Introduction
Worldwide, bladder cancer (BCa) is the
9th most common cancer and the 13th leading
cause of death due to cancer (1),
and it has attracted increasing attention from clinicians over the
past several decades. The risk factors for BCa include but are not
limited to genetic predisposition and acquired exposure, the
mechanism of which is still unclear (2–5).
Clinically, according to the pathological characteristics, BCa
includes superficial and invasive types, which are treated using
different therapeutic procedures. Briefly, the recommended
therapeutic guideline for superficial BCa is transurethral
resection of bladder tumor (TUR-bt), followed by intravesical
chemo- or immunotherapeutics. Unfortunately, 70% of superficial BCa
will inevitably progress to the invasive type followed by enhanced
tumor malignancy. For invasive BCa, cystectomy plus adjuvant or
neoadjuvant chemotherapy is accepted by clinicians and patients
(6). Thus, chemotherapy is a vital
and irreplaceable regimen for treating invasive BCa, regardless of
whether initial invasive BCa or invasive BCa progressed from the
superficial type. Many studies have indicated that cisplatin-based
regimens play effective roles in BCa therapy (7,8).
However, chemoresistance to cisplatin-containing regimens
inevitably appears in the battle against BCa, the mechanism of
which is still unknown.
Previous reports have identified the important roles
of NF-κB signaling in the initiation and progression of BCa, e.g.,
promoting the process of epithelial to mesenchymal transition (EMT)
(9), secreting MMP2/MMP9 and
inhibiting apoptosis. In addition, the promoter of MDR1 (ABCB1), a
multiple drug-resistant gene, contains an NF-κB binding site,
indicating that MDR1 was monitored by this signaling pathway
(10).
Silibinin, a polyphenolic flavonoid component
isolated from the fruits or seeds of milk thistle (Silybum
marianum), has been clinically used to treat various diseases,
and it has been suggested that this reagent exhibited protective
effects for patients with liver or heart disease. The inhibitory
effects of silibinin against cancer were also indicated (11–17)
in tumors such as breast cancer and head and neck squamous cell
carcinomas. Our previous study indicated that silibinin inhibited
BCa progression (18), leading us
to hypothesize a link between silibinin and BCa
chemoresistance.
In the present study, we hypothesized that silibinin
may play inhibitory roles in the chemoresistance of BCa cells,
possibly involving the NF-κB signaling pathway. Our results
revealed that silibinin inhibited the progression and reversed the
chemoresistance of BCa cells in an NF-κB-dependent and -independent
manner, thus providing a potential therapeutic use for silibinin in
patients with BCa.
Materials and methods
Cell culture
Human BCa cell lines T24 and J82 were obtained from
the ATCC (American Type Culture Collection, Manassas, VA, USA) and
cultured in DMEM supplemented by 10% FBS (Invitrogen, Carlsbad, CA,
USA). Cells were cultured in incubators (Thermo Scientific,
Germany) in an atmosphere with 5% CO2 at 37°C.
To obtain stable, cisplatin-resistant cell lines, we
monitored the IC50 of cisplatin and obtained values of
58 µM for T24 and 49 µM for J82. Second, the
cisplatin-resistant index (RI) was evaluated by MTT; the RIs of the
cell lines were 21.35 and 28.75 for T24 and J82, respectively. The
cultured parental T24 and J82 cells were supplemented with 20
µM cisplatin. The medium was refreshed every two days to
remove the dead cells, and the cells were washed three times with
sterile PBS (pH 7.2). This treatment was administered for more than
three months to obtain stable cisplatin-resistant T24/J82 cells
(tagged with T24R/J82R).
To inhibit NF-κB signaling, pyrrolidine
dithiocarbamate (PDTC) (Sigma-Aldrich, USA) (19), an inhibitor of the NF-κB pathway,
was used; the final concentration was 10 µM in the medium
for the last 24 h before analysis.
Wound healing assay
Wound healing assays were carried out by scratching
a 6-well dish with a 10-µl pipette tip when the dish was at
80% confluence (including parental cell and cisplatin-resistant
cell lines). The width of the scratches was compared at 0, 6, 12
and 24 h after scratching.
Western blot analysis
Pretreated cells were harvested at 80% confluency
and washed three times with cold PBS. Total cellular protein
lysates were prepared with RIPA buffer [50 mM Tris (pH 8.0), 150 mM
NaCl, 0.1% SDS, 1% NP40 and 0.5% sodium deoxycholate] containing
proteinase inhibitors [1% inhibitor cocktail and 1 mM PMSF, both
from Sigma, (St. Louis, MO, USA)]. Then, 30 µg of protein
was separated on 10% SDS-PAGE gels and transferred to
nitrocellulose membranes. The membranes were blocked at room
temperature for 1 h with 5% skim milk in Tris-buffered saline (pH
7.6, TBS). Polyclonal primary antibodies were applied at different
dilutions (Table I) in 5% skim
milk in TBS at 4°C overnight, followed by TBST (with Tween-20)
washes. Membranes were incubated with fluorescent secondary
antibodies (LI-COR, Rockford, IL, USA) coupled to the first
antibody at room temperature in the dark for 1 h, then washed with
TBST, dried with neutral absorbent paper and scanned with the
Odyssey Detection system (lI-COR). MG-132 (Sigma-Aldrich) was used
to inhibit the proteasome-dependent degradation when necessary (10
µM, 4 h before the protein harvest). GAPDH was used as a
loading control (for total cell fraction).
 | Table IInformation on the antibodies. |
Table I
Information on the antibodies.
| Gene ID | Antibody | Dilutions | Species | Supplied by |
|---|
| NM_004360.3 | E-cadherin | 1:600 | Homo | Santa Cruz |
| NM_001792.3 | N-cadherin | 1:300 | Homo | Santa Cruz |
| NM_003380.3 | Vimentin | 1:300 | Homo | Santa Cruz |
| NM_004530.4 | MMP2 | 1:400 | Homo | Santa Cruz |
| NM_004994.2 | MMP9 | 1:400 | Homo | Santa Cruz |
| NM_002046.4 | GAPDH | 1:15,000 | Homo | Abcam |
| NM_000927.4 | ABCB1 | 1:400 | Homo | Santa Cruz |
| β-actin | 1:300 | Homo | Santa Cruz |
Real-time PCR
Cellular total RNA was isolated using TRIzol reagent
(Invitrogen) and quantified by absorbance at 260 nm. RNA (2
µg) was reverse transcribed using Revert Aid™ First Strand
cDNA Synthesis kit (MBI Fermentas, St. Leon-Rot, Germany) strictly
according to the manufacturer's protocol. For real-time PCR, we
used the SYBR Premix Ex Taq™ II system (Takara Biotechnology Co.,
ltd., Dalian, China) and the Bio-Rad CFX96TM Real-time system
(Bio-Rad, CA, USA). Primers are listed in Table II. Briefly, 12.5 µl of SYBR
Premix Ex Taq II, 1 µl of primer (F and R, respectively),
200 ng of cDNA and 9.5 µl of distilled and deionized water
were mixed together, followed by two-stage, pre-denaturation at
95°C, 30 sec, one repeat; and PCR reaction, at 95°C, 5 sec followed
by 60°C, 30 sec, 30 repeats; and the third stage as dissociation,
95°C, 15 sec followed by 60°C, 30 sec, and another 95°C, 15 sec.
GAPDH was used as the loading control.
| Table IIPrimers for real-time PCR. |
Table II
Primers for real-time PCR.
| Gene ID | Gene | Primers |
|---|
| NM_002046.4 | GAPDH | F: AAC AGC GAC ACC
CAT CCT C
R: CAT ACC AGG AAA TGA GCT TGA CAA |
| NM_004360.3 |
E-cadherin | F: TGC CCA GAA AAT
GAA AAA GG
R: GTG TAT GTG GCA ATG CGT TC |
| NM_001792.3 |
N-cadherin | F: ACA GTG GCC ACC
TAC AAA GG
R: CCG AGA TGG GGT TGA TAA TG |
| NM_003380.3 |
Vimentin | F: GAG AAC TTT GCC
GTT GAA GC
R: GCT TCC TGT AGG TGG CAA TC |
| NM_004530.4 | MMP2 | F: CTC ATC GCA GAT
GCC TGG AA
R: TTC AGG TAA TAG GCA CCC TTG AAG A |
| NM_004994.2 | MMP9 | F: TGA CAG CGA CAA
GAA GTG
R: CAG TGA AGC GGT ACA TAG G |
| NM_000927.4 | ABCB1 | F: GTC CCA GGA GCC
CAT CCT
R: CCC GGC TGT TGT CTC CAT A |
Cell viability assay (MTT assay)
Cell viability was assessed using a
tetrazolium-based assay. Pretreated cells were incubated in the
absence or presence of cisplatin/doxorubicin for the indicated
times, and then washed once with PBS and incubated with 0.5 mg/ml
of MTT at 37°C for 1 h. The reagent was reduced by living cells to
form an insoluble blue formazan product. After incubation, cells
were lysed with DMSO. Colorimetric analysis using a 96-well
microplate reader was performed at a wavelength of 490 nm. The
experiments were performed in triplicate.
Cell migration/invasion assay
Migration/invasion ability was demonstrated by
Boyden chamber assay. Chambers with 8-µm-diameter pores were
obtained from Millipore (Millipore, Switzerland). For the migration
assay, 0.2 ml of FBS-free DMEM medium suspension with 10,000 cells
was added to the upper chamber in a 24-well plate, and 0.8 ml of
FBS-free DMEM was added to the lower chamber. After 12 h of
incubation, the chambers were washed with PBS (pH 7.4) three times
to remove the cells in the upper chamber, fixed with 4% formalin
for 15 min, and then stained with crystal violet (0.01% in ethanol)
for 25 min followed by washing three times with PBS. The cells were
counted using an inverted microscope, five images were randomly
taken at 200× magnification, and the average number of cells was
analyzed. For the invasion assay, the cell suspension (10,000
cells/well) in the upper chamber contained 0.2 ml mixture of
FBS-free DMEM/Matrigel at an 8/1 ratio (Matrigel, Sigma, USA).
Cells were incubated for 36 h, and the remainder of the protocol
was conducted in a similar manner to the migration assay.
Cell proliferative capacity assay
A 5-bromo-2-deoxyuridine (BrdU) incorporation assay
was used to analyze tumor proliferative ability. Briefly,
pretreated cells were plated on 8-well glass plates (Millipore)
until 50–70% confluency. BrdU was added to the medium (3
µg/ml), followed by 4 h of incubation and rinsing 3X with
PBS over 10 min to remove residual free BrdU. Cells were then fixed
with 4% paraformaldehyde for 45 min, followed by rinsing 5X with
PBS over 20 min. Then, 0.1% Triton X-100 was used to permeabilize
the cell membrane for 15 min, and 2 N HCl was added for 25 min to
separate DNA into single strands and thus allowing primary antibody
access to the incorporated BrdU. Cells were then rinsed 3X with PBS
over 10 min, and non-specific epitopes were blocked by 10% BSA for
20 min. Anti-BrdU antibody (1:200) in 10% BSA was added and
incubated overnight at 4°C. Cells were rinsed 5X with PBS, followed
by incubation with TRITC-labeled secondary antibody for 1 h at room
temperature, and finally rinsed 3X with PBS to remove the free
antibody. The fluorescence intensity of TRITC was monitored with a
SuperMicro Orifice Plate spectrophotometer (BioTek, USA) at 547
nm.
Immunofluorescence staining for nuclear
translocation of NF-κB
After the designated treatment, the pretreated cells
were washed three times with cold PBS (pH 7.4), followed by fixing
with 4% paraformaldehyde for 15 min, permeabilization in 0.5%
Triton X-100 for 10 min, and incubation in 1% BSA blocking solution
for 1 h. Fixed cells were incubated overnight at 4°C with rabbit
anti-human-P65 in 1% BSA. Cells were washed and incubated with
mouse anti-rabbit TRITC (Red) IgG antibody (Santa Cruz
Biotechnology, USA) diluted 1:100 in blocking buffer for 1 h.
Nuclei were stained with DAPI for 5 min. Cells were examined with a
fluorescence microscope equipped with narrow bandpass excitation
filters to individually select for red and blue fluorescence. Cells
were observed through the Image-Pro Plus system™ mounted on a
fluorescent microscope (Olympus, Japan). Each experiment was
repeated three times.
Statistical analysis
ANOVA test was used to analyze the statistical
discrepancy in >3 groups. Student's t-test was used to detect
any statistically significant difference between 2 groups. P-values
<0.05 were considered significant.
Results
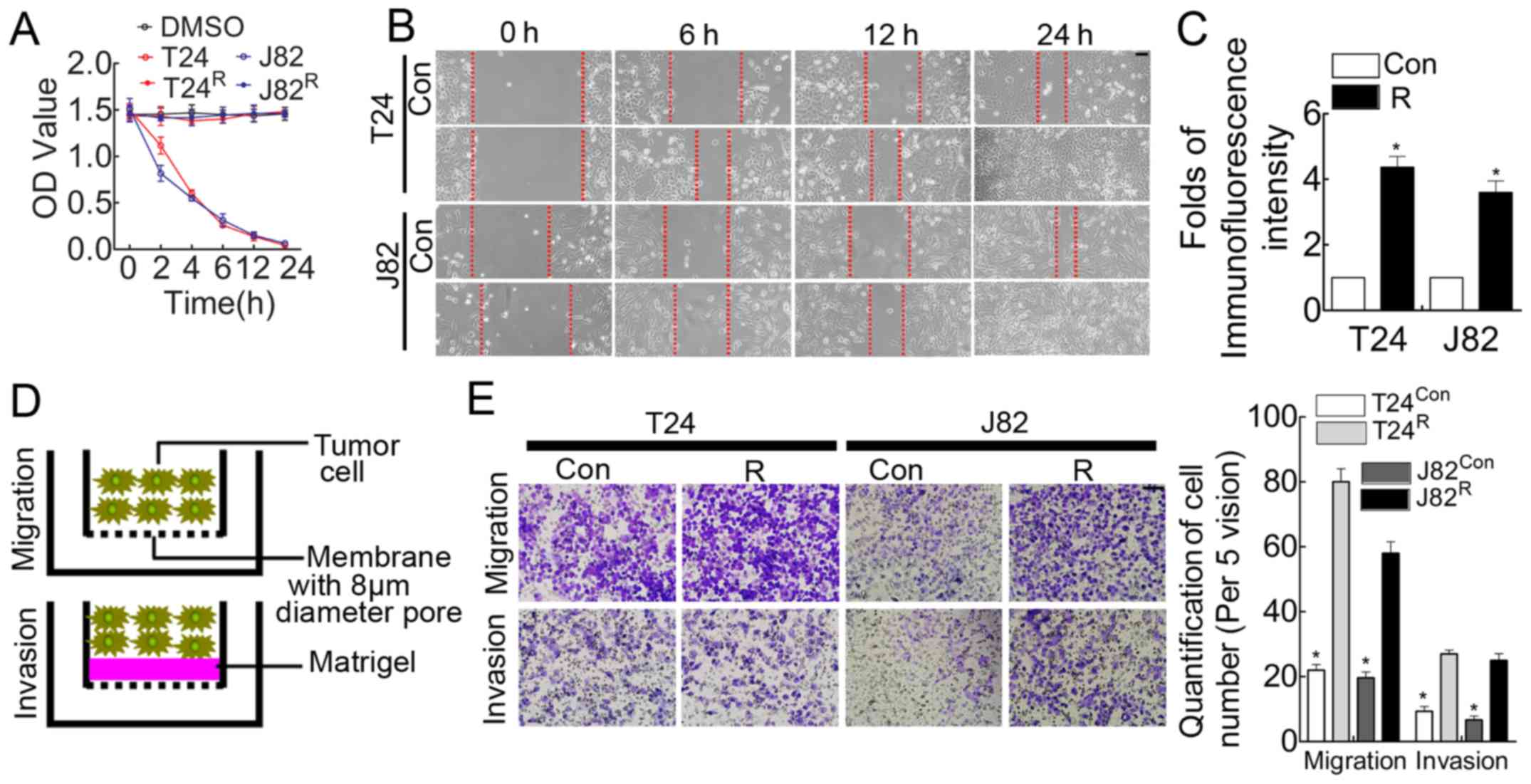
Stable chemoresistant cell lines induced
by cisplatin manifest enhanced migration/invasion and proliferation
capacity
Chemoresistance is considered a vital obstacle in
the battle against BCa and leads to the failure of BCa chemotherapy
(20). Cisplatin, which is one of
the major reagents in the chemotherapeutic regime for BCa, is
recommended as first-line treatment in the clinic (21). Therefore, cisplatin resistance is
ubiquitous in BCa patients, and dissecting the underlying mechanism
potentially brings benefits to BCa patients. Cisplatin was used to
treat BCa cell lines T24/J82 to obtain stable cisplatin-resistant
cell lines (tagged by T24R and J82R,
respectively), as indicated in the Materials and methods. Compared
with parental T24/J82, T24R/J82R manifested
cisplatin-resistance demonstrated by MTT assay (Fig. 1A). In addition, the in vitro
analysis suggested that the wound-healing time was shorter for
these cisplatin-resistant BCa cells (Fig. 1B), accompanied by enhanced
proliferation (Fig. 1C) and
migration/invasion (Fig. 1E)
ability.
EMT markers are induced in
T24R/J82R cells, accompanied by elevated
expression of ABCB1 (MDR1)
The enhanced capacity of migration/invasion and
proliferation of T24R/J82R prompted us to
monitor the expression of related genes, including EMT markers and
matrix metalloproteinase (MMP). Both western blot analysis
(Fig. 2A) and real-time PCR
(Fig. 2B) indicated the elevated
expression of EMT-related markers, e.g., N-cadherin, vimentin, MMP2
and MMP9, with decreased expression of E-cadherin. ABCB1, also
called MDR1, plays a vital role in BCa chemoresistance (20). Therefore, the expression of ABCB1
was monitored in T24R/J82R by western blot
analysis and real-time PCR. ABCB1 was elevated in
T24R/J82R vs. T24/J82, as shown by blot
analysis (Fig. 2C) and real-time
PCR (Fig. 2B).
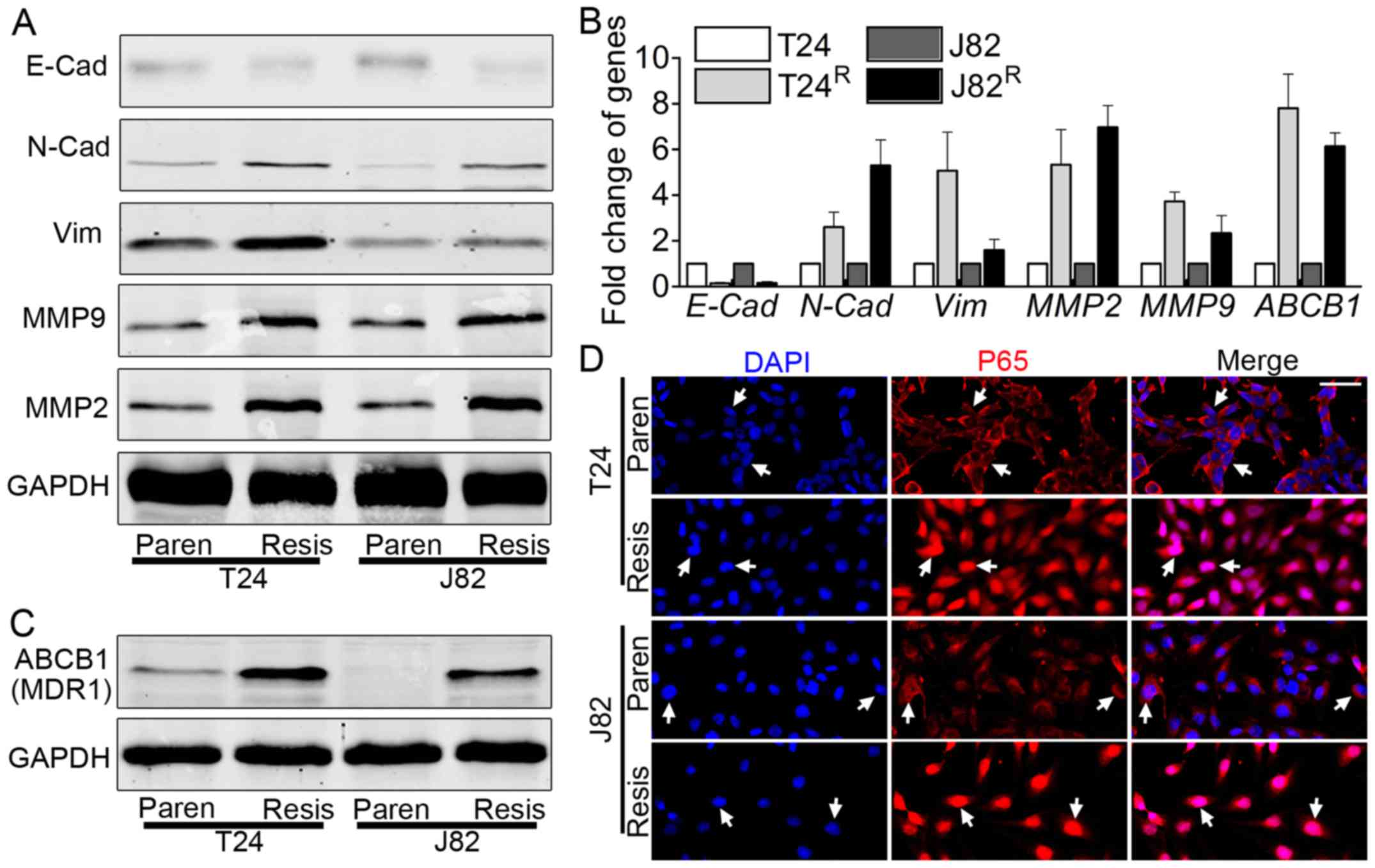 | Figure 2Epithelial to mesenchymal transition
(EMT) is enhanced in T24R/J82R cells,
accompanied by increased expression of ABCB1 and NF-κB nuclear
translocation. (A) Western blot analysis showed elevated expression
of N-cadherin, vimentin, MMP2 and MMP9 but decreased expression of
E-cadherin in T24R/J82R vs T24/J82,
indicating the process of EMT. (B) Real-time PCR results were
consistent with western blot analysis results, including elevated
expression of N-cadherin, vimentin, MMP2 and MMP9 but decreased
E-cadherin. In addition, the expression of ABCB1, the multiple
drug-resistant gene, was also elevated in
T24R/J82R vs T24/J82. (C) Western blot
analysis suggested that the expression of the multiple
drug-resistant gene, ABCB1, and also MDR1, was elevated as expected
in T24R/J82R. (D) Immunofluorescence staining
revealed the nuclear translocation of NF-κB (P65) in
T24R/J82R, as the white arrow indicates; bar,
100 µm. Paren=Parental; Resis=Resistant. |
NF-κB signaling is overactivated in
T24R/J82R
Emerging evidence noted the importance of NF-κB
signaling in tumorigenesis and cancer metastasis (22,23),
as well as chemoresistance (24),
leading us to link the cisplatin-induced phenomenon to this
signaling pathway. As indicated in Fig. 2D, immunofluorescence staining
suggested that T24R/J82R cells significantly
manifested P65 nuclear translocation, suggesting the activation of
this signaling pathway. In a parallel experiment, nuclear lysates
from T24R/J82R indicated the accumulation of
P65 in nuclei, as demonstrated by blot analysis (data not shown).
Thus, we concluded that NF-κB signaling was overactivated in
T24R/J82R cells vs. parental T24/J82
cells.
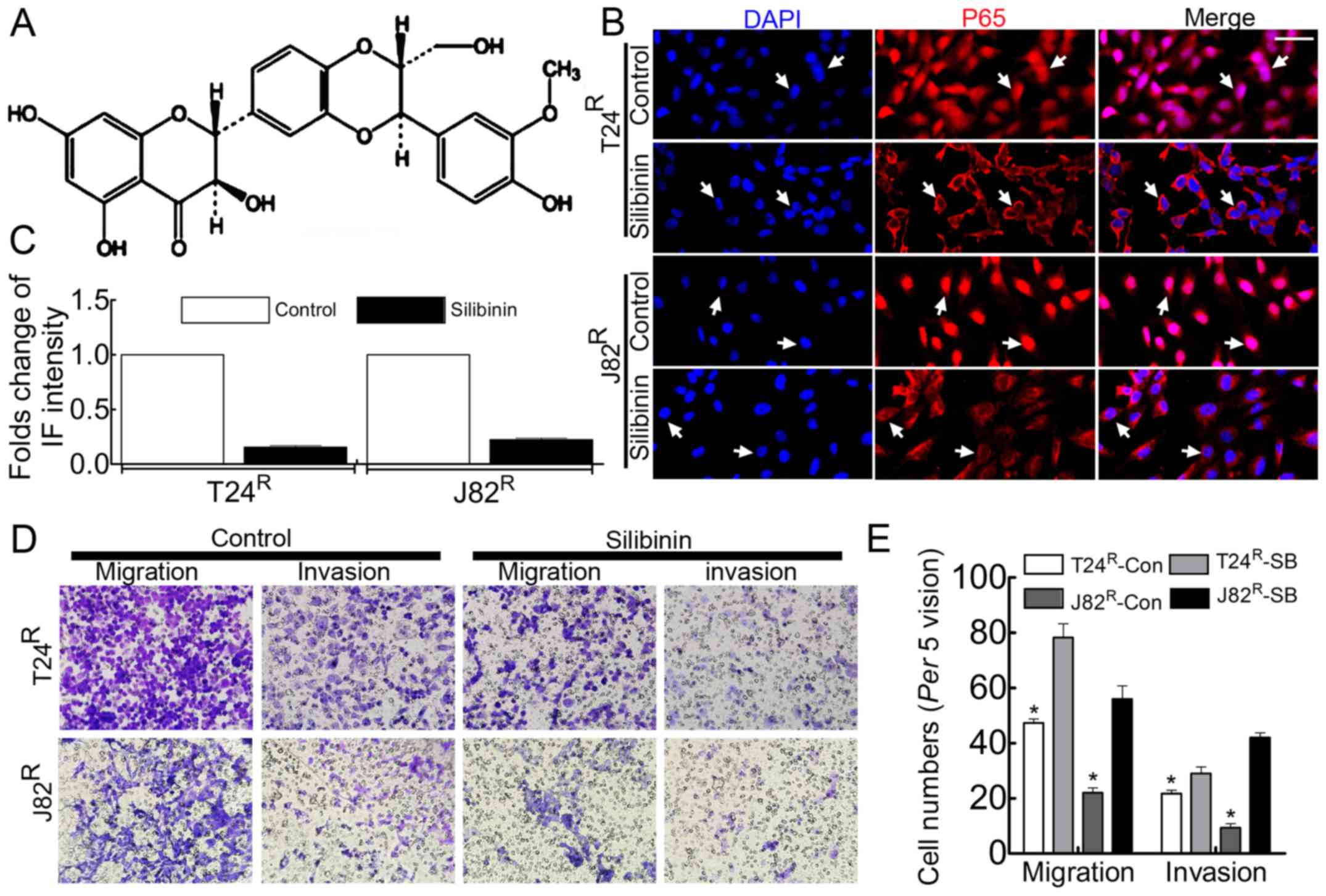
Migration/invasion ability and
proliferation of T24R/J82R are attenuated in
the present of silibinin, accompanied by inhibition of NF-κB
signaling in T24R/J82R cells
Silibinin has been used clinically to treat various
liver diseases and has been marketed as a dietary supplement
(25). Previous studies had noted
that silibinin inhibited tumor growth by suppressing MMPs (26,27),
VEGF, HIF-1α (25) and the process
of EMT (28,29). In the present study, silibinin was
used to treat BCa cell T24R/J82R, followed by
Boyden chamber assay, BrdU incorporation and immunofluorescence
staining to observe the malignant behavior. As expected, the
enhanced proliferation capacity of T24R/J82R
cells was attenuated in the presence of 100 µM silibinin
(Fig. 3B), accompanied by
decreased migration/invasion ability (Fig. 3C and E). In addition, the nuclear
translocation of NF-κB was significantly inhibited in the presence
of silibinin in T24R/J82R cells (Fig. 3D).
Silibinin suppresses the
migration/invasion and proliferation capacity of
T24R/J82R cells by inhibiting the expression
of EMT-related markers and ABCB1 in a dose-dependent manner
Previous results suggested that silibinin could
inhibit cisplatin-induced migration/invasion and proliferation. In
addition, immunofluorescence staining indicated that silibinin
could inhibit the activity of the NF-κB signaling pathway.
According to the previous studies, this phenomenon was accompanied
by an alteration of related genes and might be involved in a
related signaling pathway. Therefore, we monitored the expression
of EMT-related markers and ABCB1 in the presence of silibinin. As
indicated in Fig. 4A and B,
silibinin treatment led to significant inhibition of EMT, e.g.,
decreased expression of vimentin, N-cadherin, MMP2, and MMP9, with
increased E-cadherin expression. In addition, silibinin suppressed
the expression of ABCB1 in T24R/J82R
(Fig. 4C and D). As indicated in
Fig. 4, the effects of silibinin
on T24R/J82R manifested in a dose-dependent
manner, at doses from 100 µM, 200 µM to 400
µM, but revealed the most powerful role of PDTC.
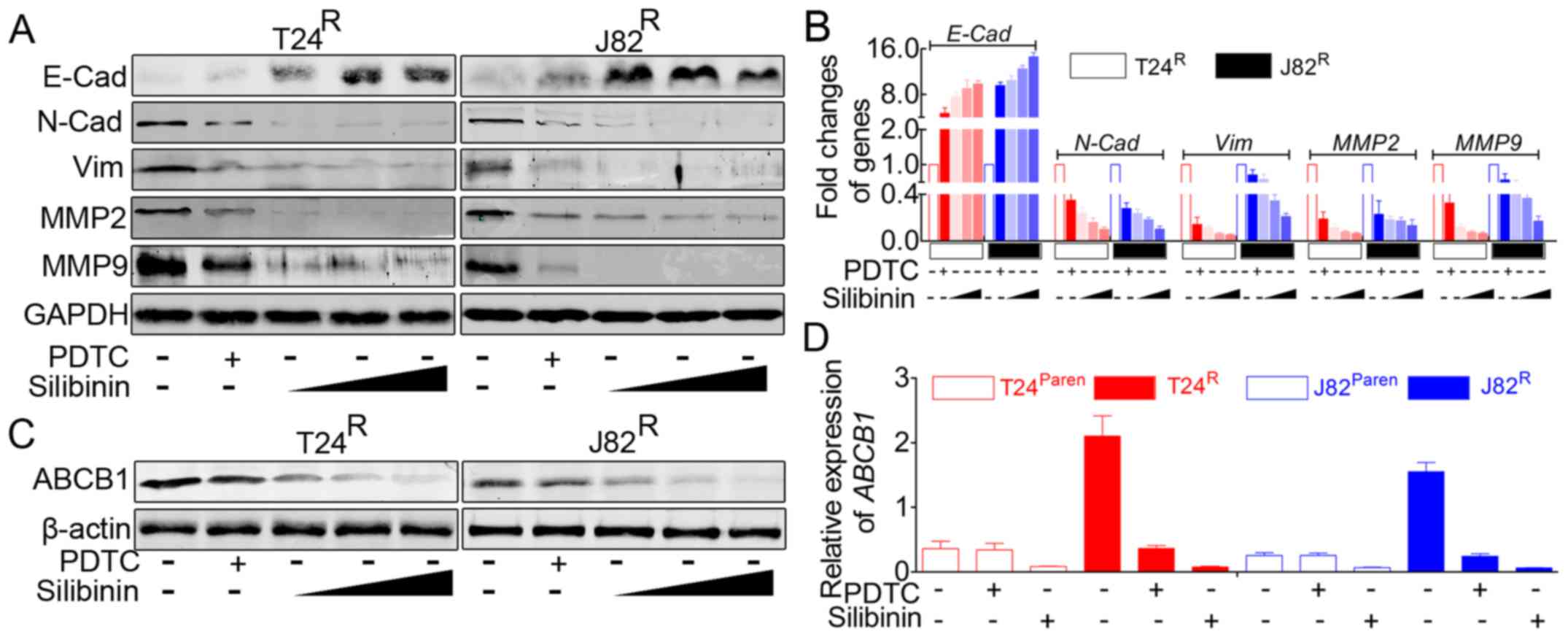 | Figure 4Silibinin inhibits
T24R/J82R malignancy in an NF-κB-dependent
and -independent manner, accompanied by attenuating the expression
of ABCB1. (A) Western blot analysis suggested that PDTC, a
classical inhibitor of NF-κB signaling, partially inhibited EMT,
e.g., elevated expression of E-cadherin accompanied by decreased
expression of N-cadherin, vimentin, MMP2 and MMP9; in addition,
silibinin led to the reversal of EMT in a dose-dependent manner.
(B) Real-time PCR indicated that the inhibition of NF-κB signaling
by PDTC led to the reversal of EMT markers in
T24R/J82R, and this EMT reversal can be
enhanced by silibinin in a dose-dependent manner. (C) Western blot
analysis showed that both PDTC and silibinin resulted in the
decreased expression of ABCB1 but that silibinin possessed more
powerful roles in a dose-dependent manner. (D) Real-time PCR
revealed that PDTC could lead to the attenuation of ABCB1 in
T24R/J82R but had no visible effects on
parental T24/J82, whereas silibinin significantly inhibited the
expression of ABCB1 in both cisplatin-resistant and parental
T24/J82 cells. |
Forced inhibition of NF-κB signaling in
T24R/J82R also leads to decreased expression
of EMT-related markers and ABCB1
Previous reports indicated that forced inhibition of
NF-κB signaling in BCa cells resulted in the reversal of EMT
(30). Herein, in parallel, we
used a specific inhibitor of NF-κB signal to repeat this process.
In accordance with previous reports, our data indicated that PDTC
effectively attenuated the activation of NF-κB signaling (data not
shown), accompanied by decreased expression of EMT-related markers
(Fig. 4A and B). In addition, as
mentioned above, ABCB1 was one of the target genes of NF-κB
signaling, and inhibiting this pathway led to the decreased
expression of ABCB1 (Fig. 4C and
D).
The suppressive roles of silibinin on
T24R/J82R cells presented NF-κB-dependent and
-independent mechanisms
The previous results suggested that silibinin
manifested more powerful inhibitory roles on
T24R/J82R cells than on parental T24/J82
cells. Moreover, as stated above, NF-κB signaling was inactivated
in parental T24/J82 cells, suggesting that PDTC had no visible
inhibitory roles on the malignancy and proliferative capacity of
parental T24/J82 cells. This led us to ask whether the inhibitory
roles of silibinin on T24R/J82R involved
NF-κB signaling or whether they were unrelated phenomena. Our
results suggested that in T24R/J82R cells,
silibinin had a stronger inhibitory role than did PDTC in the tumor
cell expression of EMT markers (Fig.
4A and B), ABCB1 (Fig. 4C),
cisplatin sensitivity (Fig. 5A and
B), proliferation capacity (Fig.
5C) and migration/invasion capacity (Fig. 5D), suggesting that the inhibitory
roles of silibinin were partially independent of NF-κB signaling.
Combined with the NF-κB signal inhibition induced by silibinin, we
concluded that silibinin suppressed BCa cell line
T24R/J82R in an NF-κB-dependent and
-independent manner.
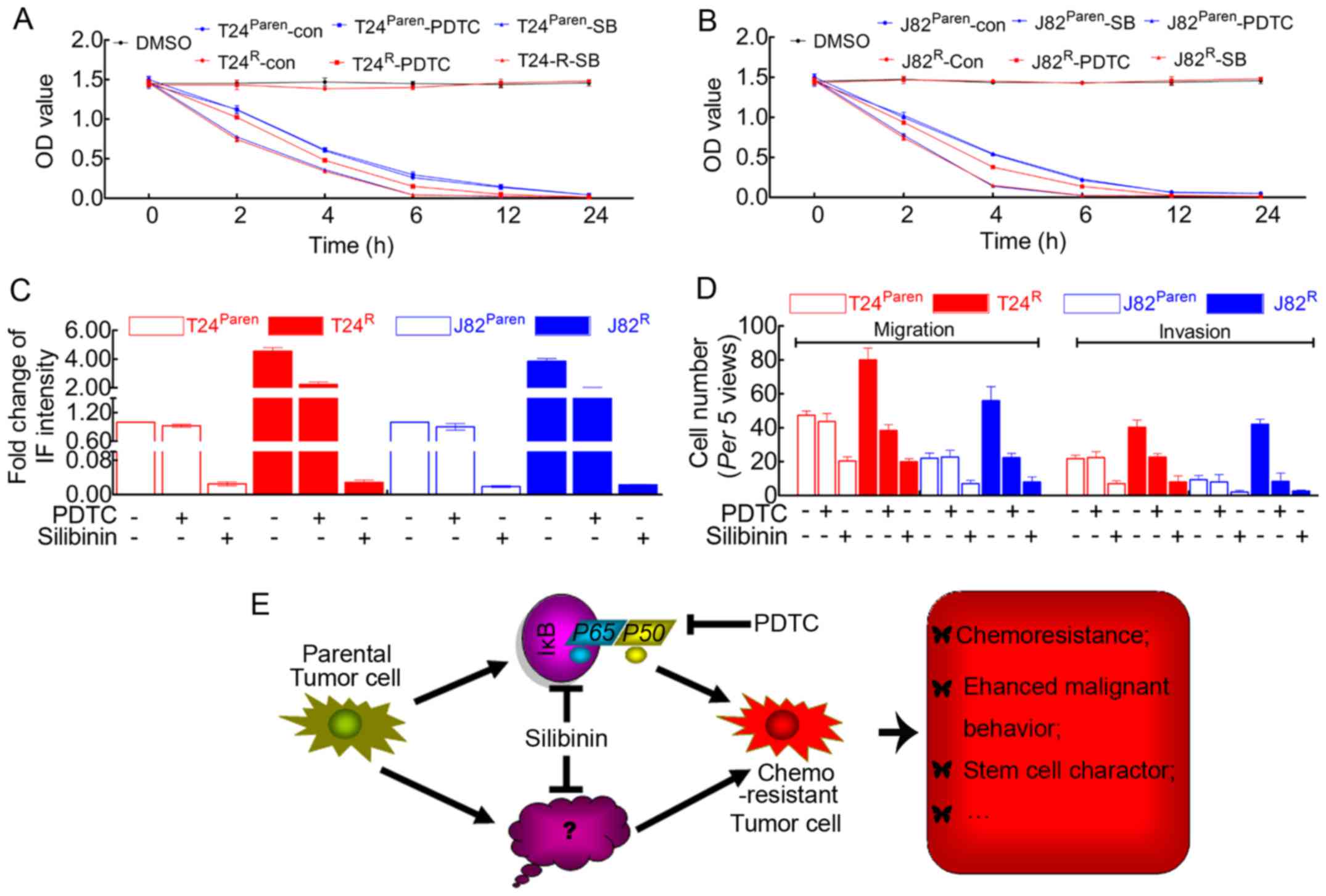 | Figure 5Silibinin exhibits more powerful
roles in inhibiting cisplatin resistance, cell proliferation and
migration/invasion. (A) MTT assay revealed that the cisplatin
resistance of T24R was destroyed in the presence of
silibinin. Silibinin induced the enhanced cisplatin sensitivity of
parental T24. In addition, PDTC attenuated the cisplatin resistance
of T24R but had no visible effect on parental T24. (B)
MTT assay revealed that the cisplatin resistance of J82R was
destroyed in the presence of silibinin. Silibinin induced the
enhanced cisplatin sensitivity of parental J82. In addition, PDTC
attenuated the cisplatin resistance of J82R but had no visible
effect on parental J82. (C) Quantification of BrdU incorporation
suggested that silibinin possessed more powerful effects on
inhibiting BCa cell proliferation in both cisplatin-resistant cell
lines and parental cell lines. However, PDTC seemed to have little
effect on parental T24/J82, although the proliferative capacity of
T24R/J82R was inhibited by this reagent. (D)
Quantification of the Boyden chamber assay suggested that silibinin
significantly attenuated the migration/invasion capacity of both
parental and cisplatin-resistant T24/J82 cells. In addition,
malignancy of T24R/J82R but not parental
T24/J82 was inhibited by PDTC. (E) In summary: The prolonged time
of cisplatin treatment inevitably resulted in drug resistance, the
mechanism of which included but was not limited to the activation
of NF-κB signaling, leading to the failure of chemotherapeutics.
Like PDTC, silibinin was able to inhibit NF-κB signaling, which
reversed the malignant behavior of drug-resistant cell lines.
However, silibinin also attenuated chemoresistance and tumor cell
malignancy in an NF-κB-independent manner, which is still unknown
[tagged by ? in (E)]. |
Forced inhibition of NF-κB signaling has
no visible inhibitory roles on parental T24/J82 cells
Our data indicated that inhibition of NF-κB
signaling by PDTC in T24R/J82R cells led to
the attenuation of malignancy and reversal of EMT. In contrast,
NF-κB signaling was suppressed in parental T24/J82 cells. In
parallel, as indicated in Fig. 5C and
D, PDTC had no visible effect on the migration/invasion and
proliferation of parental T24/J82.
Discussion
Chemoresistance, especially acquired
chemoresistance, is considered a vital obstacle in the battle
against cancer and can lead to the failure of cancer therapy
(20,31). Chemotherapy, in accordance with
radiotherapy, is the final regimen for BCa patients. In addition,
in both neoadjuvant chemotherapy and traditional chemotherapy for
BCa, cisplatin is the one irreplaceable reagent. Thus, cisplatin
resistance ubiquitously appears in BCa patients receiving
chemotherapy, which is why it is important to investigate this
mechanism further as done in recent decades. To dissect this
mechanism, the present study was performed.
First, we used cisplatin treatment to obtain
chemoresistant cell lines as described in the Materials and
methods. Our results suggested that we efficiently obtained the
stable cisplatin-resistant cell lines
T24R/J82R (Fig.
1A). In addition, in accordance with our hypothesis, these
T24R/J82R cells manifested enhanced wound
healing (Fig. 1B), proliferation
(Fig. 1C) and migration/invasion
(Fig. 1E) capacity. NF-κB
signaling was activated in these T24R/J82R
cells (Fig. 2B), accompanied by
the process of EMT (Fig. 2A) and
elevated expression of ABCB1 (Fig.
1C). Although our unpublished data partially revealed the
mechanism responsible for cisplatin-induced chemoresistance, much
work remains to be done.
Silibinin, a polyphenolic flavonoid component
isolated from the fruits or seeds of milk thistle (Silybum
marianum), has been clinically used to treat various liver
diseases and has been marketed as a dietary supplement (32). Our previous results indicate that
silibinin suppressed BCa by acting on tumor cell mitochondria
(18) or another mechanism
(33). Therefore, we hypothesized
that silibinin might play important roles in the process of EMT,
which has been previously reported in other tumors (28,29).
Our results indicate that silibinin significantly suppresses the
nuclear translocation of NF-κB, inhibiting NF-κB signaling
(Fig. 3D). In accordance with our
hypothesis, this inhibition is accompanied by an attenuated
proliferation capacity (Fig. 3B)
and migration/invasion capacity (Fig.
3C and E) and the reversal of EMT (Fig. 4A and B). The process by which
silibinin inhibits the NF-κB signal is still unknown, but this
result indicates that silibinin can potentially be used in BCa
therapy.
As indicated by our data, silibinin inhibits the
malignancy of BCa cells in a dose-dependent manner, e.g., the
concentration of silibinin ranged from 100 to 400 µM,
accompanied by decreased expression of EMT-related markers and
ABCB1 (Fig. 4). Silibinin protects
against heart disease in older patients and is used as a key
ingredient in a Chinese herbal formula for managing age-related
disease, which is also effective in a dose-dependent manner. Thus,
silibinin is suitable for older BCa patients, especially those with
heart disease.
Compared with PDTC, which is a specific inhibitor of
NF-κB signaling, silibinin manifests more powerful inhibitory roles
that affect BCa cells, especially parental T24/J82 cells (Figs. 4D and 5A–D). This indicates that the inhibitory
roles of silibinin acting upon parental BCa cells might be
independent of NF-κB signaling, with silibinin inhibiting the
malignancy of BCa cells in a more ubiquitous manner. Taken
together, these results lead us to conclude that silibinin
suppresses BCa cell malignancy in an NF-κB-dependent and
-independent manner.
In conclusion, as indicated in Fig. 5E, cisplatin treatment activates the
NF-κB signaling pathway in an unknown manner, leading to the
enhanced malignancy of BCa cells, which can be inhibited by PDTC.
However, silibinin inhibited BCa progression not only by
suppressing NF-κB signaling but also via other mechanisms,
resulting in an enhanced therapeutic effect for BCa patients.
Although the exact mechanisms are still unknown, silibinin exhibits
numerous benefits and could be incorporated into various forms of
therapy for BCa patients.
Acknowledgments
This study was supported in part by Overall
Innovation Projects of Scientific and Technological Resources of
Shaanxi Province (no. 2013KTCl03-04 to Yonggang Xu) and the
National Natural Science Foundation of China (NSFC no. 81172436 to
Sun Yi).
References
|
1
|
Skeldon SC and Larry Goldenberg S: Bladder
cancer: A portal into men's health. Urol Oncol. 33:40–44. 2015.
View Article : Google Scholar
|
|
2
|
Malats N and Real FX: Epidemiology of
bladder cancer. Hematol Oncol Clin North Am. 29:177–189. vii2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pang KH and Catto JWF: Bladder cancer.
Surgery. 31:523–529. 2013.
|
|
4
|
Turo R, Cross W and Whelan P: Bladder
cancer. Medicine. 40:14–19. 2012. View Article : Google Scholar
|
|
5
|
Kaufman DS, Shipley WU and Feldman AS:
Bladder cancer. Lancet. 374:239–249. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bellmunt J, Orsola A, Leow JJ, Wiegel T,
De Santis M and Horwich A: Bladder cancer: ESMO Practice Guidelines
for diagnosis, treatment and follow-up. Ann Oncol. 25(Suppl 3):
iii40–iii48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sternberg CN, Bellmunt J, Sonpavde G,
Siefker-Radtke AO, Stadler WM, Bajorin DF, Dreicer R, George DJ,
Milowsky MI, Theodorescu D, et al International Consultation on
Urologic Disease-European Association of Urology Consultation on
Bladder Cancer 2012: ICUD-EAU International Consultation on Bladder
Cancer 2012: Chemotherapy for urothelial carcinoma -neoadjuvant and
adjuvant settings. Eur Urol. 63:58–66. 2013. View Article : Google Scholar
|
|
8
|
Neoadjuvant chemotherapy in invasive
bladder cancer: Update of a systematic review and meta-analysis of
individual patient data advanced bladder cancer (ABC) meta-analysis
collaboration. Eur Urol. 48:202–205; discussion 205–206. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Julien S, Puig I, Caretti E, Bonaventure
J, Nelles L, van Roy F, Dargemont C, de Herreros AG, Bellacosa A
and Larue L: Activation of NF-kappaB by Akt upregulates Snail
expression and induces epithelium mesenchyme transition. Oncogene.
26:7445–7456. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Saxena M, Stephens MA, Pathak H and
Rangarajan A: Transcription factors that mediate
epithelial-mesenchymal transition lead to multidrug resistance by
upregulating ABC transporters. Cell Death Dis. 2:e1792011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kumar R, Deep G and Agarwal R: An overview
of ultraviolet B radiation-induced skin cancer chemoprevention by
silibinin. Curr Pharmacol Rep. 1:206–215. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li F, Ma Z, Guan Z, Chen Y, Wu K, Guo P,
Wang X, He D and Zeng J: Autophagy induction by silibinin
positively contributes to its anti-metastatic capacity via
AMPK/mTOR pathway in renal cell carcinoma. Int J Mol Sci.
16:8415–8429. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gu HR, Park SC, Choi SJ, Lee JC, Kim YC,
Han CJ, Kim J, Yang KY, Kim YJ, Noh GY, et al: Combined treatment
with silibinin and either sorafenib or gefitinib enhances their
growth-inhibiting effects in hepatocellular carcinoma cells. Clin
Mol Hepatol. 21:49–59. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pirouzpanah MB, Sabzichi M, Pirouzpanah S,
Chavoshi H and Samadi N: Silibilin-induces apoptosis in breast
cancer cells by modulating p53, p21, Bak and Bcl-XL pathways. Asian
Pac J Cancer Prev. 16:2087–2092. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bosch-Barrera J and Menendez JA: Silibinin
and STAT3: A natural way of targeting transcription factors for
cancer therapy. Cancer Treat Rev. 41:540–546. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Prajapati V, Kale RK and Singh RP:
Silibinin combination with arsenic strongly inhibits survival and
invasiveness of human prostate carcinoma cells. Nutr Cancer.
67:647–658. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jiang K, Wang W, Jin X, Wang Z, Ji Z and
Meng G: Silibinin, a natural flavonoid, induces autophagy via
ROS-dependent mitochondrial dysfunction and loss of ATP involving
BNIP3 in human MCF7 breast cancer cells. Oncol Rep. 33:2711–2718.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zeng J, Sun Y, Wu K, Li L, Zhang G, Yang
Z, Wang Z, Zhang D, Xue Y, Chen Y, et al: Chemopreventive and
chemotherapeutic effects of intravesical silibinin against bladder
cancer by acting on mitochondria. Mol Cancer Ther. 10:104–116.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hayakawa M, Miyashita H, Sakamoto I,
Kitagawa M, Tanaka H, Yasuda H, Karin M and Kikugawa K: Evidence
that reactive oxygen species do not mediate NF-kappaB activation.
EMBO J. 22:3356–3366. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Housman G, Byler S, Heerboth S, Lapinska
K, Longacre M, Snyder N and Sarkar S: Drug resistance in cancer: An
overview. Cancers (Basel). 6:1769–1792. 2014. View Article : Google Scholar
|
|
21
|
Herr HW, Dotan Z, Donat SM and Bajorin DF:
Defining optimal therapy for muscle invasive bladder cancer. J
Urol. 177:437–443. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li F, Zhang J, Arfuso F, Chinnathambi A,
Zayed ME, Alharbi SA, Kumar AP, Ahn KS and Sethi G: NF-κB in cancer
therapy. Arch Toxicol. 89:711–731. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gilmore TD and Wolenski FS: NF-κB: Where
did it come from and why? Immunol Rev. 246:14–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sui H, Zhu L, Deng W and Li Q:
Epithelial-mesenchymal transition and drug resistance: Role,
molecular mechanisms, and therapeutic strategies. Oncol Res Treat.
37:584–589. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jung HJ, Park JW, Lee JS, Lee SR, Jang BC,
Suh SI, Suh MH and Baek WK: Silibinin inhibits expression of HIF-1α
through suppression of protein translation in prostate cancer
cells. Biochem Biophys Res Commun. 390:71–76. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim S, Choi JH, Lim HI, Lee SK, Kim WW,
Kim JS, Kim JH, Choe JH, Yang JH, Nam SJ, et al: Silibinin prevents
TPA-induced MMP-9 expression and VEGF secretion by inactivation of
the Raf/MEK/ERK pathway in MCF-7 human breast cancer cells.
Phytomedicine. 6:573–580. 2009. View Article : Google Scholar
|
|
27
|
Kim S, Kim SH, Hur SM, Lee SK, Kim WW, Kim
JS, Kim JH, Choe JH, Nam SJ, Lee JE, et al: Silibinin prevents
TPA-induced MMP-9 expression by down-regulation of COX-2 in human
breast cancer cells. J Ethnopharmacol. 126:252–257. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu K, Zeng J, Li L, Fan J, Zhang D, Xue Y,
Zhu G, Yang L, Wang X and He D: Silibinin reverses
epithelial-to-mesenchymal transition in metastatic prostate cancer
cells by targeting transcription factors. Oncol Rep. 23:1545–1552.
2010.PubMed/NCBI
|
|
29
|
Cufí S, Bonavia R, Vazquez-Martin A,
Oliveras-Ferraros C, Corominas-Faja B, Cuyàs E, Martin-Castillo B,
Barrajón-Catalán E, Visa J, Segura-Carretero A, et al: Silibinin
suppresses EMT-driven erlotinib resistance by reversing the high
miR-21/low miR-200c signature in vivo. Sci Rep. 3:24592013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sun SC: The noncanonical NF-κB pathway.
Immunol Rev. 246:125–140. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Anreddy N, Gupta P, Kathawala RJ, Patel A,
Wurpel JN and Chen ZS: Tyrosine kinase inhibitors as reversal
agents for ABC transporter mediated drug resistance. Molecules.
19:13848–13877. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wellington K and Jarvis B: Silymarin: A
review of its clinical properties in the management of hepatic
disorders. BioDrugs. 15:465–489. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li L, Gao Y, Zhang L, Zeng J, He D and Sun
Y: Silibinin inhibits cell growth and induces apoptosis by caspase
activation, down-regulating survivin and blocking EGFR-ERK
activation in renal cell carcinoma. Cancer Lett. 272:61–69. 2008.
View Article : Google Scholar : PubMed/NCBI
|