 60 and 85 kDa], anti-PI3K (p85, 85
kDa), anti-phospho-Akt (Ser473, 60 kDa), anti-Akt (60 kDa),
anti-phospho-mTOR (Ser2448, 289 kDa), anti-mTOR (289 kDa),
anti-Nanog (42 kDa), anti-Sox2 (35 kDa), anti-Oct4 (45 kDa) and
anti-β-actin (45 kDa) antibodies were obtained from Cell Signaling
Technology (Danvers, MA, USA). Anti-CD133 (133 kDa) antibody was
obtained from Miltenyi Biotec GmbH (Bergisch Gladbach,
Germany).
Cell culture
Human glioblastoma U87MG and U373MG cell lines were
obtained from the Korean Cell Line Bank (KCLB; Seoul, Korea). The
cells were cultured in minimum essential medium (MEM; Gibco, Grand
Island, NY, USA) supplemented with 10% fetal bovine serum (FBS;
Gibco) and 1% penicillin-streptomycin-amphotericin B (Lonza,
Walkersville, MD, USA) and then maintained at 37°C in a humidified
5% CO2 incubator.
Cell growth assay
Cell growth was examined using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
colorimetric assay. Dissociated GBM cells were seeded in a 96-well
culture plate at a density of 3×103 cells/well. After a
24-h incubation, CK (6.25–100 µM) was added to each well and
incubation was resumed. After 72 h, 50 µl of MTT solution (2
mg/ml; Sigma-Aldrich) was added to each well followed by incubation
for 3 h at 37°C. To dissolve formazan crystals, the culture medium
was removed and an equal volume of DMSO was added to each well. The
absorbance of each well was determined at a wavelength of 540 nm
using a microplate reader (Thermo Fisher Scientific, Vantaa,
Finland).
Colony formation assay
To evaluate the colony forming inhibitory effect of
CK, GBM cells were seeded in a 6-well cell culture pate at a
density of 500 cells/well. After 24-h incubation, the cells were
treated with CK (10, 25 and 50 µM) for 8 days. Following
this, the colonies were fixed with 4% formaldehyde and stained with
0.5% crystal violet solution.
Wound healing assay
GBM cells were seeded in a 24-well cell culture
plate at a density of 7×104 cells/well and grown to 90%
confluence. The confluent monolayer cells were scratched using a
pipette tip and each well was washed with phosphate-buffered saline
(PBS) to remove non-adherent cells. The cells were treated with CK
(10, 25 and 50 µM) and then incubated for up to 48 h. The
perimeter of the central cell-free zone was confirmed under an
optical microscope (Olympus, Center Valley, PA, USA).
Invasion assay
Cell invasion was assayed using Transwell chamber
inserts with a pore size of 8.0 µm (Corning Costar, Acton,
MA, USA). The lower side of the polycarbonate filter was coated
with 10 µl of gelatin (1 mg/ml) and the upper side was
coated with 10 µl of Matrigel (3 mg/ml). GBM cells
(1×105) were seeded in the upper chamber of the filter
and CK (10, 25 and 50 µM) was added to the lower chamber
filled with medium, and the chamber was incubated at 37°C. After 24
h (for cancer cells) or 48 h (for cancer stem-like cells), the
cells were fixed with methanol and stained with hematoxylin/eosin.
The total number of cells that invaded the lower chamber of the
filter was counted using an optical microscope (Olympus).
Cell cycle analysis
GBM cells were seeded in a 60-mm culture dish at a
density of 5×105 cells/dish and incubated for 24 h. Next
the cells were treated with CK (50 µM) for 24 h. Following
treatment, the cells were harvested, washed with PBS, and fixed in
ice-cold 70% ethanol at −20°C for >3 h. The cells were
subsequently centrifuged and incubated with a prop-idium iodide
(PI) working solution (10 µg/ml PI and 100 µg/ml
RNase A; Sigma-Aldrich) for 30 min at room temperature in the dark.
The cell cycle distribution was analyzed using a FACScan flow
cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Apoptosis analysis
GBM cells at a density of 5×105
cells/dish were treated with CK (50 and 75 µM) and incubated
for 24 h. The cells were harvested, washed with PBS, and stained
with Annexin V-FITC and PI according to the manufacturer's
instructions for the ApopNexin Annexin V FITC Apoptosis kit (Merck
Millipore, Darmstadt, Germany). The stained cells were analyzed by
flow cytometry (BD Biosciences).
DAPI fluorescent staining
GBM cells were seeded in a 24-well culture plate at
a density of 5×104 cells/well. After treatment with CK
(25, 50 and 75 µM) for 24 h, the cells were washed with PBS,
fixed in 1% formaldehyde solution for 10 min, and then stained with
4′,6-diamidino-2-phenylindole (DAPI, 5 µg/ml; Sigma-Aldrich)
for 10 min at room temperature. The nuclear morphology of the cells
was determined by fluorescence microscopy (Optinity KI-2000F; Korea
Lab Tech, Seongmam, Korea).
Intracellular reactive oxygen species
(ROS) measurement
Intracellular reactive oxygen species (ROS) levels
were measured using a ROS-sensitive fluorescence indicator,
2′,7′-dichlorofluorescein diacetate (DCFH-DA; Sigma-Aldrich). GBM
cells were seeded in a 24-well cell culture plate at a density of
5×104 cells/well and treated with CK (25, 50 and 75
µM) for 24 h. The cells were incubated with 10 µM of
DCFH-DA for 30 min and then washed with PBS. The fluorescent images
were obtained using an Optinity KI-2000F fluorescence microscope
(Korea Lab Tech).
Mitochondrial membrane potential (MMP)
determination
The mitochondrial membrane potential (MMP) was
detected using the fluorescent, lipophilic dye, JC-1
(5,5′,6,6′-tetra-chloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine
iodide; Sigma-Aldrich). At hyperpolarized membrane potentials, this
dye forms a red fluorescent J-aggregate, whereas at depolarized
membrane potentials, this dye remains in its green fluorescent
monomeric form. GBM cells were seeded in a 24-well cell culture
plate at a density of 5×104 cells/well. After treatment
with CK (25, 50 and 75 µM) for 24 h, the cells were
incubated with 10 µg/ml of JC-1 for 20 min and washed with
PBS. The images were obtained using an Optinity KI-2000F
fluorescence microscope.
Western blot analysis
Cell lysates were separated by 10% SDS-PAGE and the
separated proteins were transferred to polyvinylidene difluoride
membranes (Millipore, Billerica, MA, USA) using standard
electroblotting procedures. The blots were blocked and
immunolabeled with primary antibodies against MMP-2, MMP-9, cyclin
D1, cyclin D3, PARP, cleaved caspase-3, cleaved caspase-9,
phospho-PI3K, PI3K, phospho-Akt, Akt, phospho-mTOR, mTOR, CD133,
Nanog, Sox2, Oct4 and β-actin overnight at 4°C. Immunolabeling was
detected with an enhanced chemiluminescence (ECL) kit (Bio-Rad
Laboratories, Hercules, CA, USA), according to the manufacturer's
instructions.
GSC culture
To propagate GSCs, U87MG and U373MG cells grown in
the serum-based media were cultured in Dulbecco's modified Eagle's
medium/nutrient mixture F-12 (DMEM/F12; Gibco) containing 1× B-27
serum-free supplement (Gibco), 5 µg/ml heparin
(Sigma-Aldrich), 2 mM l-glutamine (Gibco), 20 ng/ml epidermal
growth factor (EGF; Gibco), 20 ng/ml basic fibroblast growth factor
(bFGF; KOMA Biotech, Seoul, Korea) and 1% penicillin/streptomycin
(Gibco). Neurospheres grown in the serum-free media were
subcultured every 7 days by dissociating with Accutase (Millipore,
Temecula, CA, USA) and maintained at 37°C in a humidified 5%
CO2 incubator.
GSC growth assay
GSC growth was evaluated using the WST-1
colorimetric assay, a water-soluble tetrazolium salt method.
Dissociated neurosphere cells were seeded in a 96-well cell culture
plate at a density of 3×103 cells/well using the
serum-free media with EGF and bFGF. After 7 days of CK treatment
(6.25–100 µM), 10 µl of WST-1 reagent solution
(DoGen, Seoul, Korea) was added to each well and the cells were
incubated for an additional 3 h at 37°C. The absorbance was
measured at a wavelength of 450 nm using a microplate reader
(Thermo Fisher Scientific).
Neurosphere formation assay
Dissociated neurosphere cells were seeded in a
96-well cell culture plate at a density of 200 cells/well using the
serum-free media with EGF and bFGF. After 9–12 days of CK treatment
(10, 25 and 50 µM), the number of formed neurospheres in
each well was counted under a microscope.
Statistical analysis
The results are expressed as the mean ± standard
error (SE). The Student's t-test was used to determine statistical
significance between the control and test groups. A P<0.05 was
considered statistically significant.
Results
Compound K suppresses the growth of
glioblastoma cells
To investigate the effect of CK on GBM cell growth,
U87MG and U373MG cells were treated at various concentrations
(0–100 µM) of CK for 72 h. Cell growth was then assessed
using the MTT assay. As shown in Fig.
1B, a significant decrease in the growth of both cells was
observed for 50 µM CK. The growth inhibitory effect of CK
against GBM cells was further assessed using a colony formation
assay. U87MG and U373MG cells were treated with CK at
concentrations of 0–50 µM for 8 days. As shown in Fig. 1C, colony formation for both cell
types was remarkably suppressed by treatment with 25 and 50
µM CK, indicating that CK inhibited the proliferation of GBM
cells.
Compound K inhibits the migration and
invasion of glioblastoma cells
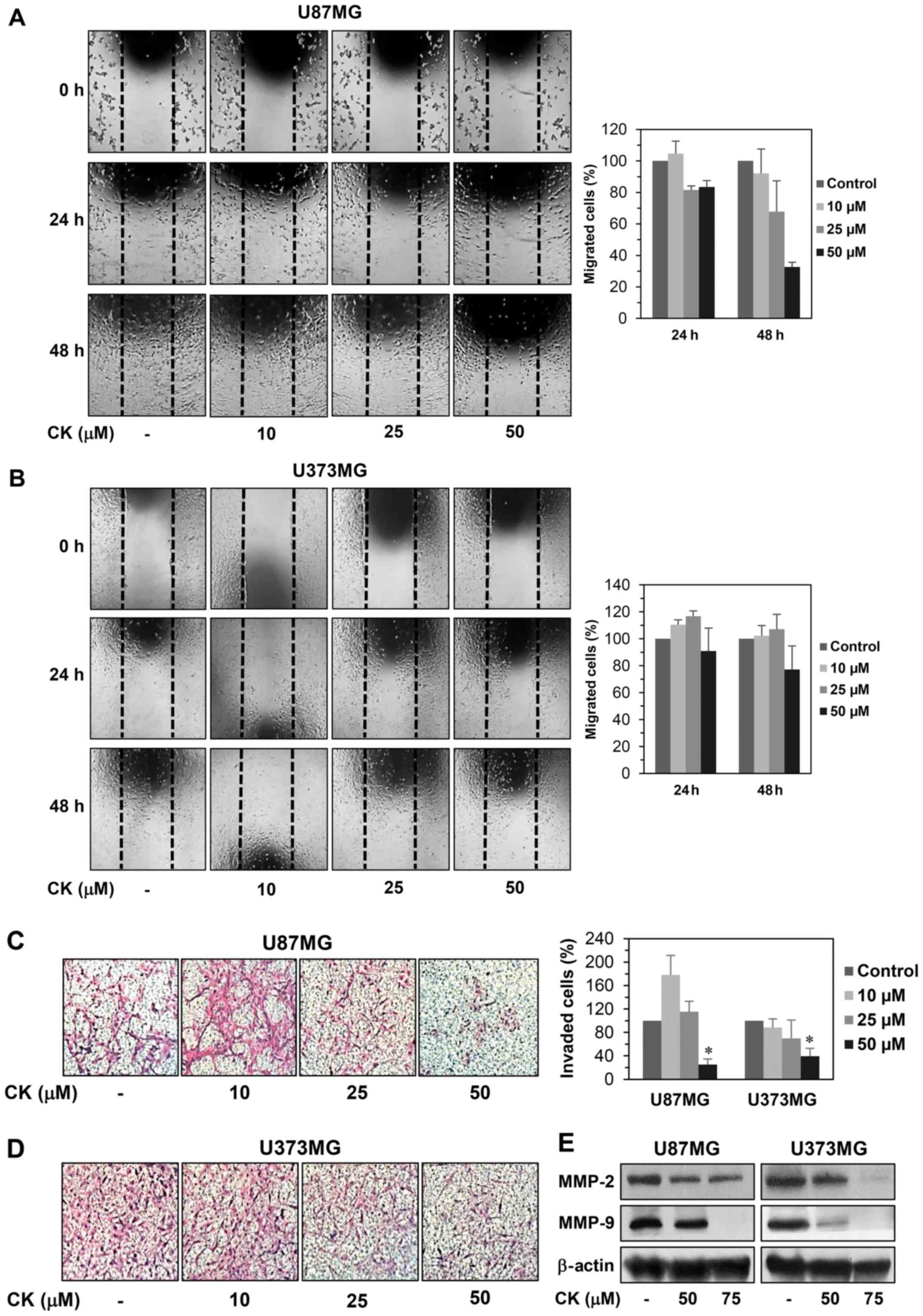
Cell motility is an important factor for cancer
metastasis (20). We therefore
examined whether CK could inhibit the migration and invasion of GBM
cells. A monolayer wound healing assay was performed to evaluate
the effect of CK on the migration of U87MG and U373MG cells. CK at
a concentration of 50 µM decreased the migration of both
cell types at 24 and 48 h after treatment when compared with the
control condition (Fig. 2A and B).
For the Transwell invasion assay, U87MG and U373MG cells were
treated with CK at the indicated concentrations for 24 h, reducing
the invasiveness of both cell types in a dose-dependent manner
(Fig. 2C and D). Notably, the
inhibitory effect of CK on GBM cell invasion was very potent at 50
µM.
A characteristic of metastatic cancer cells is the
capacity to degrade extracellular matrix (ECM) through the
upregulation of matrix metalloproteinases (MMPs) (21). In particular, MMP-2 and MMP-9 have
been shown to play a critical role in cancer metastasis (22). To define the mechanism by which CK
reduces GBM cell migration and invasion, the protein expression
levels of MMP-2 and MMP-9 in U87MG and U373MG cells were
investigated. As shown in Fig. 2E,
CK effectively suppressed the expression of MMP-2 and MMP-9,
suggesting that CK may inhibit the migration and invasion of GBM
cells by decreasing MMP-2 and MMP-9 expression.
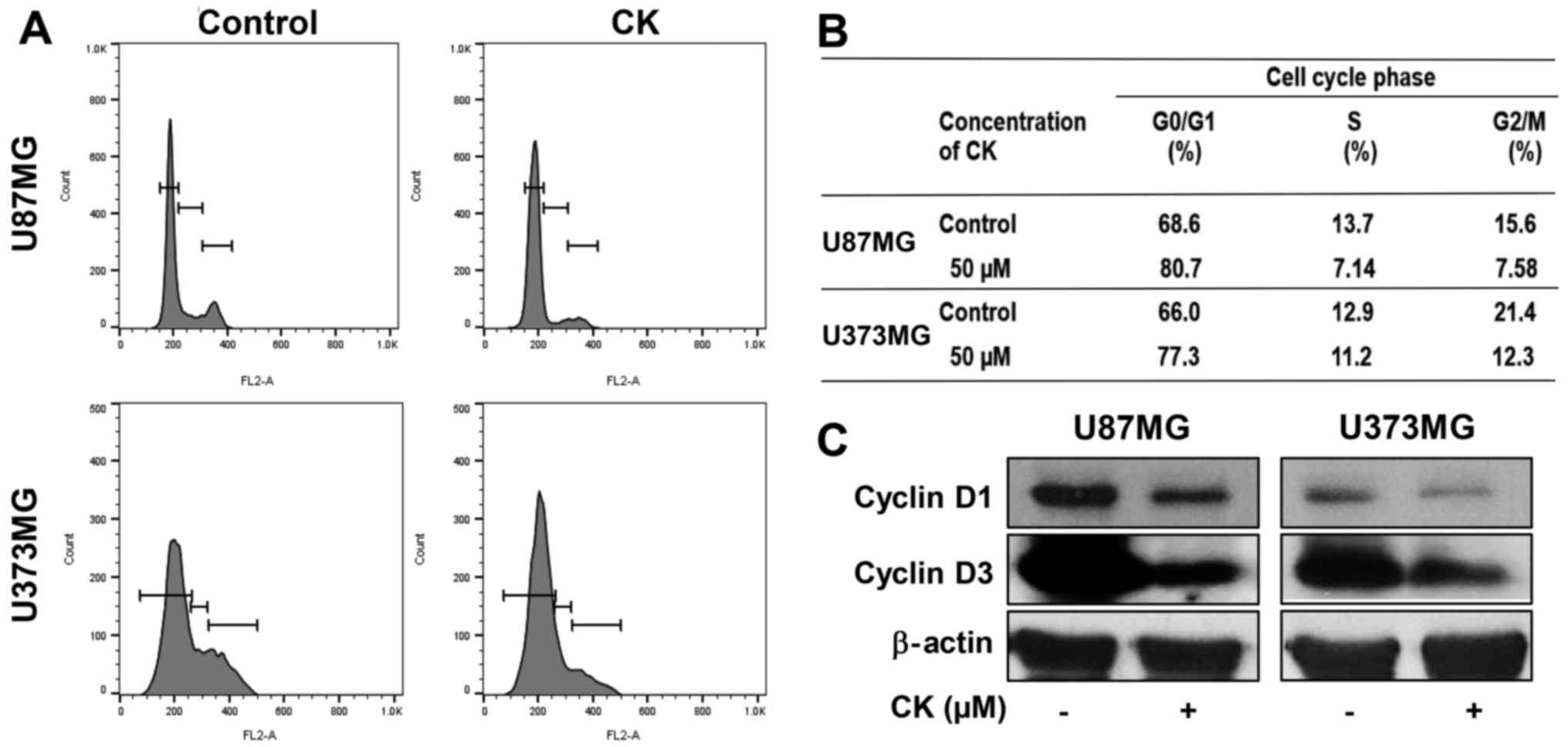
Compound K induces G0/G1 phase arrest in
glioblastoma cells
To determine whether the antiproliferative effect of
CK on GBM cells was caused by cell cycle arrest, the effect of CK
on the cellular cell cycle distribution was quantified using flow
cytometric analysis. U87MG and U373MG cells were treated with 50
µM of CK for 24 h. As shown in Fig. 3A and B, CK induced G0/G1 phase
arrest (an increase in the proportion of arrested cells from 68.6
to 80.7% for U87MG cells and from 66.0 to 77.3% for U373MG cells)
along with a decrease of S and G2/M phases when compared with the
control cells.
The D-type cyclins (cyclins D1, D2 and D3) are
important regulators for the transition from G0/G1 phase to S phase
of the cell cycle (23,24). Therefore, the effect of CK on the
expression of cell cycle regulators was assessed. As shown in
Fig. 3C, 50 µM of CK led to
a significant decrease in the protein levels of cyclin D1 and D3 in
both U87MG and U373MG cells. This demonstrates that CK blocked cell
cycle progression at the G0/G1 phase, thereby inhibiting GBM cell
proliferation.
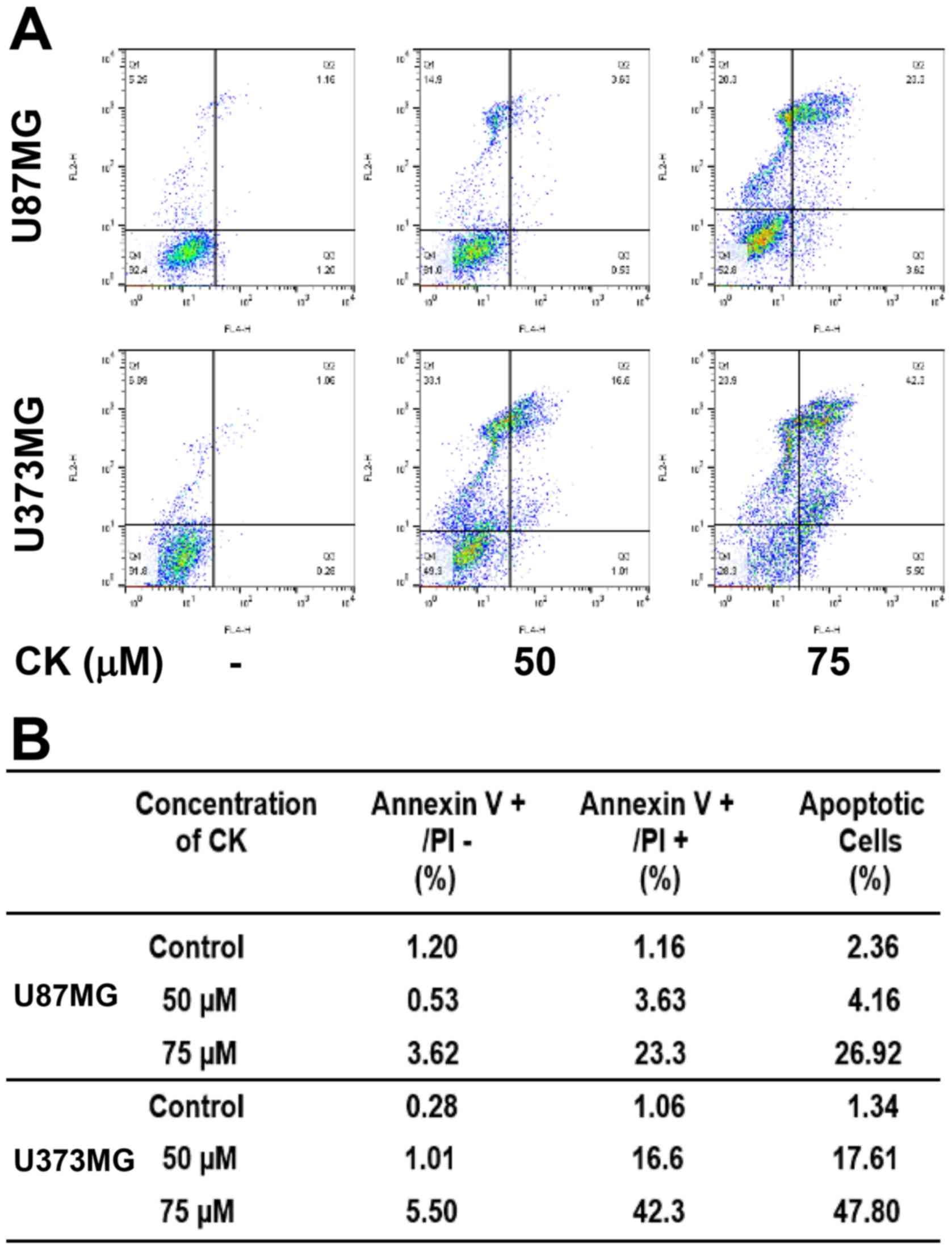
Compound K induces apoptosis in
glioblastoma cells
To further elucidate the anticancer effect of CK in
GBM cells, cellular apoptosis was quantitatively measured using
flow cytometric analysis following Annexin V-FITC and PI dual
labeling. Annexin V is a marker of early apoptosis and PI is a
marker of late apoptosis and necrosis (25,26).
When U87MG and U373MG cells were treated with CK at either 50 or 75
µM for 24 h, the total amount of early and late apoptotic
cells were markedly increased in a dose-dependent manner after CK
treatment in comparison with controls (from 2.36 to 26.92% for
U87MG cells and from 1.34 to 47.8% for U373MG cells; Fig. 4). This indicates that CK induced
apoptotic cell death in GBM cells.
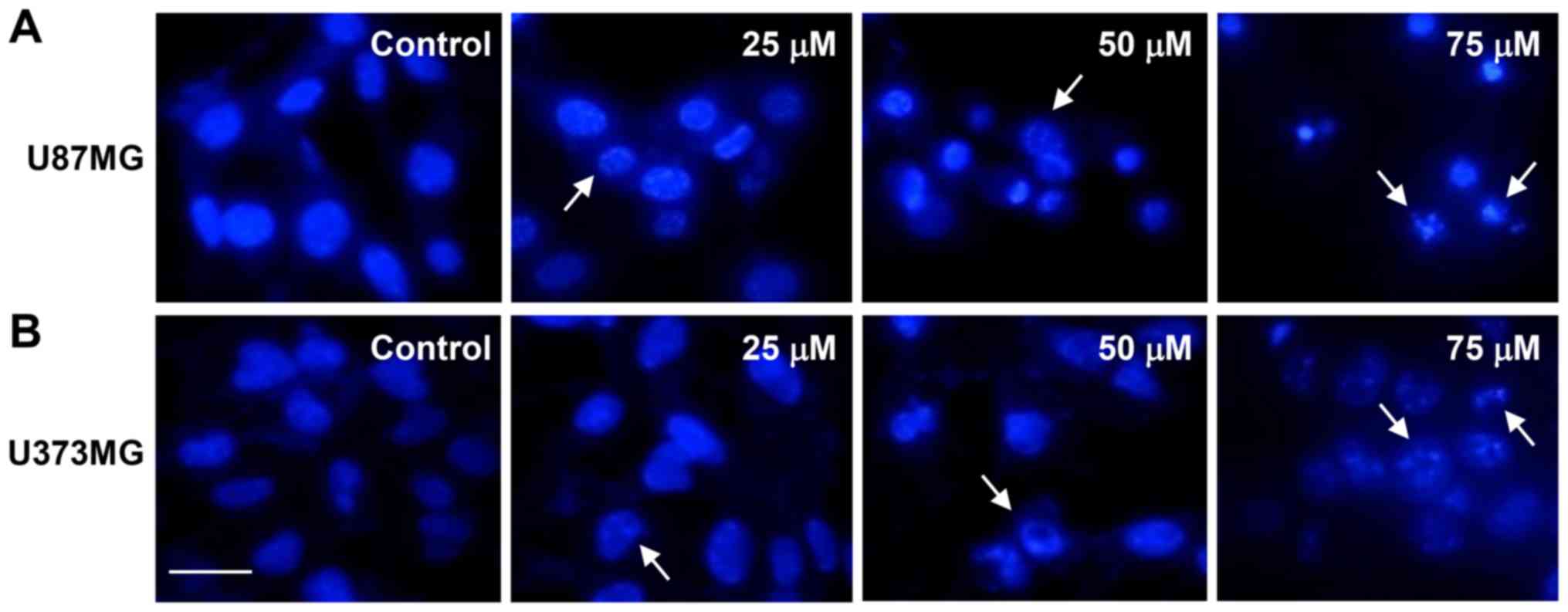
Next, we investigated whether CK causes nuclear
apoptotic changes in GBM cells. DAPI staining revealed that CK
induced the nuclear condensation and fragmentation of U87MG and
U373MG cells in a dose-dependent manner (as indicated by the arrows
in Fig. 5).
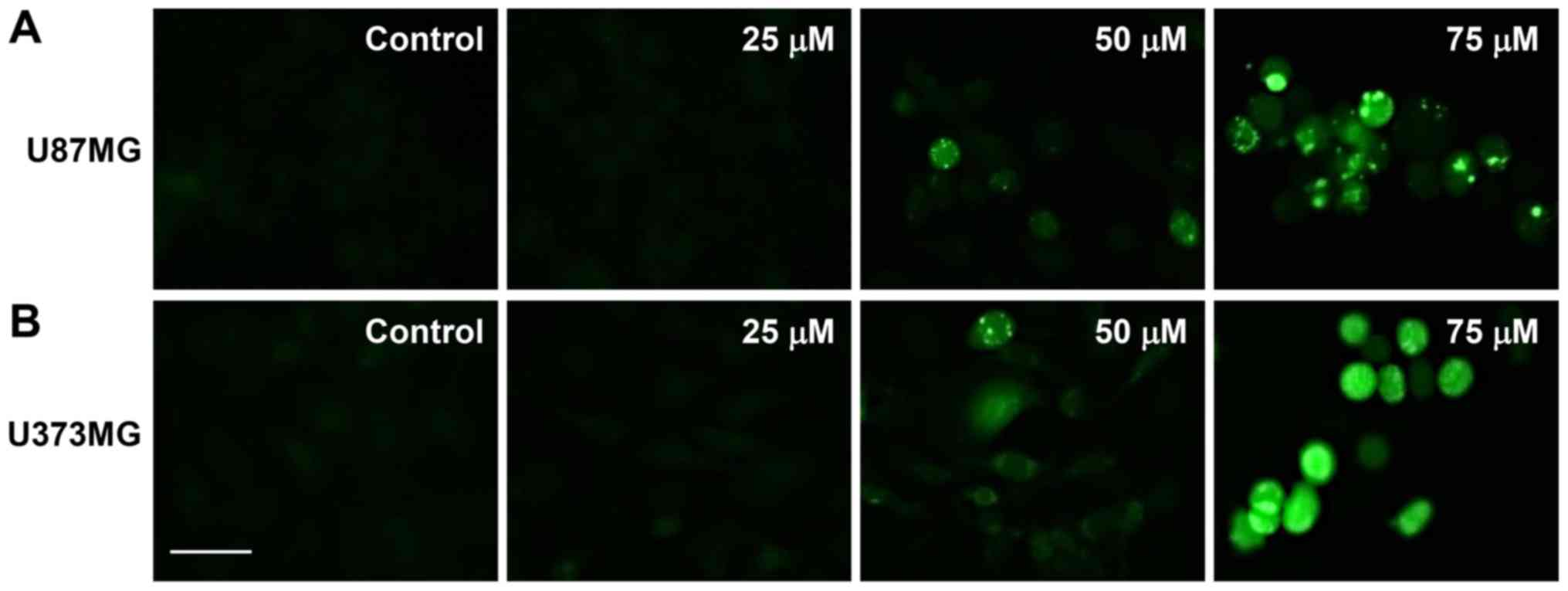
The elevated formation of intracellular ROS plays an
important role in mediating apoptotic processes (27). Therefore, to determine whether ROS
are involved in the regulation of apoptosis induced by CK, the
intracellular generation of ROS in GBM cells was observed using the
fluorescent DCFH-DA product. As shown in Fig. 6, CK dose-dependently elevated the
production of ROS in U87MG and U373MG cells in comparison with
control cells.
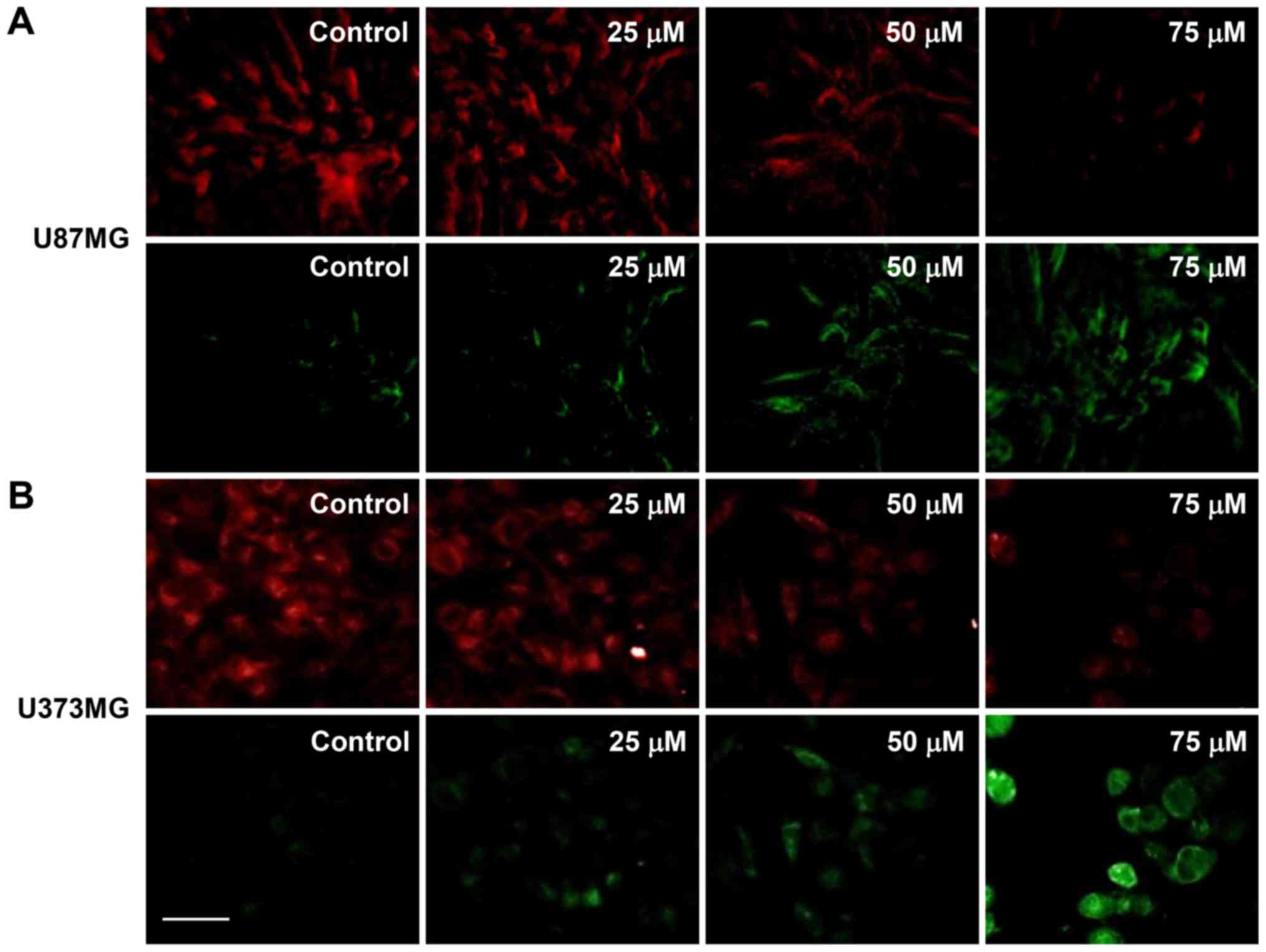
Mitochondrial dysfunction is an early occurring
event during apoptosis and includes a change in the MMP (28). Therefore, changes in the MMP of
U87MG and U373MG cells after CK treatment was investigated using
JC-1. As shown in Fig. 7, the
control cells exhibited a low level of green fluorescence and a
high red fluorescence (i.e., a large negative MMP), whereas
treatment with CK led to an increase in green fluorescence and a
decrease in red fluorescence in a dose-dependent manner (i.e., a
loss of MMP). These data demonstrate that mitochondrial dysfunction
may be closely related to CK-induced apoptosis of GBM cells.
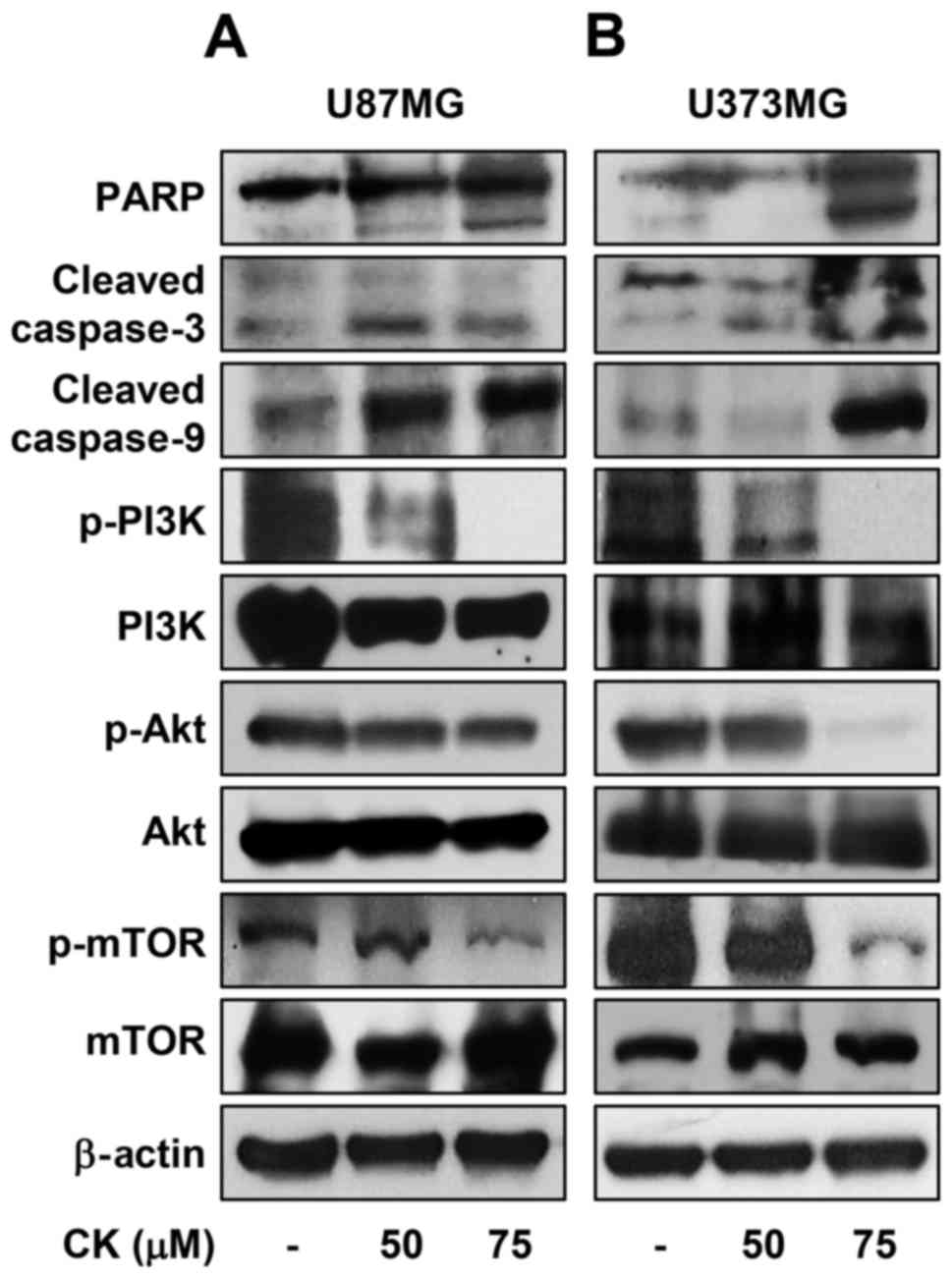
Subsequently, the effect of CK on caspase activity
was assessed to determine whether CK induces caspase-dependent
apoptosis in GBM cells. Western blot analysis indicated that
treatment with CK resulted in cleavage of PARP as well as
activation of caspase-3 and -9 in U87MG and U373MG cells (Fig. 8). These results therefore suggest
that CK induces apoptosis in a caspase-dependent manner in GBM
cells.
The oncogenic phosphatidylinositol 3-kinase
(PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR)
signaling axis plays a key role in the regulation of proliferation
and death of various cancer cells (29). The effect of CK on the
PI3K/Akt/mTOR activation was therefore investigated in GBM cells.
CK significantly inhibited the phosphorylation of PI3K, Akt and
mTOR in both U87MG and U373MG cells (Fig. 8), indicating that CK exhibits
anti-proliferative and apoptotic effects in GBM cells by negatively
regulating the PI3K/Akt/mTOR signaling axis.
Compound K suppresses self-renewal
capacity and invasiveness of glioblastoma stem-like cells
The stem cell subpopulation in GBM has been proposed
to be a central driver of tumor initiation, progression, recurrence
and therapeutic resistance (30).
To assess the therapeutic effect of CK against cancer stem
cell-like phenotypes of GBM cells, spheroid cultures for the
expansion of cancer stem cell populations from U87MG and U373MG
cells was examined.
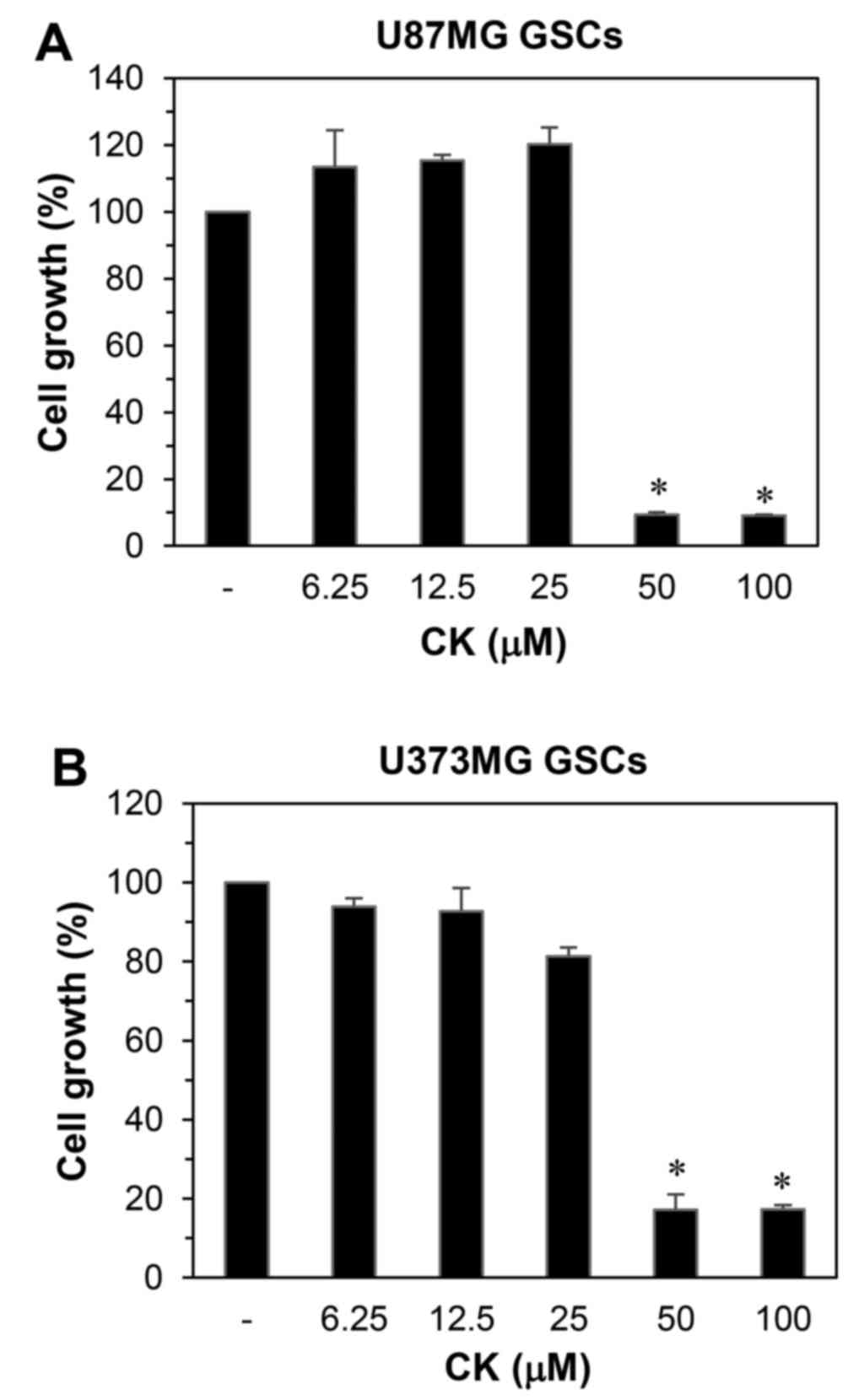
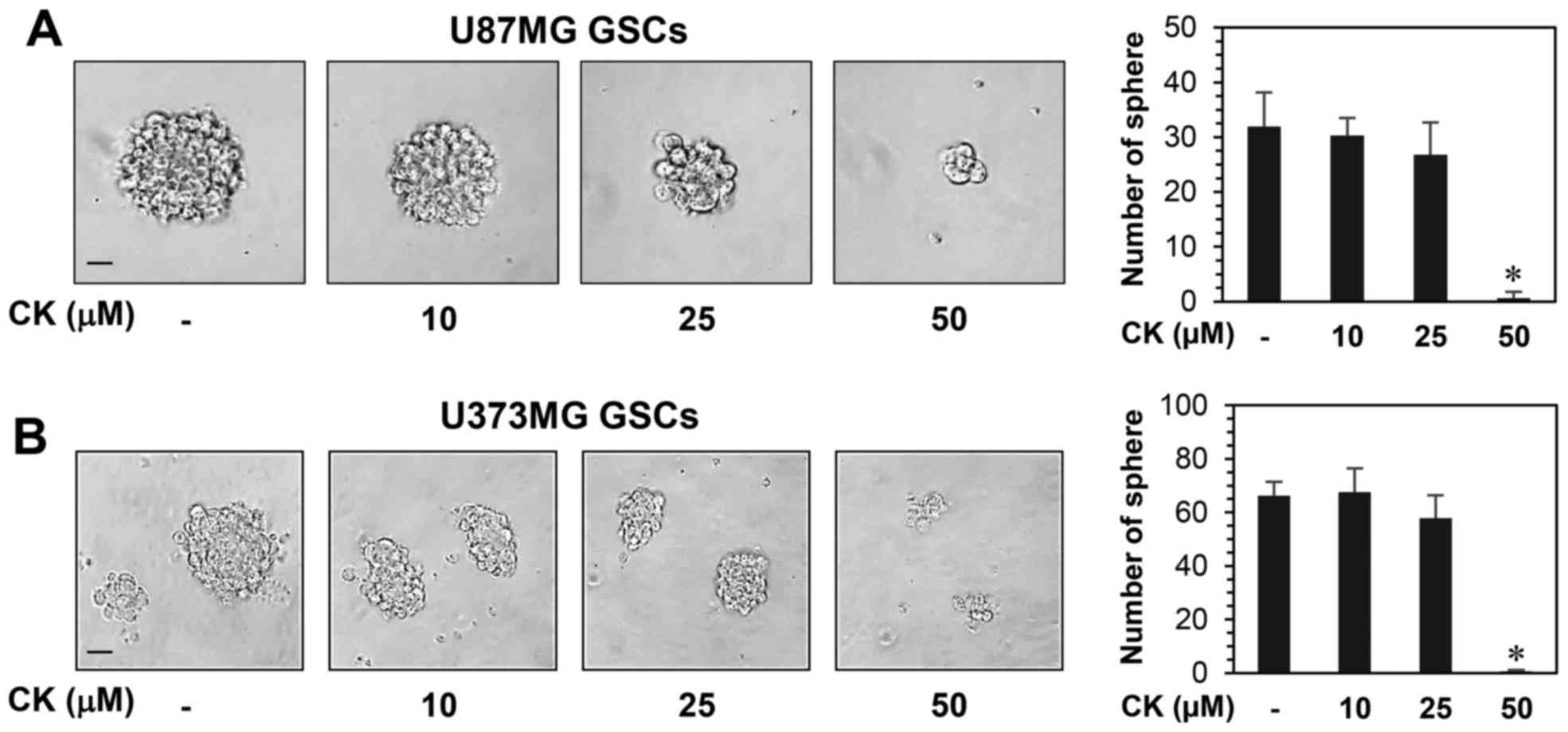
The effect of CK on the growth of GSCs derived from
U87MG and U373MG cells was determined using a water-soluble
tetrazolium salt method. As shown in Fig. 9, CK remarkably inhibited the growth
of GSCs at 50 µM. Furthermore, the neurosphere formation for
both GSC types were markedly suppressed by treatment with 50
µM CK (Fig. 10).
Therefore, CK treatment demonstrated a reduction in the
self-renewal capacity of GSCs, including cell growth and
clonogenicity.
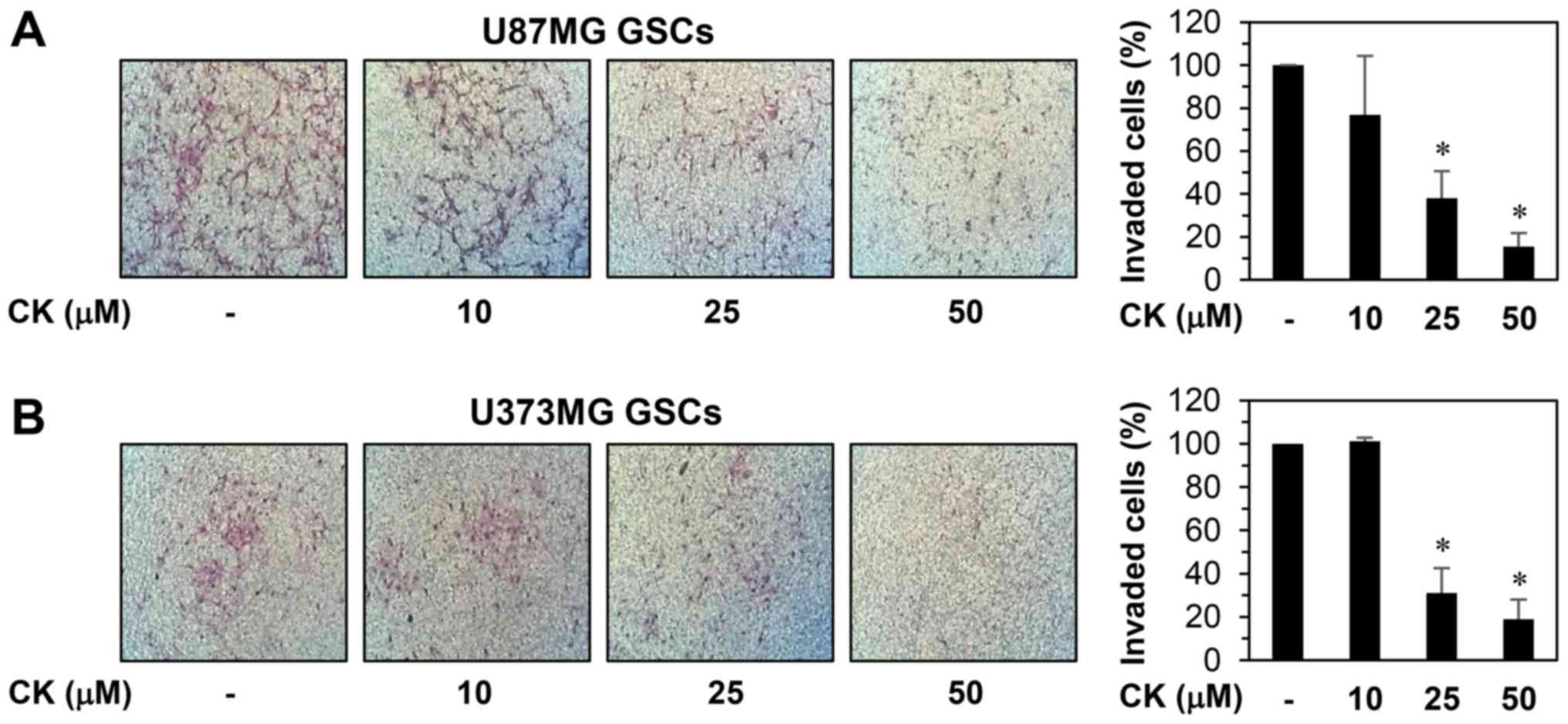
The invasion capabilities of GSCs also contributes
to the initiation of cancer metastasis (31). As such, the anti-invasive potential
of CK on GSCs from U87MG and U373MG cells was evaluated. The
Matrigel invasion assay revealed that CK resulted in a
dose-dependent reduction in the invasiveness of both GSCs (Fig. 11).
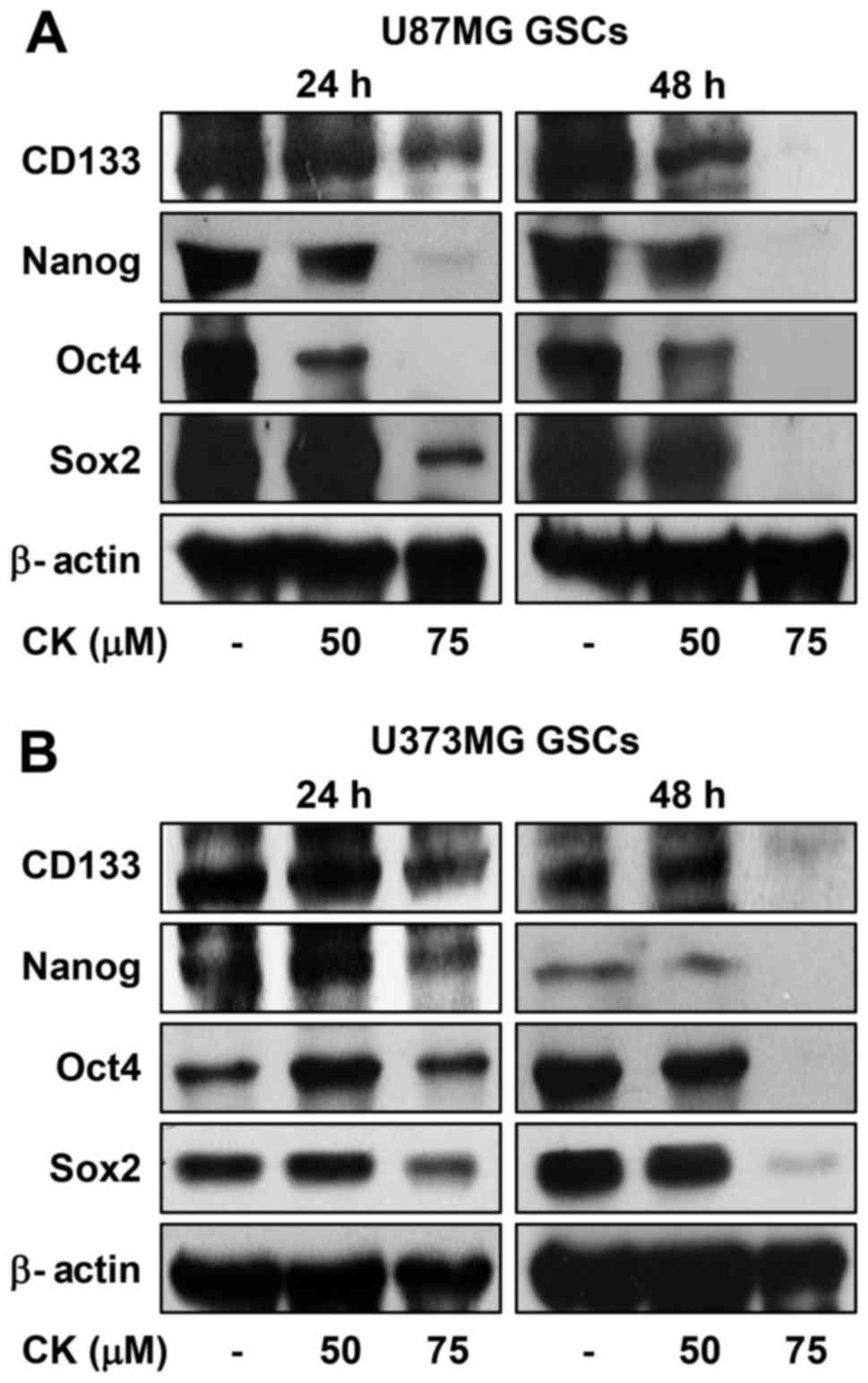
The increased expression of transcription factors,
such as Sox2, Oct4 and Nanog, has been reported to induce stem-like
properties (32). Accordingly, CK
treatment reduced the expression levels of the key stemness
transcription factors, as well as CD133, a cell surface marker for
GSCs, suggesting that the inhibitory effect of CK against GSCs may
be associated with the downregulation of stemness regulators in GBM
(Fig. 12).
Discussion
CK is an active ginsenoside metabolite that has been
used worldwide for preventative and therapeutic purposes (33,34).
A number of studies have shown that CK possesses anticancer
activity against a variety of cancer cells in vitro and
in vivo through the inhibition of oncogenic signaling
pathways (35). However, the
anticancer effects and underlying mechanisms of CK in GBM is not
fully understood.
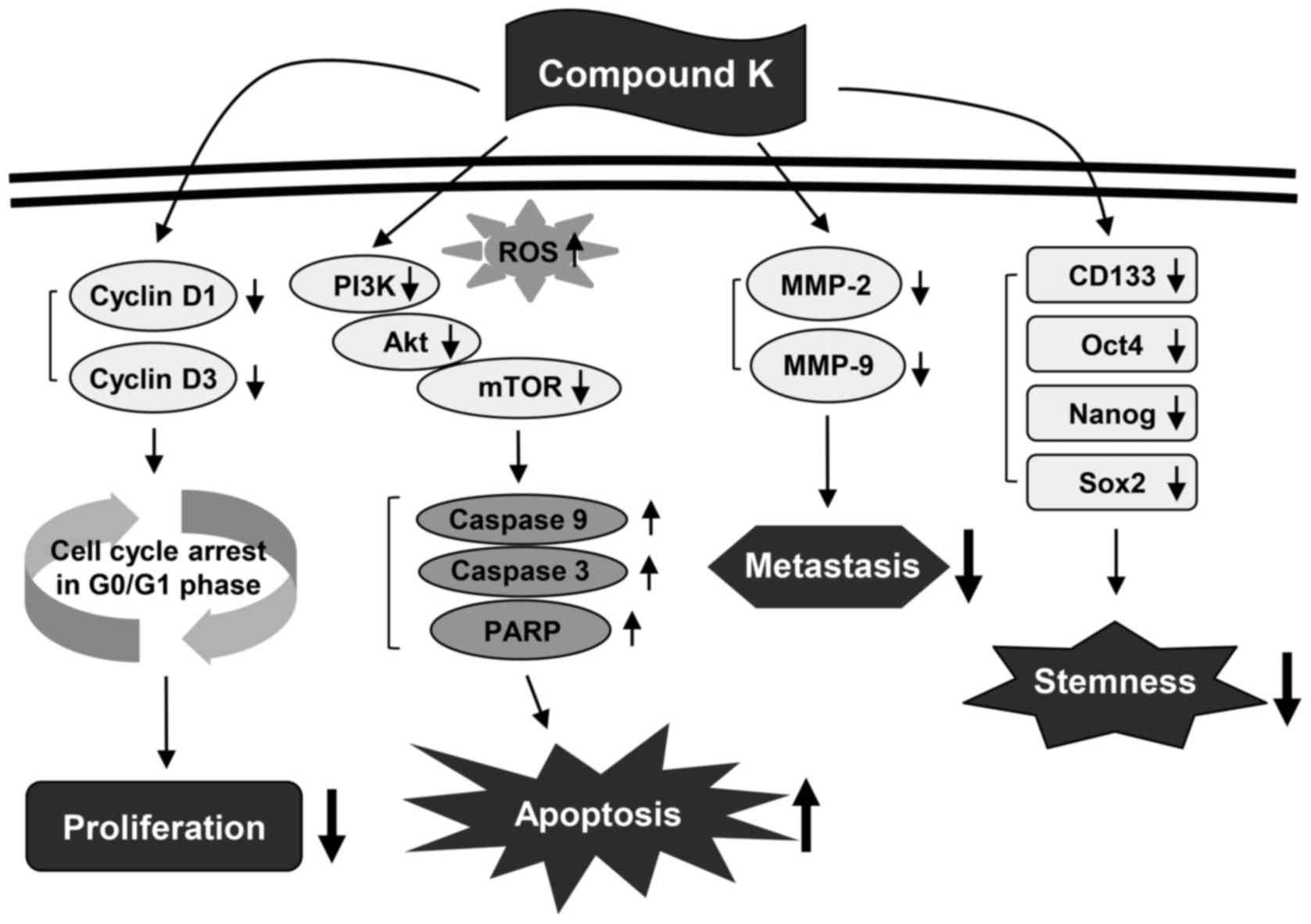
The present study assessed the chemotherapeutic
ability of CK against GBM. Our results demonstrated that CK
significantly inhibits the growth, metastatic potential, and
stemness of GBM cells (Fig. 13).
As detected by MTT and colony forming assays, CK suppressed the
growth of U87MG and U373MG cells. The antiproliferative effect of
CK on GBM cells was caused by arresting cell cycle progression at
the G0/G1 phase and inducing apoptotic cell death. CK treatment
resulted not only in the downregulation of cyclin D1 and cyclin D3
expression, but also the activation of caspase-3, caspase-9 and
PARP in both GBM cell types. In addition, CK suppressed the
phosphorylation of PI3K, Akt and mTOR, suggesting that CK might
promote G0/G1 cell cycle arrest and caspase-dependent apoptosis
through the blockade of PI3K/Akt/mTOR-mediated pathways in human
GBM cells.
Tumor metastasis is promoted by the increased
activity of proteolytic enzymes that are involved in the
destruction of the ECM (36).
Proteolytic enzymes, including MMPs, have been overexpressed during
tumor progression (37). Notably,
elevated levels of MMP-2 and MMP-9 have been closely associated
with the migration and invasion of human GBM cells (38). In the present study, CK also
inhibited the migration and invasion of U87MG and U373MG cells by
downregulating the expression of MMP-2 and MMP-9. Taken together,
these findings indicate that CK possesses promising anticancer
activity against GBM cells via the suppression of cell growth and
metastasis.
Traditional therapies for cancer, such as surgical
resection, chemotherapy and radiotherapy, have several limitations
that lead to cancer recurrence (39). Causes of cancer relapse include
incomplete resection, a high proliferative ability and resistance
to chemotherapy and radiotherapy (40). In recent studies, cancer stem cells
(CSCs) have been proposed as central drivers of tumor initiation,
progression, recurrence and therapeutic resistance (41). CSCs, a subpopulation of tumor
cells, have the ability to increase in number through
self-regeneration and differentiate into various cell types
(42). Increasing evidence has
revealed that GBM also contains CSCs that contribute to tumor
progression and treatment resistance (43). Therefore, targeting GSCs will
improve outcomes for patients with GBM. In the present study, we
determined the inhibitory effect of CK against the cancer stem
cell-like phenotypes of U87MG and U373MG cells that were propagated
through spheroid culture in serum-free media (44). CK treatment significantly reduced
both the self-renewal capacity, including cell growth and
clonogenicity, and the invasive potential of GSCs derived from
U87MG and U373MG cells. Furthermore, CK inhibited the expression of
key stemness markers for GSCs, such as CD133, Nanog, Oct4 and Sox2,
which contribute to self-renewal, multilineage capabilities and
heterogeneity in GSCs (45).
Therefore, these results suggest that CK has the potential to
eradicate GSCs via downregulation of cell surface glycoproteins and
stemness regulatory transcription factors in GSCs.
In conclusion, the present study provides novel
insights into the molecular mechanisms involved in the anticancer
effect of CK in GBM for both cancer cells and cancer stem-like
cells. The blood-brain barrier (BBB) excludes many therapeutic
compounds and thus makes GBM treatment more difficult. Therefore,
the ability of the drug to pass through the BBB is very important
for efficient treatment of GBM. Although the capability of CK to
cross the BBB is unclear, it has been reported to have
neuroprotective and cognition enhancing effects (46). In addition, highly lipophilic
ginsenoside Rd can diffuse across the BBB in an energy deficient
environment (47). In light of the
above, CK, as a metabolite of Rd, may cross the BBB. Together, our
findings suggest that CK could potentially be useful in GBM
treatment.
However, the precise mechanisms regarding how CK
regulates multiple signaling pathways remain unclear. Several
studies have demonstrated that CK inhibits colorectal cancer cell
growth and induces apoptosis by downregulating histone deacetylase
(HDAC) and DNA methyltransferase (DNMT) (17,48).
CK also reduces proliferation and increases apoptosis through the
inhibition of epidermal growth factor receptor (EGFR) and
fibroblast growth factor receptor (FGFR) activation in colon cancer
and myeloma cells respectively (49,50).
Therefore, CK may regulate multiple downstream signaling pathways
that are important for proliferation, apoptosis, metastasis, and
stemness in GBM by targeting growth factor receptors, such as EGFR,
insulin-like growth factor receptor (IGFR), FGFR, platelet-derived
growth factor receptor (PDGFR) and hepatocyte growth factor
receptor (c-Met), as well as key enzymes mediating the epigenetic
regulation of gene expression, such as HDAC and DNMT. Further
studies to identify the upstream cellular target proteins for CK
will be needed to understand the exact mechanisms controlling the
anticancer activity of CK against GBM.
Acknowledgments
The present study was carried out with the support
of 'Cooperative Research Program for Agriculture Science and
Technology Development (Project No. PJ01188001)' Rural Development
Administration, Republic of Korea, the Basic Science Research
Program through the National Research Foundation of Korea (NRF)
funded by the Ministry of Education (NRF-2016R1D1A1B03932956), and
the Brain Korea 21 Plus Project, Republic of Korea. The authors
thank Ilhwa Co., Ltd., (Guri, Republic of Korea) for providing
compound K.
References
|
1
|
Pazhouhi M, Sariri R, Rabzia A and Khazaei
M: Thymoquinone synergistically potentiates temozolomide
cytotoxicity through the inhibition of autophagy in U87MG cell
line. Iran J Basic Med Sci. 19:890–898. 2016.PubMed/NCBI
|
|
2
|
Bak DH, Kang SH, Choi DR, Gil MN, Yu KS,
Jeong JH, Lee NS, Lee JH, Jeong YG, Kim DK, et al: Autophagy
enhancement contributes to the synergistic effect of vitamin D in
temozolomide-based glioblastoma chemotherapy. Exp Ther Med.
11:2153–2162. 2016.PubMed/NCBI
|
|
3
|
Safari M and Khoshnevisan A: Cancer stem
cells and chemo-resistance in glioblastoma multiform: A review
article. J Stem Cells. 10:271–285. 2015.
|
|
4
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al European Organisation for Research and Treatment of Cancer
Brain Tumor and Radiotherapy Groups; National Cancer Institute of
Canada Clinical Trials Group: Radiotherapy plus concomitant and
adjuvant temozolomide for glioblastoma. N Engl J Med. 352:987–996.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Park D and Yoon M: Compound K, a novel
ginsenoside metabolite, inhibits adipocyte differentiation in
3T3-L1 cells: Involvement of angiogenesis and MMPs. Biochem Biophys
Res Commun. 422:263–267. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Attele AS, Wu JA and Yuan CS: Ginseng
pharmacology: Multiple constituents and multiple actions. Biochem
Pharmacol. 58:1685–1693. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cho CW, Kim YC, Kang JH, Rhee YK, Choi SY,
Kim KT, Lee YC and Hong HD: Characteristic study on the chemical
components of Korean curved ginseng products. J Ginseng Res.
37:349–354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guo J, Chang L, Zhang X, Pei S, Yu M and
Gao J: Ginsenoside compound K promotes β-amyloid peptide clearance
in primary astrocytes via autophagy enhancement. Exp Ther Med.
8:1271–1274. 2014.PubMed/NCBI
|
|
9
|
Qi LW, Wang CZ, Du GJ, Zhang ZY, Calway T
and Yuan CS: Metabolism of ginseng and its interactions with drugs.
Curr Drug Metab. 12:818–822. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li W, Zhang M, Gu J, Meng ZJ, Zhao LC,
Zheng YN, Chen L and Yang GL: Hypoglycemic effect of
protopanaxadiol-type ginsenosides and compound K on Type 2 diabetes
mice induced by high-fat diet combining with streptozotocin via
suppression of hepatic gluconeogenesis. Fitoterapia. 83:192–198.
2012. View Article : Google Scholar
|
|
11
|
Kim DY, Yuan HD, Chung IK and Chung SH:
Compound K, intestinal metabolite of ginsenoside, attenuates
hepatic lipid accumulation via AMPK activation in human hepatoma
cells. J Agric Food Chem. 57:1532–1537. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim K, Park M, Lee YM, Rhyu MR and Kim HY:
Ginsenoside metabolite compound K stimulates glucagon-like
peptide-1 secretion in NCI-H716 cells via bile acid receptor
activation. Arch Pharm Res. 37:1193–1200. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Akao T, Kanaoka M and Kobashi K:
Appearance of compound K, a major metabolite of ginsenoside Rb1 by
intestinal bacteria, in rat plasma after oral administration -
measurement of compound K by enzyme immunoassay. Biol Pharm Bull.
21:245–249. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shin KO, Seo CH, Cho HH, Oh S, Hong SP,
Yoo HS, Hong JT, Oh KW and Lee YM: Ginsenoside compound K inhibits
angiogenesis via regulation of sphingosine kinase-1 in human
umbilical vein endothelial cells. Arch Pharm Res. 37:1183–1192.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hasegawa H, Sung JH and Huh JH: Ginseng
intestinal bacterial metabolite IH901 as a new anti-metastatic
agent. Arch Pharm Res. 20:539–544. 1997. View Article : Google Scholar
|
|
16
|
Zhang K and Li Y: Effects of ginsenoside
compound K combined with cisplatin on the proliferation, apoptosis
and epithelial mesenchymal transition in MCF-7 cells of human
breast cancer. Pharm Biol. 54:561–568. 2016. View Article : Google Scholar
|
|
17
|
Kang KA, Piao MJ, Kim KC, Zheng J, Yao CW,
Cha JW, Kim HS, Kim DH, Bae SC and Hyun JW: Compound K, a
metabolite of ginseng saponin, inhibits colorectal cancer cell
growth and induces apoptosis through inhibition of histone
deacetylase activity. Int J Oncol. 43:1907–1914. 2013.PubMed/NCBI
|
|
18
|
Zhang Z, Du GJ, Wang CZ, Wen XD, Calway T,
Li Z, He TC, Du W, Bissonnette M, Musch MW, et al: Compound K, a
ginsenoside metabolite, inhibits colon cancer growth via multiple
pathways including p53-p21 interactions. Int J Mol Sci.
14:2980–2995. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zheng ZZ, Ming YL, Chen LH, Zheng GH, Liu
SS and Chen QX: Compound K-induced apoptosis of human
hepatocellular carcinoma MHCC97-H cells in vitro. Oncol Rep.
32:325–331. 2014.PubMed/NCBI
|
|
20
|
Aroui S, Aouey B, Chtourou Y, Meunier AC,
Fetoui H and Kenani A: Naringin suppresses cell metastasis and the
expression of matrix metalloproteinases (MMP-2 and MMP-9) via the
inhibition of ERK-38-JNK signaling pathway in human glioblastoma.
Chem Biol Interact. 244:195–203. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zou M, Zhang X and Xu C: IL6-induced
metastasis modulators p-STAT3, MMP-2 and MMP-9 are targets of
3,3′-diindolyl-methane in ovarian cancer cells. Cell Oncol (Dordr).
39:47–57. 2016. View Article : Google Scholar
|
|
22
|
Yang N, Hui L, Wang Y, Yang H and Jiang X:
SOX2 promotes the migration and invasion of laryngeal cancer cells
by induction of MMP-2 via the PI3K/Akt/mTOR pathway. Oncol Rep.
31:2651–2659. 2014.PubMed/NCBI
|
|
23
|
Chen BB, Glasser JR, Coon TA and
Mallampalli RK: F-box protein FBXL2 exerts human lung tumor
suppressor-like activity by ubiquitin-mediated degradation of
cyclin D3 resulting in cell cycle arrest. Oncogene. 31:2566–2579.
2012. View Article : Google Scholar
|
|
24
|
Lin JT, Li HY, Chang NS, Lin CH, Chen YC
and Lu PJ: WWOX suppresses prostate cancer cell progression through
cyclin D1-mediated cell cycle arrest in the G1 phase. Cell Cycle.
14:408–416. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qian C, Wang JQ, Song CL, Wang LL, Ji LN
and Chao H: The induction of mitochondria-mediated apoptosis in
cancer cells by ruthenium(II) asymmetric complexes. Metallomics.
5:844–854. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chou YC, Chang MY, Wang MJ, Harnod T, Hung
CH, Lee HT, Shen CC and Chung JG: PEITC induces apoptosis of Human
Brain Glioblastoma GBM8401 Cells through the extrinsic- and
intrinsic signaling pathways. Neurochem Int. 81:32–40. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ray T, Chakrabarti MK and Pal A:
Hemagglutinin protease secreted by V. cholerae induced apoptosis in
breast cancer cells by ROS mediated intrinsic pathway and regresses
tumor growth in mice model. Apoptosis. 21:143–154. 2016. View Article : Google Scholar
|
|
28
|
Liu Y, Jiao R, Ma ZG, Liu W, Wu QQ, Yang
Z, Li FF, Yuan Y, Bian ZY and Tang QZ: Sanguinarine inhibits
angiotensin II-induced apoptosis in H9c2 cardiac cells via
restoring reactive oxygen species-mediated decreases in the
mitochondrial membrane potential. Mol Med Rep. 12:3400–3408.
2015.PubMed/NCBI
|
|
29
|
Yu XS, Du J, Fan YJ, Liu FJ, Cao LL, Liang
N, Xu DG and Zhang JD: Activation of endoplasmic reticulum stress
promotes autophagy and apoptosis and reverses chemoresistance of
human small cell lung cancer cells by inhibiting the PI3K/AKT/mTOR
signaling pathway. Oncotarget. 7:76827–76839. 2016.PubMed/NCBI
|
|
30
|
Bischof J, Westhoff MA, Wagner JE,
Halatsch ME, Trentmann S, Knippschild U, Wirtz CR and Burster T:
Cancer stem cells: The potential role of autophagy, proteolysis,
and cathepsins in glioblastoma stem cells. Tumour Biol.
39:10104283176922272017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jung EH, Lee HN, Han GY, Kim MJ and Kim
CW: Targeting ROR1 inhibits the self-renewal and invasive ability
of glioblastoma stem cells. Cell Biochem Funct. 34:149–157. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo Y, Liu S, Wang P, Zhao S, Wang F, Bing
L, Zhang Y, Ling EA, Gao J and Hao A: Expression profile of
embryonic stem cell-associated genes Oct4, Sox2 and Nanog in human
gliomas. Histopathology. 59:763–775. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim H, Roh HS, Kim JE, Park SD, Park WH
and Moon JY: Compound K attenuates stromal cell-derived growth
factor 1 (SDF-1)-induced migration of C6 glioma cells. Nutr Res
Pract. 10:259–264. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Choi K, Kim M, Ryu J and Choi C:
Ginsenosides compound K and Rh2 inhibit tumor necrosis
factor-alpha-induced activation of the NF-kappaB and JNK pathways
in human astroglial cells. Neurosci Lett. 421:37–41. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang XD, Yang YY, Ouyang DS and Yang GP: A
review of biotransformation and pharmacology of ginsenoside
compound K. Fitoterapia. 100:208–220. 2015. View Article : Google Scholar
|
|
36
|
Gutschalk CM, Yanamandra AK, Linde N,
Meides A, Depner S and Mueller MM: GM-CSF enhances tumor invasion
by elevated MMP-2, -9, and -26 expression. Cancer Med. 2:117–129.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kalhori V and Törnquist K: MMP2 and MMP9
participate in S1P-induced invasion of follicular ML-1 thyroid
cancer cells. Mol Cell Endocrinol. 404:113–122. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chou YC, Chang MY, Wang MJ, Yu FS, Liu HC,
Harnod T, Hung CH, Lee HT and Chung JG: PEITC inhibits human brain
glioblastoma GBM 8401 cell migration and invasion through the
inhibition of uPA, Rho A, and Ras with inhibition of MMP-2, -7 and
-9 gene expression. Oncol Rep. 34:2489–2496. 2015.PubMed/NCBI
|
|
39
|
Lathia JD, Mack SC, Mulkearns-Hubert EE,
Valentim CL and Rich JN: Cancer stem cells in glioblastoma. Genes
Dev. 29:1203–1217. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cho DY, Lin SZ, Yang WK, Lee HC, Hsu DM,
Lin HL, Chen CC, Liu CL, Lee WY and Ho LH: Targeting cancer stem
cells for treatment of glioblastoma multiforme. Cell Transplant.
22:731–739. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cruceru ML, Neagu M, Demoulin JB and
Constantinescu SN: Therapy targets in glioblastoma and cancer stem
cells: Lessons from haematopoietic neoplasms. J Cell Mol Med.
17:1218–1235. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gatti M, Pattarozzi A, Bajetto A, Würth R,
Daga A, Fiaschi P, Zona G, Florio T and Barbieri F: Inhibition of
CXCL12/CXCR4 autocrine/paracrine loop reduces viability of human
glioblastoma stem-like cells affecting self-renewal activity.
Toxicology. 314:209–220. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ashizawa T, Miyata H, Iizuka A, Komiyama
M, Oshita C, Kume A, Nogami M, Yagoto M, Ito I, Oishi T, et al:
Effect of the STAT3 inhibitor STX-0119 on the proliferation of
cancer stem-like cells derived from recurrent glioblastoma. Int J
Oncol. 43:219–227. 2013.PubMed/NCBI
|
|
44
|
Kim B, Jung N, Lee S, Sohng JK and Jung
HJ: Apigenin inhibits cancer stem cell-like phenotypes in human
glioblastoma cells via suppression of c-Met signaling. Phytother
Res. 30:1833–1840. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Safa AR, Saadatzadeh MR, Cohen-Gadol AA,
Pollok KE and Bijangi-Vishehsaraei K: Emerging targets for
glioblastoma stem cell therapy. J Biomed Res. 30:19–31. 2015.
|
|
46
|
Oh J and Kim JS: Compound K derived from
ginseng: Neuroprotection and cognitive improvement. Food Funct.
7:4506–4515. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ye R, Zhang X, Kong X, Han J, Yang Q,
Zhang Y, Chen Y, Li P, Liu J, Shi M, et al: Ginsenoside Rd
attenuates mitochondrial dysfunction and sequential apoptosis after
transient focal ischemia. Neuroscience. 178:169–180. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kang KA, Kim HS, Kim DH and Hyun JW: The
role of a ginseng saponin metabolite as a DNA methyltransferase
inhibitor in colorectal cancer cells. Int J Oncol. 43:228–236.
2013.PubMed/NCBI
|
|
49
|
Dougherty U, Mustafi R, Wang Y, Musch MW,
Wang CZ, Konda VJ, Kulkarni A, Hart J, Dawson G, Kim KE, et al:
American ginseng suppresses Western diet-promoted tumorigenesis in
model of inflammation-associated colon cancer: Role of EGFR. BMC
Complement Altern Med. 11:1112011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Choi HH, Jong HS, Park JH, Choi S, Lee JW,
Kim TY, Otsuki T, Namba M and Bang YJ: A novel ginseng saponin
metabolite induces apoptosis and down-regulates fibroblast growth
factor receptor 3 in myeloma cells. Int J Oncol. 23:1087–1093.
2003.PubMed/NCBI
|