Introduction
The erythropoietin receptor (EPOR) is a
transmembrane protein from the type I cytokine receptor superfamily
(1). The majority of EPOR is
expressed on erythropoietic progenitor cells in the hematopoietic
tissue of the bone marrow (reviewed in ref. 2). After erythropoietin (EPO) binding,
the preformed EPOR is activated and triggers several downstream
signaling pathways, including those of Janus kinase 2/signal
transducer and activator of transcription 5 (JAK2/STAT5),
phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT), guanine
nucleotide binding protein (3)/mitogen-activated protein kinase
(MAPK), and protein kinase C (PKC) (reviewed in ref. 4). The activation of this signaling not
only results in erythroid proliferation and differentiation, but
also in protection of erythroid progenitors against apoptosis
(5).
Subsequently, the presence of functional EPOR was
confirmed in a number of non-hematopoietic cells, including neurons
(6), endothelial cells (7), kidney cells (8), and myoblasts (9), as well as in various tumor cells.
Reports have emerged over the last 6 years that have described
active EPOR signaling in head and neck squamous cell carcinomas
(10), cervical cancer cells
(11), glioma cells (12), renal carcinoma cells (13), and breast cancer cells (14). These findings of functional EPOR
for numerous body tissues then led to the description of novel
physiological effects of EPO/EPOR signaling, in addition to the
regulation of erythropoiesis (15). The presence of EPOR in tumor
tissues and cell lines raises the question of possible adverse
effects on tumor-cell proliferation and growth, and on inhibition
of apoptosis. These effects might be induced by recombinant human
EPO or its analogs (e.g., erythropoiesis-stimulating agents) that
are used in the treatment of patients with cancers who suffer from
chemotherapy-induced anemia (reviewed in ref. 16).
Breast cancer is the second most frequent type of
cancer worldwide, with over 1.5 million new cases per year. Only
lung cancer accounts for more cancer deaths in women. Breast cancer
cells can express a variety of growth factor receptors, which
determine the molecular classification of the disease (17). The most commonly identified of
these growth factor receptors is the estrogen receptor (ER)
(18), which can provide important
prognostic and predictive information of tumor responses to
endocrine-based therapies (19).
The standard therapies for ER-positive (ER+) breast
cancer include the administration of selective ER-activity
modulators or inhibitors of estrogen biosynthesis.
Treatment with the chemopreventive agent and
selective ER-activity modulator tamoxifen was the first targeted
therapy for mammary adenocarcinomas (20). Tamoxifen was approved by the US
Food and Drug Administration in 1998 for the reduction of breast
cancer risk in premenopausal and postmenopausal women in the USA.
Tamoxifen and some other selective ER-activity modulators can have
estrogen-like agonistic effects as well as anti-estrogen-like
antagonistic effects that are tissue-selective or
cell-type-selective. In mammary adenocarcinoma cells, tamoxifen
prevents estrogen binding to the ER, which thus reduces cell
proliferation and tumor growth (21). Tamoxifen has also been shown to
induce apoptosis and G1 cell cycle arrest in human breast cancer
cells, and to inhibit ER-independent and MAPK-induced cell
proliferation (22–24).
However, some ER+ breast tumors fail to
respond to tamoxifen, or develop resistance with prolonged
tamoxifen treatment (25).
Tamoxifen resistance is rarely caused by mutations in ER (26). The alterations in signaling
pathways that have been implicated in acquired tamoxifen resistance
are those that affect cell survival and proliferation, the cell
cycle, apoptosis, and epithelial-to-mesenchymal transition of tumor
cells (27,28). In this regard, tamoxifen-resistant
breast cancer phenotypes are promoted by changes in AKT activity
(29).
Responses of breast cancer cells to tamoxifen have
also been correlated to EPOR expression. Larsson et al
(30) indicated that high EPOR
expression is a negative prognostic factor for recurrence-free
survival of tamoxifen-treated patients with
ER+/progesterone receptor (PR)+ breast
tumors. On the contrary, recurrence-free survival of non-treated
ER+/EPOR+ breast cancer patients was
significantly improved. Their study also revealed associations
between EPOR, ER and PR in breast cancer cells and patients, while
Volgger et al (32) further
showed positive association between EPOR/ER/PR status and increased
local cancer recurrence.
In terms of breast cancer cell lines, EPOR
expression has been associated with expression of G-protein-coupled
ER (GPER) (33). Silencing of EPOR
expression via EPOR knock-down resulted in decreased
proliferation of cultured EPOR+/ERα+ breast
cancer cells, but not of ERα− cells. Recombinant human
EPO stimulation of cultured cells also had no effects on cell
proliferation (34).
The objective of the present study was to assess the
effects of EPOR on the growth of breast cancer cells, and on their
sensitivity to tamoxifen, in the absence of stimulation with
recombinant human EPO. For this purpose, we compared two rat
mammary epithelial cell lines that show different levels of EPOR
expression. RAMA 37 cells are a benign non-invasive cell line that
shows low EPOR expression, while a RAMA 37 clone, known as RAMA
37-28 cells, stably expresses higher levels of human EPOR (35). Herein, both of these cell lines
were characterized for EPOR and ER expression and compared to each
other in terms of their proliferation capacity, response to
tamoxifen-induced cytotoxicity, and activation of protein
signaling.
Materials and methods
Cell lines and cell culture
The parental non-metastatic benign tumor-derived rat
mammary epithelial cell line, RAMA 37 cells, and its derived stably
transformed cell subclone, RAMA 37-28 cells, were used as the model
system. Transfection was carried out with the pcDNA3.1 expression
vector that contained wild-type human EPOR, followed by selection
of modified cells with 1.0 mg/ml geneticin (35). The cells were cultured in
Dulbecco's modified Eagle's medium supplemented with high glucose
(4.5 g/l), L-glutamine (GE Healthcare, Little Chalfont, UK) and 10%
fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA). The cell cultures were maintained at 37°C in a humidified
5% CO2 (v/v) atmosphere. Cell numbers were determined
using a Coulter counter (model ZF; Coulter Electronics, Luton, UK),
and total cell viability was analyzed by staining with 0.15% eosin,
followed by light microscopy.
Gene expression analysis
RNA isolation
Total RNA was extracted using the TRI reagent
(Sigma-Aldrich, St. Louis, MO, USA), and treated with DNase I
(Roche Diagnostic, Basel, Switzerland), according to the
manufacturer instructions. The quality of the RNA samples was
determined using a bioanalyzer (Agilent), to assure that all RIN
values were >9.8. Total RNA (1 µg) was transcribed to
cDNA using SuperScript III reverse transcriptase (Invitrogen,
Carlsbad, CA, USA), according to the manufacturer's
instructions.
Quantitative real-time reverse
transcriptase-PCR
The PrimerExpress software (Applied Biosystems,
Foster City, CA, USA) was used for the design of forward and
reverse primers for EPO, EPOR, ESR1, ESR2, GPER, RPLP0 and
CYCA that spanned intron-exon junctions. These were checked
for their specificities with the BLAST algorithm. RPLP0 and
CYCA were selected as the reference genes, and were used for
normalization of the expression levels for the genes of interest.
Quantitative real-time reverse transcriptase was performed using
SybrGreen chemistry (Roche Diagnostic) on a 384-well platform of a
LightCycler 480 Real-time PCR System (Roche Diagnostic).
Amplification of the specific PCR products was performed in
triplicate in a total reaction mixture of 5 µl, which
contained 0.75 µl of the cDNA template. Gene expression
normalization factors were calculated using the GeNorm algorithm
for each sample, based on the geometric means of the selected
reference genes (31). The minimum
information for publication of quantitative real-time PCR
experiments (MIQE) was followed in the performance and
interpretation of the qPCR reactions (36). The data are presented as relative
expression levels for each gene of interest ± SD, from three
biological replicates.
Cell proliferation
MTT assay
The cells (12.5×103) in the medium
supplemented with 1% or 10% fetal bovine serum were seeded as
quintuplicates in 96-well plates, and left to adhere for 24 h. Cell
proliferation was determined using
3-(4.5-dimethylthiazol-2-yl)-2.5-diphenyltetrazolium bromide (MTT
reagent; Sigma-Aldrich), and was measured for five sequential days.
Each day, MTT reagent was added to the wells of one plate, which
was then incubated for 4 h under the standard culture conditions.
Viable cells with active metabolism can convert MTT into an
insoluble formazan product, which can be dissolved by addition of
20% sodium dodecyl sulfate (in HCl). Absorbance was measured at 570
nm using a spectrophotometer (Epoch; BioTek, Winooski, VT, USA).
After the blank subtraction, the fold-changes in cell proliferation
were determined for each day, by normalization to the proliferation
levels after 24 h of culturing. The data are presented as mean
fold-changes in cell proliferation ± SD, of three independent
experiments.
Clonogenic assays
The cells were seeded in 6-well plates in triplicate
at 100 cells/well and cultured for 14 days. The culture medium was
changed every 5 days. Colony quantification was carried out
manually and using the UviPro software after crystal violet
staining (0.5%; Sigma-Aldrich). Colonies were classified as small
if they contained <100 cells, and otherwise they were classified
as big. The data are presented as mean colony numbers ± SD, of
three independent experiments.
Cell viability assays
xCELLigence
The experiments were performed using an xCELLigence
dual-plate real-time cell analyzer (Roche Diagnostic), which was
placed in a humidified incubator at 37°C and 5% CO2.
Cell proliferation and cytotoxicity were determined using 16-well
plates (E-plates; Roche Diagnostic) with electrodes attached to the
bottom for impedance-based detection of cell attachment, spreading,
and proliferation. The impedance of each well was automatically
monitored using the xCELLigence system, which was expressed as the
Cell Index. Initially, 100 µl cell-free media (with 10%
fetal bovine serum) was added to the wells and incubated for 30 min
at room temperature. This was following by addition of 50 µl
cell suspension (for 1,250 cells/well). The cells were left to
adhere for 30 min at room temperature, and then placed on the
real-time cell analyzer for 72 h. The Cell Index was monitored
every hour. After the initial 72 h of culture, the cells were
treated with tamoxifen (Sigma-Aldrich) at 25, 37, 50, 75, 100 or
150 µM (in ethanol). Ethanol-treated cells and cells without
tamoxifen treatment were used as controls. Each tamoxifen
concentration was tested in duplicate within the same experiment,
and the Cell Index was again monitored every hour, to 48 h of
tamoxifen treatment.
Normalized Cell Index
The normalized Cell Index (37) was calculated by its normalization
with the Cell Index at the point of tamoxifen treatment (i.e., 72
h). The data are presented as the mean normalized Cell Index curves
± SD, of three independent experiments.
IncuCyte ZOOM system
Experiments were performed using an IncuCyte ZOOM
system (Essen BioScience, Ann Arbor, MI, USA), which consists of a
microscope gantry in a humidified incubator at 37°C and 5%
CO2, and a networked external controller hard drive that
gathers and processes image data. The cells were seeded in 96-well
plates in sextuplicates at 2,000 cells/well (as 100 µl cell
suspension/well) and placed in the IncuCyte ZOOM system. After the
initial 24 h of culture, the cells were treated with 25 µM
tamoxifen (dissolved in ethanol). Ethanol-treated cells and those
growing in media without tamoxifen were used as controls. The
IncuCyte ZOOM system automatically monitored the cell confluence in
each well through a ×20 objective (Nikon) every 2 h, until 72 h of
tamoxifen treatment. The experiment was performed three times.
Western blotting
Western blotting was carried out according to
standard protocols. Using the MTT assay, 50 µM tamoxifen was
defined as the optimal concentration for analysis of protein
signaling. The cells were grown in the standard growth media for 24
h and then exposed to 50 µM tamoxifen for 0, 5, 10, 15, 30
and 60 min. The cells were then lysed for 10 min in lysis buffer on
ice, as described by Kutuk et al (38), and the soluble proteins were
recovered in the supernatant following centrifugation at 12,000 × g
for 10 min. Protein samples were separated using 12% SDS-PAGE gels
and equal loading confirmed by detection of β-actin. This was
followed by electroblotting onto Immobilon-P transfer membranes
(Millipore, Billerica, MA, USA), which were then incubated
overnight with the primary antibodies, as specified in Table I. The membranes were then incubated
with the secondary horseradish-peroxidase-conjugated antibody
(Table I) for 1 h, and the
antibody reactivity was visualized using ECL Western blotting
substrate (Pierce, Waltham, MA, USA) and Kodak Biomax film
(Sigma-Aldrich). The data are shown as a representative figure and
in Table as means of the densities recorded from three independent
experiments.
 | Table IPrimary and secondary antibodies used
in the western blotting. |
Table I
Primary and secondary antibodies used
in the western blotting.
| Type | Antibody | Specificity | Dilution | Manufacturer | Cat. no. |
|---|
| Primary | Anti-p44/42 MAP
kinase (ERK1/2) | Rabbit | 1:1000 | Cell Signaling
Technology | 9102 |
|
Anti-phospho-p44/42 | Rabbit | 1:1000 | Cell Signaling
Technology | 9101 |
| MAP kinase
(pERK1/2) | | | | |
| Anti-AKT | Rabbit | 1:1000 | Cell Signaling
Technology | 9272 |
|
Anti-phospho-AKT | Rabbit | 1:1000 | Cell Signaling
Technology | 9271 |
| Anti-STAT5 | Rabbit | 1:2000 | R&D
Systems | AF2168 |
| Anti-phospho-STAT5
(Y694/Y699) | Rabbit | 1:1000 | R&D
Systems | AF4190 |
| Anti-EPOR
(M-20) | Rabbit | 1:400 | Santa Cruz
Biotechnology | sc697 |
| Anti-ERα
(F-10) | Mouse | 1:2000 | Santa Cruz
Biotechnology | sc-8002 |
| Anti-ERβ
(H-150) | Rabbit | 1:1000 | Santa Cruz
Biotechnology | sc-8974 |
| Anti-GPER | Rabbit | 1:500 | Sigma
Chemicals | HPA027052 |
| Anti-β-actin | Mouse | 1:5000 | Sigma
Chemicals | A5441 |
| | Rabbit | 1:2000 | Sigma
Chemicals | A2066 |
| Secondary | Anti-mouse IgG | Rabbit | 1:2000 | Pierce | 31452 |
| | Goat | 1:5000 | Jackson
ImmunoResearch | 115-035-068 |
| | | | Laboratories | |
| Anti-rabbit
IgG | Goat | 1:2000 | Pierce | 31461 |
| | Goat | 1:5000 | Jackson
ImmunoResearch | 111-035-003 |
| | | | Laboratories | |
Statistical analysis
The significance of the differences in gene
expression and colony formation capacity between the RAMA 37 and
RAMA 37-28 cells were determined using Student's t-tests.
Statistical significance for cell proliferation or in response to
tamoxifen-induced cytotoxicity was determined using two-way ANOVA
tests, while one-way ANOVA was used to assess the statistical
differences in the protein signaling. P<0.05, p<0.01 or
p<0.001 were considered as statistically significant.
Results
Expression of erythropoietin and estrogen
receptor
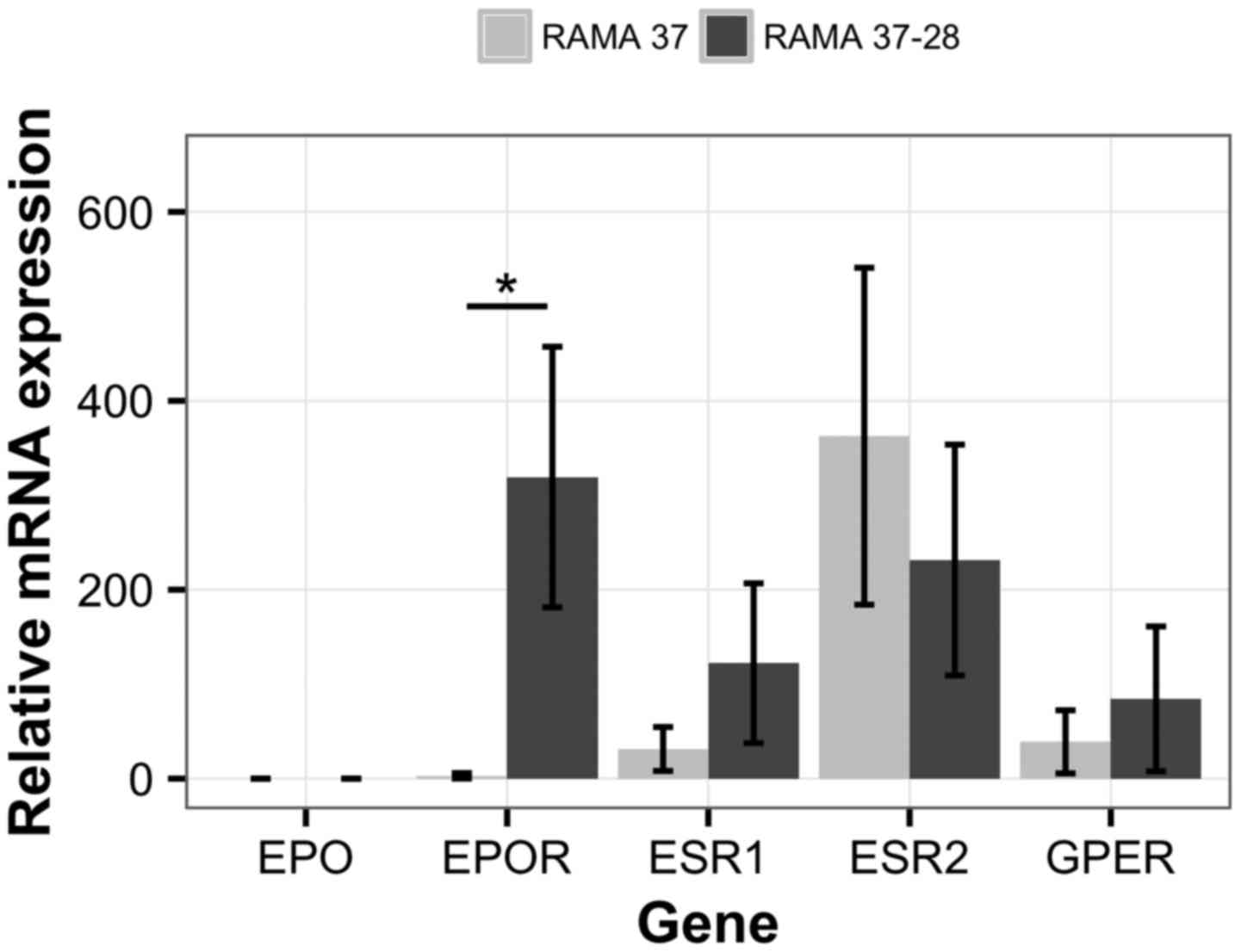
Real-time PCR analysis of EPO, EPOR, ESR1,
ESR2, and GPER was performed to determine the level of
endogenous EPO mRNA and the differences in receptor status
between the RAMA 37 and RAMA 37-28 cells (Fig. 1). Endogenous EPO expression
was not confirmed in the tested cell lines. EPOR mRNA levels
were not detectable in the parental RAMA 37 cells, while relatively
high expression was seen for the RAMA 37-28 cells, which had been
stably transfected with human EPOR (35). ESR1 expression was low in
both cell lines, while ESR2 was slightly higher in RAMA 37
cells. However, the difference in ESR2 expression between
the two cell lines was not statistically significant. The
expression level of GPER was low in both cell lines.
The EPO, EPOR, and ER expression was evaluated at
the protein level. The expression of ERβ (i.e., the protein product
of ESR2), EPOR, and GPER was confirmed in both cell lines
(Fig. 2). Low levels of EPOR were
detected in the parental RAMA 37 cells, with higher expression in
the RAMA 37-28 cells. EPO and ERα (i.e., the protein product of
ESR1) were not detected at the protein level.
Effects of EPOR overexpression on cell
growth
The RAMA 37 and RAMA 37-28 cells were compared in
terms of cell growth and proliferation, and their clonogenic
characteristics, to evaluate the effects of EPOR overexpression on
the cell physiology (Figs. 3 and
4). Over 5 days, RAMA 37 cells
showed higher proliferation levels compared to RAMA 37-28 cells in
media supplemented with 1% or 10% fetal bovine serum (Fig. 3). Statistically significant
differences (p<0.05) were confirmed for days 4–5 in 1% fetal
bovine serum, and for days 3–5 in 10% fetal bovine serum.
Colony numbers were determined according to colony
size (Fig. 4). Colonies were
considered small if they containing <100 cells, or they were
considered big at >100 cells. The number of big colonies was
comparable for both of these cell lines. However, the number of
small colonies was significantly higher for RAMA 37-28 cells.
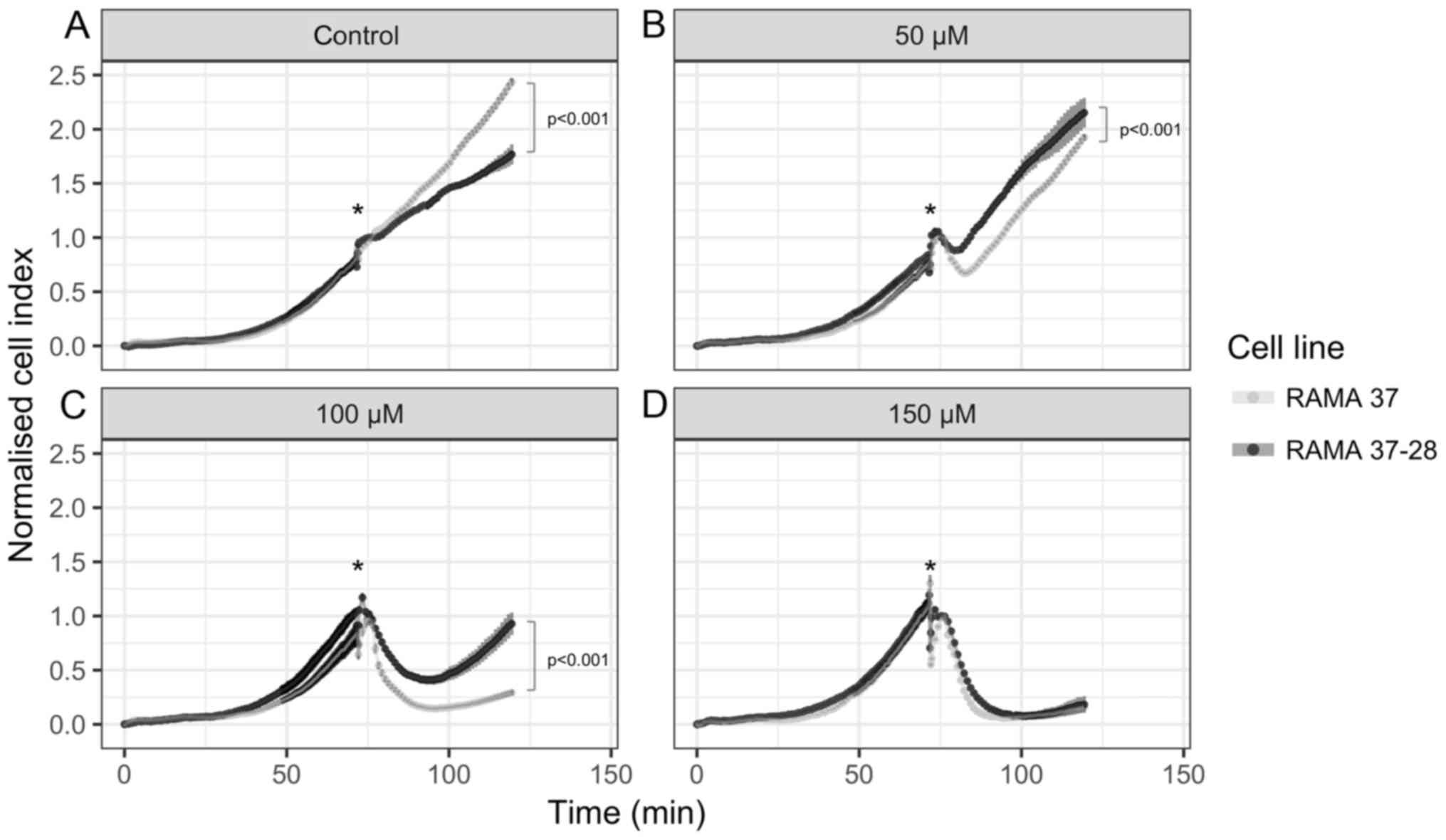
Effects of EPOR on tamoxifen
resistance
The proliferation of RAMA 37 and RAMA 37-28 cells
and their responses to tamoxifen treatment were screened using the
xCELLigence system (Fig. 5). The
cells were plated and left to proliferate for 72 h, and then they
were treated with tamoxifen for 48 h (i.e., to 120 h). The
proliferation capacity of RAMA 37 cells under the control
conditions was significantly higher that for the RAMA 37-28 cells
(Fig. 5A). However, the RAMA 37-28
cells were more resistant to the tamoxifen treatment, as they
showed faster proliferation in the presence of tamoxifen (Fig. 5B and C). The highest tamoxifen
concentration (150 µM) was lethal for both of these cell
lines (Fig. 5D).
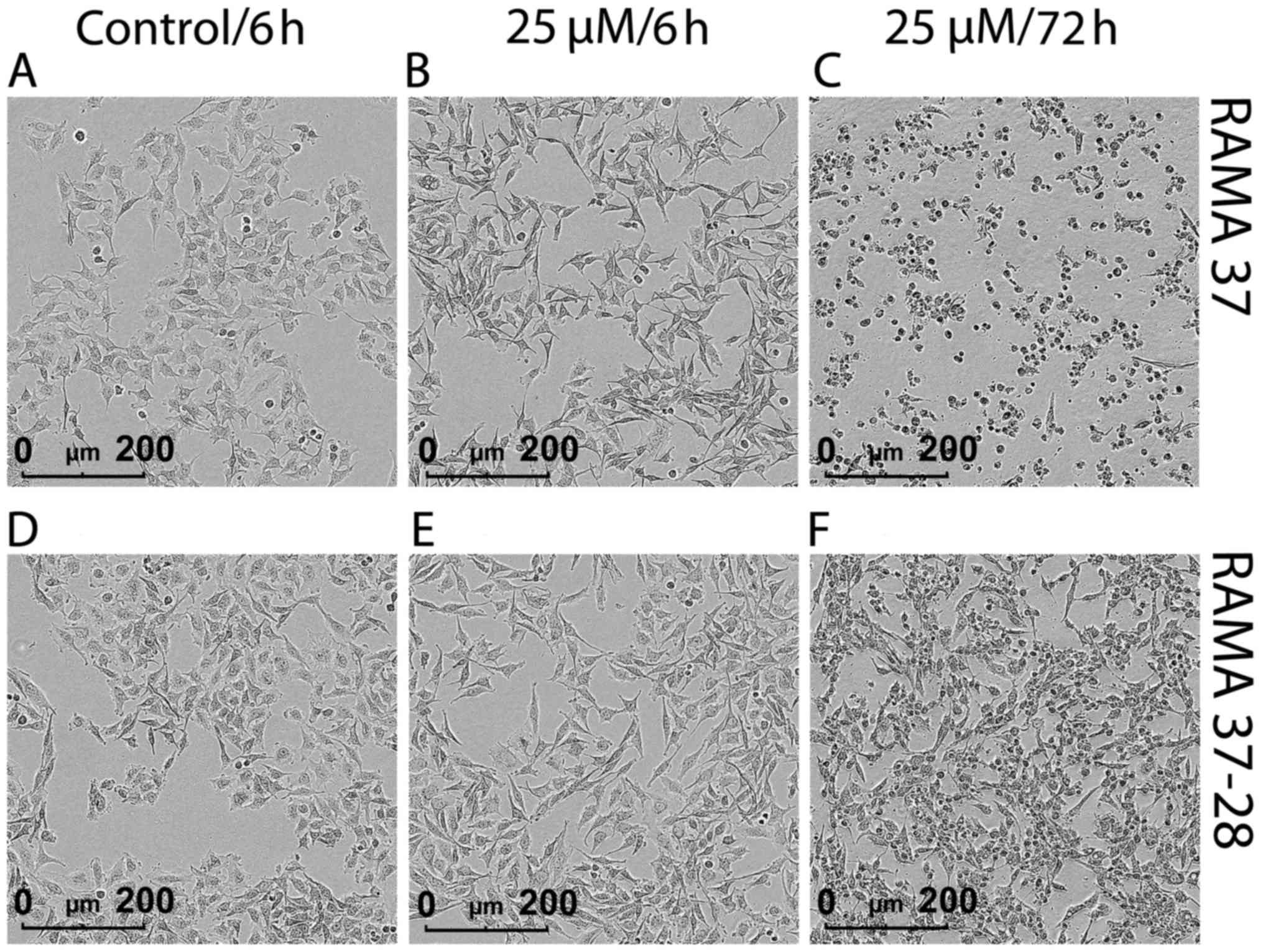
The morphological changes of the RAMA 37 and RAMA
37-28 cells upon tamoxifen treatment were assessed using the
IncuCyte ZOOM system, which monitors cell proliferation by
determining the area occupied by the cells (i.e., the percent
confluence) in images taken over time (Fig. 6). The cells were plated and left to
proliferate for 24 h, followed by 25 µM tamoxifen treatment
for 72 h. This tamoxifen treatment promoted morphological changes
in both cell lines, although they were more evident and more rapid
in onset for RAMA 37 cells (Fig.
6A–C). Short-term tamoxifen treatment (6 h) promoted the
formation of nanotube-like structures (i.e., protrusions) that were
more pronounced and longer for RAMA 37 cells (Fig. 6B). These morphological changes
might be indicative of a pre-apoptosis stage. Moreover, prolonged
treatment with tamoxifen (72 h) promoted greater cell death for
RAMA 37 cells (Fig. 6C), with RAMA
37-28 cells not affected to the same extent (Fig. 6F). Movies demonstrating the
differences in the tamoxifen responsiveness of RAMA 37 and RAMA
37-28 cells are available at http://ibk.mf.uni-lj.si/people/debeljak/RAMA.
Modified signal activation during
tamoxifen treatment
The effects of tamoxifen treatment on signal
transduction were determined using Western blotting for ERK1/2,
AKT, and STAT5a/b phosphorylation (Fig. 7 and Table II). Upon tamoxifen treatment, no
STAT5 phosphorylation (pSTAT5) was observed in RAMA 37 cells or in
the EPOR-overexpressing RAMA 37-28 cells. The PI3K/AKT signaling
pathway was activated already in both of these cell lines prior the
tamoxifen treatment, as they both showed low levels of endogenous
AKT phosphorylation (pAKT). Tamoxifen treatment then changed the
AKT phosphorylation patterns in a cell-type-dependent manner. The
pAKT levels were reduced in RAMA 37 cells (e.g., 15 min of
tamoxifen treatment), but increased in RAMA 37-28 cells. These
increased pAKT levels in RAMA 37-28 cells were still seen after 60
min of tamoxifen treatment, which suggests that the activity of the
PI3K/AKT signaling pathway is increased in this cell line.
Tamoxifen treatment also resulted in changes in the RAS/MAPK
signaling pathway, with increased phosphorylation levels of ERK1/2
(pERK1/2) were seen. ERK1/2 phosphorylation was a little faster and
stronger in RAMA 37 cells, but still comparable to that in RAMA
37-28 cells.
| Table IIEffect of tamoxifen treatment on AKT
and ERK1/2 phosphorylation in RAMA 37 and RAMA 37-28 cells. |
Table II
Effect of tamoxifen treatment on AKT
and ERK1/2 phosphorylation in RAMA 37 and RAMA 37-28 cells.
| Cell type | Target
ratioa | Ratio according to
tamoxifen treatment (min)
|
|---|
| 0 | 5 | 10 | 15 | 30 | 60 |
|---|
| RAMA 37 | pAKT | 1.00±0.00 | 1.42±0.44 | 0.86±0.12 | 0.53±0.02d | 0.32±0.03d | 0.30±0.06d |
| pERK1/2 | 1.00±0.00 | 1.97±0.27 | 2.27±1.00 | 2.32±0.71b | 2.24±0.34b | 2.74±0.86b |
| RAMA 37-28 | pAKT | 1.00±0.00 | 1.38±0.30 | 1.03±0.18 | 0.83±0.09 | 1.15±0.30 | 1.16±0.30 |
| pERK1/2 | 1.00±0.00 | 1.55±0.85 | 1.40±0.27 | 1.89±0.30b | 2.50±0.50c | 2.42±0.40c |
Discussion
Many studies have suggested that EPOR has a role in
tumor progression (30) through
the stimulation of cell proliferation and/or inhibition of
apoptosis of cancer cells upon EPO binding (10,14).
In contrast, other studies have argued that although EPOR is
present in cancer cells, it is not biologically active and is not
essential for tumor growth (2,39),
as stimulation with exogenous EPO does not stimulate tumor cell
proliferation (34). However,
cancer cells are believed to undergo a continuous cycle of
selection changes, with only the more adapted cells favored for the
passing on of their genetic information. These cells are not likely
to retain biologically inactive metabolic and regulatory pathways,
and therefore it is reasonable to expect that EPOR expression in
cancer cells is not redundant, but is instead involved in the tumor
biology.
In the present study, we examined the effects of
EPOR on the growth of breast cancer cells, and its role in the
tamoxifen response. RAMA 37 and RAMA 37-28 cells differ only in
their EPOR expression levels, which we confirmed with the
expression analysis at the mRNA and protein levels. Indeed, there
were no significant differences in the expression of ERα (i.e., the
product of ESR1), ERβ (i.e., the product of ESR2) or
GPER between these two cell lines, indicating that they represent a
good model for the evaluation of EPOR effects on cell
physiology.
The ERα status is used as a prognostic marker in
breast cancer evaluation, and it is the primary target of endocrine
therapies. Generally, patients with ERα-negative tumors cannot
benefit from tamoxifen therapy, although a fraction of these tumors
do appear to be sensitive to tamoxifen (28,40).
Furthermore, phosphorylation of ERα by protein kinase A has been
shown to convert the antagonist tamoxifen into an agonist, thereby
reversing its effects on tumor cell growth. Activation of protein
kinase A can occur through GPER or adenylyl cyclase, although it is
difficult to achieve constitutive protein kinase A activation due
to desensitization of both of these receptors upon activation
(41). Additionally, low levels of
ERβ are associated with tamoxifen resistance, and as such this
might serve as an additional independent predictive marker
(42). As we demonstrated
comparable expression levels of ERα, ERβ, and GPER in these RAMA 37
and RAMA 37-28 cells, we propose that (an)other molecule(s) or
signaling pathway(s) contribute to the tamoxifen-resistant
phenotype that is observed for RAMA 37-28 cells.
We showed that EPOR overexpression in these cancer
cells can influence cell proliferation and resistance even in the
absence of EPO. The greater proliferation capacity of RAMA 37 cells
indicates that EPOR lowered the ability of RAMA 37-28 cells to
divide. When comparing the clonogenic potential of RAMA 37 and RAMA
37-28 cells, the RAMA 37-28 cells were more clonogenic. Although
the numbers of big colonies (>100 cells) were comparable across
both of these cell lines, RAMA 37-28 cells formed a significantly
greater number of small colonies (<100 cells). This greater
clonogenic potential of RAMA 37-28 cells has also been previously
indicated (35). Reinbothe et
al (34) suggested a role for
EPOR in the proliferation control of ERα breast cancer cells, as
EPOR knock-down in ERα-positive breast cancer cell lines resulted
in cell growth inhibition. Furthermore, a study on A2780 ovarian
cancer cell line reported that inhibition of EPOR expression
abrogated growth of its tumor xenograft (39).
Tamoxifen induces apoptosis in human breast cancer
cell lines and inhibits cell proliferation in human ovarian cancer
cell lines through a mechanism that is independent of overall ER
expression (22–24). We observed here that while under
the control conditions the proliferation potential of RAMA 37 cells
was higher than that of RAMA 37-28 cells, the treatment with
tamoxifen affected RAMA 37 cells more and resulted in a greater
block of proliferation and a higher rate of cell death than for
RAMA 37-28 cells. These data therefore suggest that EPOR can
protect cells against tamoxifen-induced cell death even in the
absence of EPO.
Previously, Reinbothe et al (34) showed that EPOR knock-down resulted
in a more efficient tamoxifen-induced inhibition of the ERα
activity. Herein, tamoxifen treatment caused morphological changes
to RAMA 37 and RAMA 37-28 cells that corresponded to a
pre-apoptotic stage. These morphology changes were manifested in
the form of pronounced protrusions that were especially obvious in
RAMA 37 cells, which correlated with their higher sensitivity to
tamoxifen-induced cytotoxicity. Protrusions were less obvious in
RAMA 37-28 cells and might correlate with the higher survival rate
(greater resistance) of these RAMA 37-28 cells. Based on our
knowledge, such EPOR-modulated cell morphology changes in response
to tamoxifen treatment have not been described previously for
breast cancer cells, and thus we will further investigate this in
detail.
A correlation between signaling through AKT and
tamoxifen resistance was recently shown in several studies. Shi
et al (35) demonstrated
the role of EPO/EPOR-induced persistent AKT activation in the
transformation of RAMA 37-28 cells from a benign to a tumorigenic
phenotype. Furthermore, Paragh et al (39) showed the presence of phosphorylated
EPOR signaling components in A2780 human ovarian adenocarcinoma
cells, even when the cells were not exposed to exogenous EPO. EPOR
knock-down in breast cancer cell lines reduces pAKT levels, which
suggests its involvement in transmission of signals, including
phosphorylation and activation of AKT (34). Furthermore, MCF7 cell transfection
with AKT showed tamoxifen resistance, as tamoxifen showed reduced
inhibition of the growth of these transformed cells (43). Previous studies have also shown
that simultaneous treatment of breast cancer cell lines with
tamoxifen and the PI3K inhibitor, LY294002, reduced
tamoxifen-induced AKT phosphorylation and significantly increased
the pro-apoptotic effects of tamoxifen (29). In the present study, RAMA 37 and
RAMA 37-28 cells showed similar phosphorylation status for the key
EPOR signaling proteins. The tamoxifen treatment induced comparable
changes in pERK1/2 for these cell lines, and had no effects on the
phosphorylation of STAT5a/b. On the other hand, tamoxifen treatment
caused prolonged AKT activation (pAKT) in RAMA 37-28 cells, but not
in RAMA 37 cells. Given these results, we propose modified AKT
signaling in RAMA 37-28 cells as the potential mechanism that leads
to this increased cell resistance to tamoxifen for RAMA 37-28
cells.
In conclusion, the present study investigated the
effects of EPOR expression on breast cancer cell growth and the
sensitivity of these cells toward tamoxifen. The study was
conducted using benign non-invasive mammary epithelial cells that
express low EPOR levels, compared to the counterpart cell line with
greater EPOR expression. Despite the absence of EPO in these cells,
we indicated differences in cell growth, morphology, EPOR-induced
signal transduction, and tamoxifen resistance across these two cell
lines. The data show that tamoxifen treatment induces prolonged
activation of AKT signaling in RAMA 37-28 cells but not in RAMA 37
cells. Therefore, we suggest that sustained signaling via AKT
renders RAMA 37-28 cells more resistant to tamoxifen. Moreover, we
report here the first evidence of EPOR-modulated cell morphology
changes upon tamoxifen treatment, which result in increased
formation of cell protrusions and subsequent cell death. The
observed putative link between EPOR expression and the responses of
breast cancer cells to tamoxifen might have clinical relevance. The
detailed mechanisms that alter this cell growth, morphology and
tamoxifen resistance need to be investigated further.
Acknowledgments
The RAMA 37 and RAMA 37-28 cell lines were the
subject of a Material Transfer Agreement between Queen's University
Belfast and the University of Ljubljana. This study was supported
by a Young Researcher grant to N.T., a J3-0124 grant to N.D., and
P1-0390 Research Programme to R.K, all from the Slovenian Research
Agency (ARRS). This study was also supported by a VEGA 1/0394/15
grant to P.S. from the Scientific Grant Agency of the Ministry of
Education, Science, Research and Sport of the Slovak Republic, and
internal scientific grants VVGS-2014-190 to L.I. and
VVGS-PF-2016-72617 to E.S. from Pavol Jozef Šafárik University,
Slovakia. The authors thank Dr Chris Berrie for critical appraisal
and editing of the manuscript.
References
|
1
|
Bazan JF: Haemopoietic receptors and
helical cytokines. Immunol Today. 11:350–354. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Elliott S and Sinclair AM: The effect of
erythropoietin on normal and neoplastic cells. Biologics.
6:163–189. 2012.PubMed/NCBI
|
|
3
|
Fraser M, Bai T and Tsang BK: Akt promotes
cisplatin resistance in human ovarian cancer cells through
inhibition of p53 phosphorylation and nuclear function. Int J
Cancer. 122:534–546. 2008. View Article : Google Scholar
|
|
4
|
Richmond TD, Chohan M and Barber DL:
Turning cells red: Signal transduction mediated by erythropoietin.
Trends Cell Biol. 15:146–155. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koury MJ and Bondurant MC: The molecular
mechanism of erythropoietin action. Eur J Biochem. 210:649–663.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Masuda S, Nagao M, Takahata K, Konishi Y,
Gallyas F Jr, Tabira T and Sasaki R: Functional erythropoietin
receptor of the cells with neural characteristics. Comparison with
receptor properties of erythroid cells J Biol Chem.
268:11208–11216. 1993.
|
|
7
|
Anagnostou A, Liu Z, Steiner M, Chin K,
Lee ES, Kessimian N and Noguchi CT: Erythropoietin receptor mRNA
expression in human endothelial cells. Proc Natl Acad Sci USA.
91:3974–3978. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Westenfelder C, Biddle DL and Baranowski
RL: Human, rat, and mouse kidney cells express functional
erythropoietin receptors. Kidney Int. 55:808–820. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ogilvie M, Yu X, Nicolas-Metral V, Pulido
SM, Liu C, Ruegg UT and Noguchi CT: Erythropoietin stimulates
proliferation and interferes with differentiation of myoblasts. J
Biol Chem. 275:39754–39761. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Abhold E, Rahimy E, Wang-Rodriguez J,
Blair KJ, Yu MA, Brumund KT, Weisman RA and Ongkeko WM: Recombinant
human erythropoietin promotes the acquisition of a malignant
phenotype in head and neck squamous cell carcinoma cell lines in
vitro. BMC Res Notes. 4:5532011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lopez TV, Lappin TR, Maxwell P, Shi Z,
Lopez-Marure R, Aguilar C and Rocha-Zavaleta L: Autocrine/paracrine
erythropoietin signalling promotes JAK/STAT-dependent proliferation
of human cervical cancer cells. Int J Cancer. 129:2566–2576. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pérès EA, Valable S, Guillamo JS, Marteau
L, Bernaudin JF, Roussel S, Lechapt-Zalcman E, Bernaudin M and
Petit E: Targeting the erythropoietin receptor on glioma cells
reduces tumour growth. Exp Cell Res. 317:2321–2332. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu P, Zhang N, Wang X, Zhang C, Li T, Ning
X and Gong K: The erythropoietin/erythropoietin receptor signaling
pathway promotes growth and invasion abilities in human renal
carcinoma cells. PLoS One. 7:e451222012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou B, Damrauer JS, Bailey ST, Hadzic T,
Jeong Y, Clark K, Fan C, Murphy L, Lee CY, Troester MA, et al:
Erythropoietin promotes breast tumorigenesis through
tumor-initiating cell self-renewal. J Clin Invest. 124:553–563.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arcasoy MO: Non-erythroid effects of
erythropoietin. Haematologica. 95:1803–1805. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Debeljak N, Solár P and Sytkowski AJ:
Erythropoietin and cancer: The unintended consequences of anemia
correction. Front Immunol. 5:5632014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schnitt SJ: Classification and prognosis
of invasive breast cancer: From morphology to molecular taxonomy.
Mod Pathol. 23(Suppl 2): S60–S64. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Anderson WF, Chatterjee N, Ershler WB and
Brawley OW: Estrogen receptor breast cancer phenotypes in the
Surveillance, Epidemiology, and End Results database. Breast Cancer
Res Treat. 76:27–36. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ali S and Coombes RC: Endocrine-responsive
breast cancer and strategies for combating resistance. Nat Rev
Cancer. 2:101–112. 2002. View
Article : Google Scholar
|
|
20
|
Jensen EV and Jordan VC: The estrogen
receptor: A model for molecular medicine. Clin Cancer Res.
9:1980–1989. 2003.PubMed/NCBI
|
|
21
|
Jordan VC and Koerner S: Tamoxifen (ICI
46,474) and the human carcinoma 8S oestrogen receptor. Eur J
Cancer. 11:205–206. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen H, Tritton TR, Kenny N, Absher M and
Chiu JF: Tamoxifen induces TGF-beta 1 activity and apoptosis of
human MCF-7 breast cancer cells in vitro. J Cell Biochem. 61:9–17.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ellis PA, Saccani-Jotti G, Clarke R,
Johnston SR, Anderson E, Howell A, A'Hern R, Salter J, Detre S,
Nicholson R, et al: Induction of apoptosis by tamoxifen and ICI
182780 in primary breast cancer. Int J Cancer. 72:608–613. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mabuchi S, Ohmichi M, Kimura A, Ikebuchi
Y, Hisamoto K, Arimoto-Ishida E, Nishio Y, Takahashi K, Tasaka K
and Murata Y: Tamoxifen inhibits cell proliferation via
mitogen-activated protein kinase cascades in human ovarian cancer
cell lines in a manner not dependent on the expression of estrogen
receptor or the sensitivity to cisplatin. Endocrinology.
145:1302–1313. 2004. View Article : Google Scholar
|
|
25
|
Jordan VC: Tamoxifen (ICI46,474) as a
targeted therapy to treat and prevent breast cancer. Br J
Pharmacol. 147(Suppl 1): S269–S276. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hopp TA and Fuqua SA: Estrogen receptor
variants. J Mammary Gland Biol Neoplasia. 3:73–83. 1998. View Article : Google Scholar
|
|
27
|
Milani A, Geuna E, Mittica G and Valabrega
G: Overcoming endocrine resistance in metastatic breast cancer:
Current evidence and future directions. World J Clin Oncol.
5:990–1001. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Viedma-Rodríguez R, Baiza-Gutman L,
Salamanca-Gómez F, Diaz-Zaragoza M, Martínez-Hernández G, Ruiz
Esparza-Garrido R, Velázquez-Flores MA and Arenas-Aranda D:
Mechanisms associated with resistance to tamoxifen in estrogen
receptor-positive breast cancer (Review). Oncol Rep. 32:3–15.
2014.PubMed/NCBI
|
|
29
|
Clark AS, West K, Streicher S and Dennis
PA: Constitutive and inducible Akt activity promotes resistance to
chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol
Cancer Ther. 1:707–717. 2002.PubMed/NCBI
|
|
30
|
Larsson AM, Jirström K, Fredlund E,
Nilsson S, Rydén L, Landberg G and Påhlman S: Erythropoietin
receptor expression and correlation to tamoxifen response and
prognosis in breast cancer. Clin Cancer Res. 15:5552–5559. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Van de Sompele J, De Preter K, Pattyn F,
Poppe B, Van Roy N, De Paepe A and Speleman F: Accurate
normalization of real-time quantitative RT-PCR data by geometric
averaging of multiple internal control genes. Genome Biol.
3:Research0034. 2002.
|
|
32
|
Volgger B, Kurz K, Zöschg K, Theurl I,
Ciresa-König A, Marth C and Weiss G: Importance of erythropoetin
receptor expression in tumour tissue for the clinical course of
breast cancer. Anticancer Res. 30:3721–3726. 2010.PubMed/NCBI
|
|
33
|
Trošt N, Hevir N, Rižner TL and Debeljak
N: Correlation between erythropoietin receptor(s) and estrogen and
progesterone receptor expression in different breast cancer cell
lines. Int J Mol Med. 31:717–725. 2013.
|
|
34
|
Reinbothe S, Larsson AM, Vaapil M, Wigerup
C, Sun J, Jögi A, Neumann D, Rönnstrand L and Påhlman S:
EPO-independent functional EPO receptor in breast cancer enhances
estrogen receptor activity and promotes cell proliferation. Biochem
Biophys Res Commun. 445:163–169. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shi Z, Hodges VM, Dunlop EA, Percy MJ,
Maxwell AP, El-Tanani M and Lappin TR: Erythropoietin-induced
activation of the JAK2/STAT5, PI3K/Akt, and Ras/ERK pathways
promotes malignant cell behavior in a modified breast cancer cell
line. Mol Cancer Res. 8:615–626. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bustin SA, Benes V, Garson JA, Hellemans
J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL,
et al: The MIQE guidelines: Minimum information for publication of
quantitative real-time PCR experiments. Clin Chem. 55:611–622.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Simoncini T, Genazzani AR and De Caterina
R: Towards a molecular understanding of the atheroprotective
effects of estrogens: A review of estrogen effects on endothelial
activation. Ital Heart J. 1:104–107. 2000.PubMed/NCBI
|
|
38
|
Kutuk O, Arisan ED, Tezil T, Shoshan MC
and Basaga H: Cisplatin overcomes Bcl-2-mediated resistance to
apoptosis via preferential engagement of Bak: Critical role of
Noxa-mediated lipid peroxidation. Carcinogenesis. 30:1517–1527.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Paragh G, Kumar SM, Rakosy Z, Choi SC, Xu
X and Acs G: RNA interference-mediated inhibition of erythropoietin
receptor expression suppresses tumor growth and invasiveness in
A2780 human ovarian carcinoma cells. Am J Pathol. 174:1504–1514.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
McGuire WL: Current status of estrogen
receptors in human breast cancer. Cancer. 36:638–644. 1975.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Michalides R, Griekspoor A, Balkenende A,
Verwoerd D, Janssen L, Jalink K, Floore A, Velds A, van't Veer L
and Neefjes J: Tamoxifen resistance by a conformational arrest of
the estrogen receptor alpha after PKA activation in breast cancer.
Cancer Cell. 5:597–605. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Esslimani-Sahla M, Simony-Lafontaine J,
Kramar A, Lavaill R, Mollevi C, Warner M, Gustafsson JA and
Rochefort H: Estrogen receptor beta (ER beta) level but not its ER
beta cx variant helps to predict tamoxifen resistance in breast
cancer. Clin Cancer Res. 10:5769–5776. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Campbell RA, Bhat-Nakshatri P, Patel NM,
Constantinidou D, Ali S and Nakshatri H: Phosphatidylinositol
3-kinase/AKT-mediated activation of estrogen receptor alpha: A new
model for anti-estrogen resistance. J Biol Chem. 276:9817–9824.
2001. View Article : Google Scholar : PubMed/NCBI
|