Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a highly
lethal malignancy with an estimated 5-year survival rate <5%,
and by 2020, it is estimated to be the second leading cause of
cancer-related mortality (1). Due
to the lack of early detection methods and effective preventive
strategies, PDAC is usually diagnosed at an advanced stage when it
is deemed unresectable and exhibits poor response to
chemotherapeutic drugs, including gemcitabine and 5-fluorouracil
(2). Thus, it is essential to
understand the underlying mechanisms for the chemoresistance of
PDAC.
Krüppel-like factor 4 (KLF4) is a transcription
factor that serves as an important regulator in cell
differentiation, cell cycle, cell survival and apoptosis (3). KLF4 exerts seemingly paradoxical
functions in different cell types. KLF4 is down-regulated in
bladder and gastric cancer cells, and enforced expression of KLF4
promotes apoptosis in these cells (4). In PDAC, KLF4 also functions as a
tumor-suppressor (5). Conversely,
KLF4 exhibits oncogenic functions in oral, skin squamous carcinoma
cells and breast ductal carcinoma (6). To date, little is known about the
precise role of KLF4 in gemcitabine resistance of PDAC.
The epithelial-mesenchymal transition
(EMT)-activator ZEB1 is a transcriptional repressor of epithelial
genes, such as E-cadherin (7).
ZEB1 has been widely implicated in the invasiveness and metastasis
of solid tumors, including lung adenocarcinoma, colorectal and
breast cancers (8–10). Ectopic expression of ZEB1 also
promotes drug-resistant phenotypes (7). However, the role of ZEB1 in
gemcitabine resistance of PDAC remains unexplored.
In this study, we showed that gemcitabine treatment
inhibited KLF4 expression and promoted ZEB1 expression. Further
investigation demonstrated that KLF4 suppressed ZEB1 expression and
gemcitabine resistance by directly regulating the expression of
miR-200b and miR-183. In addition, ZEB1 knockdown attenuated
gemcitabine resistance in PDAC cells.
Materials and methods
Antibodies and reagents
The following monoclonal antibodies (mAbs) and
polyclonal antibodies (pAbs) were used: anti-KLF4 (pAb, ab106629),
and anti-ZEB1 (pAb, ab155249) were purchased from Abcam (Cambridge,
MA, USA); anti-GAPDH (pAb) was purchased from Anbo Biotechnology
Co. (San Francisco, CA, USA); Cell counting kit-8 was purchased
from Dojindo Laboratories (Tokyo, Japan). Gemcitabine was purchased
from Eli Lilly (Indianapolis, IN, USA). ZEB1 siRNA, miR-200b and
miR-183 were synthesized by Biosune Biotechnology (Shanghai,
China).
Cell culture
The human PDAC cell lines BxPC-3, Panc-1 and
MIApaca-2 were purchased from the American Type Culture Collection
(Manassas, VA, USA). Cells were cultured in RPMI-1640 medium
(Hyclone, USA) supplemented with 10% fetal bovine serum (Hyclone),
100 U/ml penicillin, 100 U/ml streptomycin and 0.03% L-glutamine at
37°C in 5% CO2.
Cell viability assay
PDAC cells were seeded at a density of
1.0×104 cells/well in 96-well microplates with 100
µl RPMI-1640 medium. After grown to ~80% confluence, the
cells were incubated with the indicated concentration of
gemcitabine. After treatment for 72 h, the cells were incubated
with 10% WST-8 dye for 2 h at 37°C. The absorbance was detected at
450 nm using a SpectraMax M2. IC50 values were
determined using GraphPad Prism 6.0.
Flow cytometric analysis
PDAC cells were seeded at a density of
2×105 cells/well in 6-well microplates. After treatment,
flow cytometric analysis was performed to determine cell apoptosis.
In brief, after treatment with gemcitabine, the cells were digested
using trypsin lacking EDTA and phenol red, and were incubated in
binding buffer containing Annexin V-FITC (2.5 mg/ml) and
propidiumiodide (5 mg/ml) for 10 min in the dark at room
temperature. Subsequently, the labeled cells were detected by a
flow cytometer (Beckman Coulter, Chicago, IL, USA).
Lentiviruses construction
For KLF4 knockdown, the short hairpin RNA (shRNA)
targeting the coding sequence of KLF4 (TACCCATCCTTCCTGCCCGAT) was
digested using the restriction enzymes AgeI and EcoRI
(New England Biolabs UK Ltd., UK), and was then cloned into
pGCSIL-GFP plasmid. For KLF4 overexpression, the full coding region
of KLF4 was amplified using specific primers (forward,
5′-CGCGGATCCGCGATGGCTGTCAGCGACGCG-3′; reverse,
5′-GGGTACCGGTCGCCACCTTCTCTTCTGGCAGTGTGGG-3′). The product was
digested using the restriction enzymes BamHI and
AgeI, and was then linked to p-GC365-EGFP plasmid. The
plasmids were transformed into E. coli DH5a. Positive clones
were harvested and identified by sequencing. The recombinant
plasmids were transfected to 293T cells by Lipofectamine 2000
(Invitrogen). The infection efficiency was determined by measuring
fluorescence intensity under microscopy (Olympus 3.3RTV, Japan).
After 48 h of transfection, the supernatant was collected by
centrifugation at 4,000 g for 10 min at 4°C, followed by
ultrafiltration centrifugation at 4000 g for 12 min. The
concentrated solution was stored at −80°C for future
experiments.
Gene transfection
BxPC-3, Panc-1 and MIApaca-2 cells were seeded at a
density of 2×105 cells/well in 6-well microplates. After
growing overnight, the cells were transfected with lentivirus
carrying shRNAs against KLF4 or KLF4 cDNA (GeneChem, Shanghai,
China). For ZEB1 knockdown, PDAC cells were transfected with siRNA
against ZEB1 (GGTAGA TGGTAATGTAATA) using Lipofectamine 3000
(Invitrogen).
Western blot analysis
Western blot analysis was performed as previously
described (11). Equal amounts of
protein were loaded to sodium dodecyl sulfate polyacrylamide gels.
The protein was subsequently transferred onto PVDF membranes. After
blocked with 5% BSA, membranes were incubated with the indicated
primary antibodies overnight at 4°C, followed by treatment with
horseradish peroxidase-conjugated secondary antibodies for 1 h at
room temperature. The protein signals were detected using the ECL
kit and quantified using an Alpha Imager 2200 (Alpha Innotech
Corp., USA).
Northern blot analysis
The total RNA was extracted using TRIzol reagent
(Invitrogen). RNA (20 µg) was subjected to 15%
urea-polyacrylamide gels, and was then transferred to nylon
membrane. The expression of miR-200b, miR-183 and U6 were detected
using biotin-labeled oligonucleotide probes
(5′-TCATCATTACCAGGCAGTATTA-3′, miR-200b;
5′-CAGTGAATTCTACCAGTGCCAT-3′, miR-183 and
5′-ATATGGAACGCTTCACGAATT-3′, U6). The signals of blots were
measured using the Fujifilm LAS-4000 imaging system.
Luciferase reporter assay
Panc-1 and MIApaca-2 cells were co-transfected with
increasing concentrations of pcDNA3.1 carrying KLF4 and 0.2
µg of luciferase reporter plasmid containing the miR-200b or
miR-183 promoter using Lipofectamine 3000 (Roche); the pRL-SV40
plasmid (Promega) was transfected as a normalization control. After
24-h incubation, cell lysates were collected to determine
luciferase activity using the Dual-Luciferase assay (Promega)
according to the manufacturer's instructions. The primers were used
as follows: 5′-ATATGCGGCCGCGTTTTGGCTTCGTTTCTTCT-3′ and
5′-GCTTGGTACCGACCCCATCTGTTCTTTGATT-3′ for KLF4;
5′-AGAAAGGGTGGGAAGGAGGACA-3′ and 5′-GGACTCGCTGGGAAGCTCAGTA-3′ for
miR-200b; 5′-ATTGTAGTAAGGGAAACTGAGGC-3′ and
5′-GCAGAAGTGGGTAAGGTGCT-3′ for miR-183. The sequence of ZEB1-3′UTR
containing the miR-200b seed regions were amplified using specific
primers (forward, 5′-CGAGCTCATTGTTTTATCTTATCAGTATTATC-3′; reverse,
5′-CCGCTCGAGAACTAAAAGAAATAAAATAATACTG-3′) SacI and
XhoI restriction sites. The PCR product was digested using
the restriction enzymes SacI and XhoI, and was
subsequently linked to pHSA-MIR-Report (Ambion). Mutations in the
miR-200b seed regions of the ZEB1-3′UTR were generated using the
QuikChange Multi-site-directed mutagenesis kit (Stratagene). For
miR-183, two targeting sequences locating at position 880-886,
1801-1807 of ZEB1-3′UTR were amplified using forward
(5′-CGAGCTCAGTGCCATTTCTCAGTATTTTCAAG-3′) and reverse
(5′-CCGCTCGAGAGTGCCATTTCTCAGTATTTTCAAG-3′) primers containing
SacI and XhoI restriction sites. The corresponding
mutagenesis was generated using specific primers (forward,
5′-CGAGCTCACACGGATTTCTCAGTATTTTCAAG-3′; reverse,
5′-CCGCTCGAGAGACGGATTTCTCAGTATTTTCAAG-3′).
Chromatin immunoprecipitation assay
(ChIP)
ChIP was performed using a ChIP assay kit
(Millipore) according to the manufacturer's instructions. In brief,
Panc-1 cells were crosslinked with fresh 1% formaldehyde, followed
by incubation in SDS lysis buffer containing 1% protease
inhibitors. The cell lysates were sonicated to shear crosslinked
DNA to 200-1,000 bp in length. Protein/DNA complexes were
immunoprecipitated with 3 µg anti-ZEB1 antibody or control
IgG. The complexes were eluted from the antibodies, and were
dissociated with 5 M NaCl. ChIP samples were analyzed with
real-time quantitative PCR using primers specific for the promoters
of miR-200b and miR-183. The primers are listed in Tables I and II.
 | Table IPrimers for miR-200b. |
Table I
Primers for miR-200b.
| Sites | Forward primer | Reverse primer |
|---|
| 1 |
5′-TCAGACCCCAGACCAGCA-3′ |
5′-CCACCTGGAGACCCCTCT-3′ |
| 2 |
5′-GTGGCGGGGACCGTTCTGT-3′ |
5′-TCTGAGGCAGGGGACAA-3′ |
| 3 |
5′-CCTTGTCCCCTGCCTCA-3′ |
5′-CCTTCCCTCTGGGTGGTC-3′ |
| 4 |
5′-GCCTTCCTATGGGACCACC-3′ |
5′-CGAGTCCAGCAGCCACCAG-3′ |
| 5 |
5′-GGCAGAGGGCCCGTGTCA-3′ |
5′-GGGTTGCATGGGACTCGCT-3′ |
| Table IIPrimers for miR-183. |
Table II
Primers for miR-183.
| Sites | Forward primer | Reverse primer |
|---|
| 1 |
5′-CAAGCTGGATTGTCCTCTGG-3′ |
5′-CCTCTTGGCGATGTTACCC-3′ |
| 2 |
5′-GCTCCCTCCAAGCCACCT-3′ |
5′-CCCCAGGAACAAACCGAAT-3′ |
| 3 |
5′-TGTTCCTGGGGCTCTGTTT-3′ |
5′-GGACCATCCATCTCCTTGC-3′ |
| 4 |
5′-GTCTGGGTGATGTGGAGGG-3′ |
5′-AGCGGCTGCTACGGTCT-3′ |
| 5 |
5′-TAGCAGCCGCTGCTGAGG-3′ |
5′-GCCCACGATGCCCGTT-3″ |
Quantitative RT-PCR
Total RNA was extracted using TRIzol reagent
(Invitrogen). The expression of miR-200b and miR-183 were analyzed
using TaqMan miRNA assays (Applied Biosystems, CA, USA) according
to the manufacturer's instructions. U6 snRNA was used as an
internal control.
Immunohistochemistry
Immunohistochemistry was performed as previously
described (12). Briefly, tumor
sections were fixed in formalin, embedded with paraffin,
deparaffinized in xylene, and hydrated with ethanol. After antigen
retrieval using microwave, the tissue sections were treated with 3%
hydrogen peroxide and blocked with 10% goat serum. The sections
were then stained with anti-KLF4 and anti-ZEB1 antibodies, followed
by treatment with a biotinylated secondary antibody. The images
were captured using a fluorescence microscope (Leica DM IRE2).
Mouse xenograft models
The orthotopic PDAC nude mouse model was established
as previously described by us (12), with a minor modification. Briefly,
4- to 6-week-old BALB/c nude mice of both sexes were obtained from
Weitonglihua Animal Center (Beijing, China) and maintained under
specific pathogen-free conditions in the animal facility. All
procedures were approved by the Institutional Laboratory Animal
Care and Use Committee at Shandong Provincial Hospital. Panc-1
cells (1×107) in 100 µl PBS/Matrigel (1:1, v/v,
BD Biosciences, USA) transfected with control lentivirus or
lentivirus carrying KLF4 cDNA were injected into both flanks of the
mice. One week after injection, gemcitabine (80 mg/kg; q3 days) was
administered via intraperitoneal injection for six weeks. The long
(L) and short axes (S) of the tumors were measured weekly using
Vernier calipers, and the tumor volumes were calculated as follows:
V = (L x S2) x π/6. The mice were sacrificed 24 h after
the last injection, and the tumors were harvested for subsequent
analysis.
Statistical analysis
Statistical analysis was carried out using the SPSS
18.0 software package. All data are presented as the mean ±
standard deviation from at least three independent experiments.
Student's t-test was used for comparisons between two groups.
One-way ANOVA was used for comparisons among multiple groups.
p<0.05 was considered statistically significant.
Results
Gemcitabine inhibits the expression of
KLF4, miR-200b and miR-183 and promotes ZEB1 expression
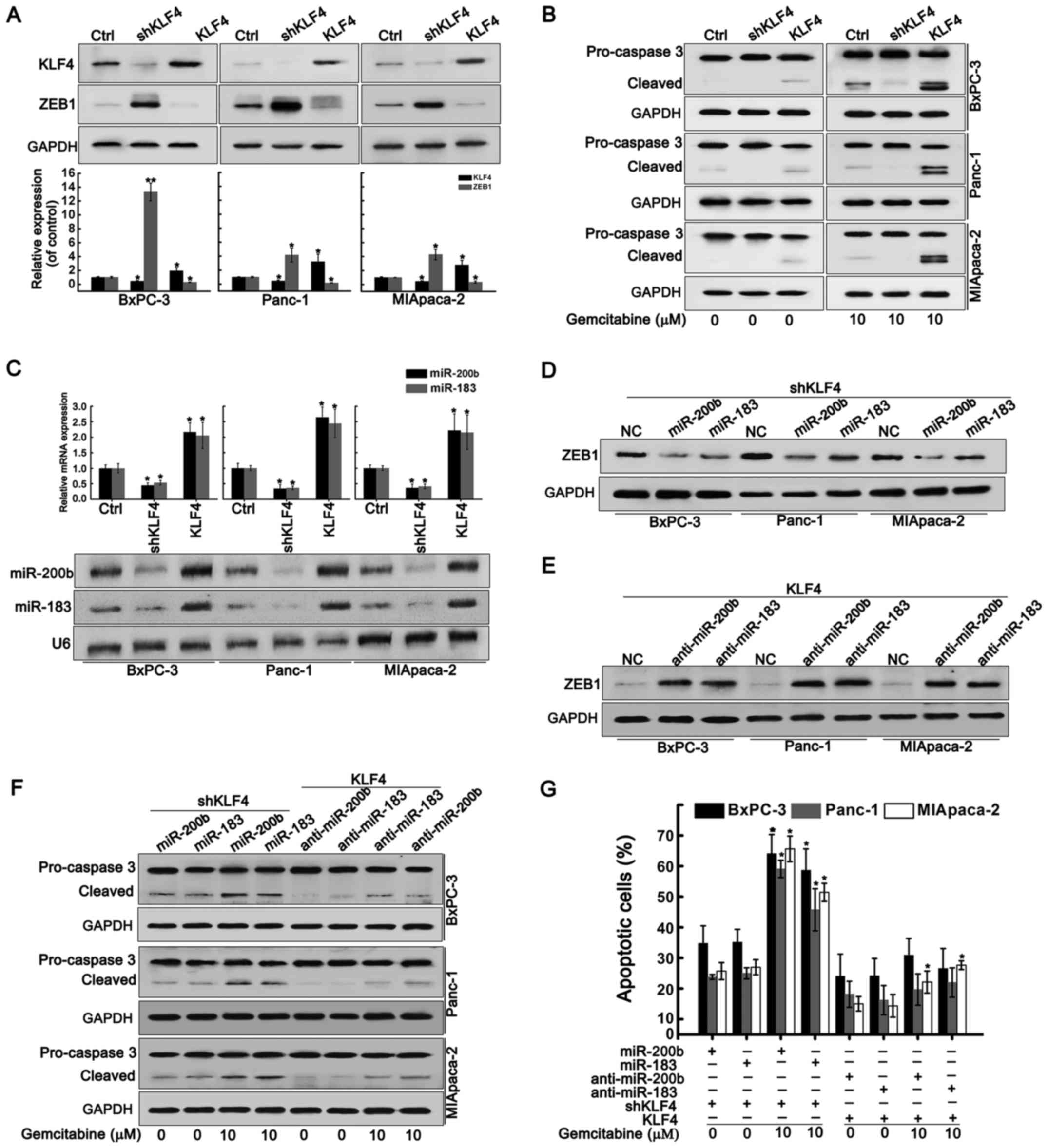
First, we measured the expression of KLF4 and ZEB1
in BxPC-3, Panc-1 and MIApaca-2 cells. Fig. 1A showed a higher level of KLF4 in
BxPC-3 cells than those in Panc-1 and MIApaca-2 cells while lower
levels of ZEB1 in BxPC-3 cells than those in Panc-1 and MIApaca-2
cells. Exposure of BxPC-3 cells to gemcitabine caused more apparent
decline in cell viability (IC50=3.942 µM)
compared to Panc-1 (IC50=9.383 µM) and MIApaca-2
cells (IC50=7.904 µM) (Fig. 1B). Flow cytometry analysis
demonstrated that treatment with 10 µM gemcitabine induced
the death of 68.34% of the BxPC-3 cells, 50.45% of the Panc-1 cells
and 51.02% of the MIApaca-2 cells (Fig. 1C), suggesting that BxPC-3 cells
were more sensitive to gemcitabine than Panc-1 and MIApaca-2 cells.
Further investigation found that gemcitabine treatment reduced the
expression of KLF4, miR-200b and miR-183 in a dose-dependent
manner, whereas led to a dose-dependent increase in ZEB1 expression
(Fig. 1D and E).
KLF4 attenuates ZEB1 expression and
gemcitabine resistance by upregulation of miR-200b and miR-183
Next, we examined the correlation between KLF4 and
ZEB1. KLF4 knockdown significantly promoted ZEB1 expression while
KLF4 overexpression inhibited ZEB1 expression (Fig. 2A). KLF4 depletion inhibited
gemcitabine-induced activation of caspase 3 while KLF4
overexpression promoted the activation of caspase 3 even in the
absence of gemcitabine (Fig. 2B),
suggesting that KLF4 downregulation conferred gemcitabine
resistance of PDAC cells. KLF4 knockdown restrained the expression
of miR-200b and miR-183 (Fig. 2C).
In contrast, KLF4 overexpression elevated the levels of miR-200b
and miR-183 (Fig. 2C). In
KLF4-depleted cells, forced expression of miR-200b and miR-183
repressed the ZEB1 expression again (Fig. 2D). In KLF4-overexpressing cells,
forced expression of anti-miR-200b and anti-miR-183 restored the
ZEB1 expression (Fig. 2E).
Restoration of miR-200b and miR-183 activated caspase 3 in
KLF4-depleted cells whereas anti-miR-200b and anti-miR-183
significantly reduced caspase 3 activation in KLF-overexpressing
cells (Fig. 2F). Overexpression of
miR-200b and miR-183 in KLF-4-depleted cells promoted cell
apoptosis in the presence of gemcitabine, but inhibition of
miR-200b and miR-183 in KLF-4-overexpressing cells blocked cell
apoptosis induced by gemcitabine (Fig.
2G). These results suggested that KLF4 knockdown contributed to
ZEB1 expression and gemcitabine resistance by suppressing miR-200b
and miR-183.
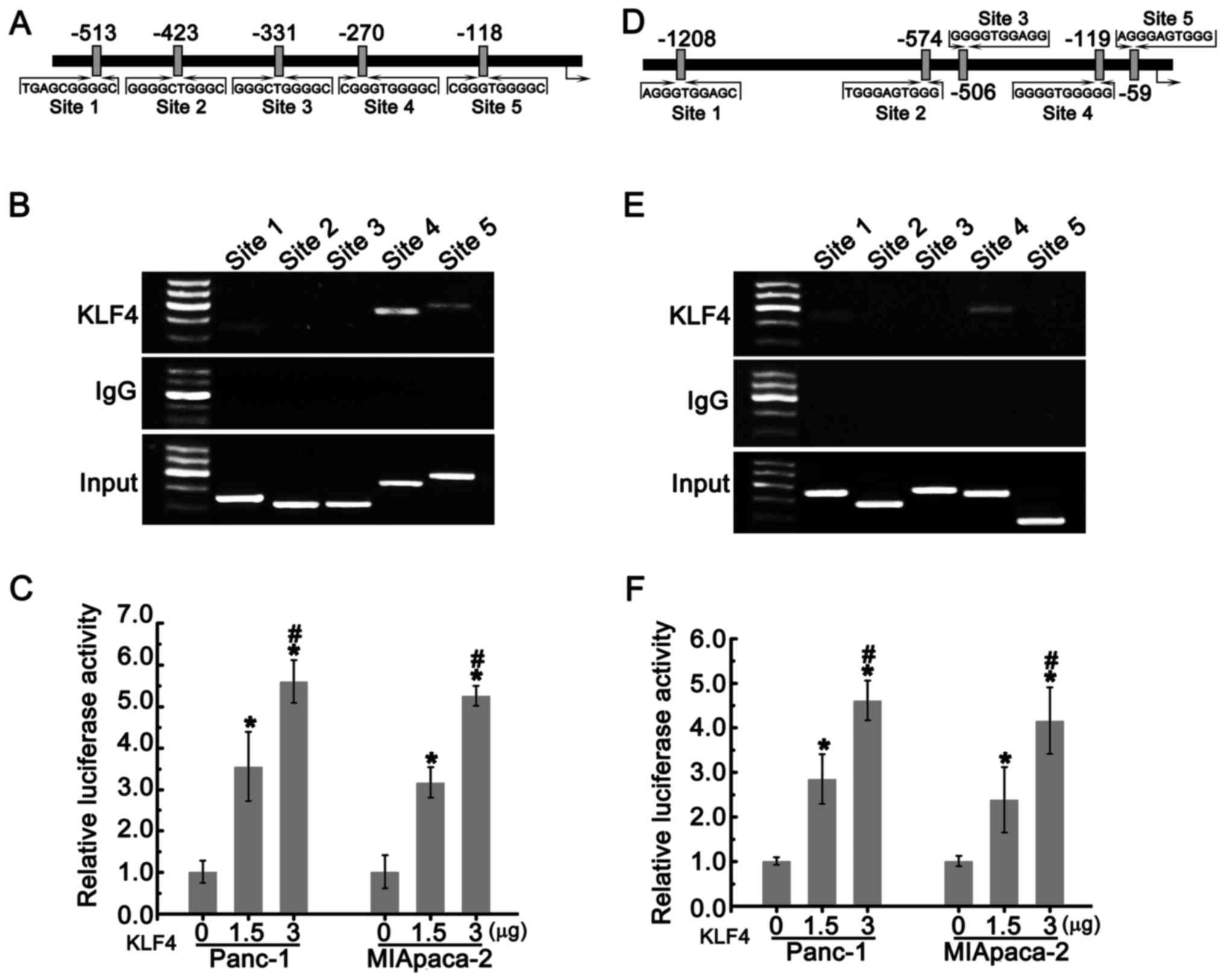
KLF4 positively regulates the expression
of miR-200b and miR-183 in PDAC cells
Since KLF4 is a transcriptional factor that
regulates the expression of multiple genes, we hypothesized that
KLF4 directly regulated the expression of miR-200b and miR-183. The
putative binding sites of KLF4 located on the promoters of miR-200b
and miR-183 were shown in Fig. 3A and
D. Chromatin immunoprecipitation (ChIP) assays revealed that
KLF4 could interact with site 4 and 5 in the miR-200b promoter and
the site 4 in the miR-183 promoter (Fig. 3B and E). We subsequently
constructed the pGL3-200-luc and pGL3-183-luc plasmids by inserting
these sequences into the pGL3 luciferase reporter. Panc-1 and
MIApaca-2 cells were co-transfected with 0.2 µg pGL3-200-luc
or pGL3-183-luc and the indicated concentration of pcDNA3.1
carrying KLF4 together with 100 ng pRL-SV40. As shown in Fig. 3C and F, with the increasing dose,
KLF4 substantially enhanced luciferase activities. These data
suggested that KLF4 positively regulated the expression of miR-200b
and miR-183 by directly binding to their promoters.
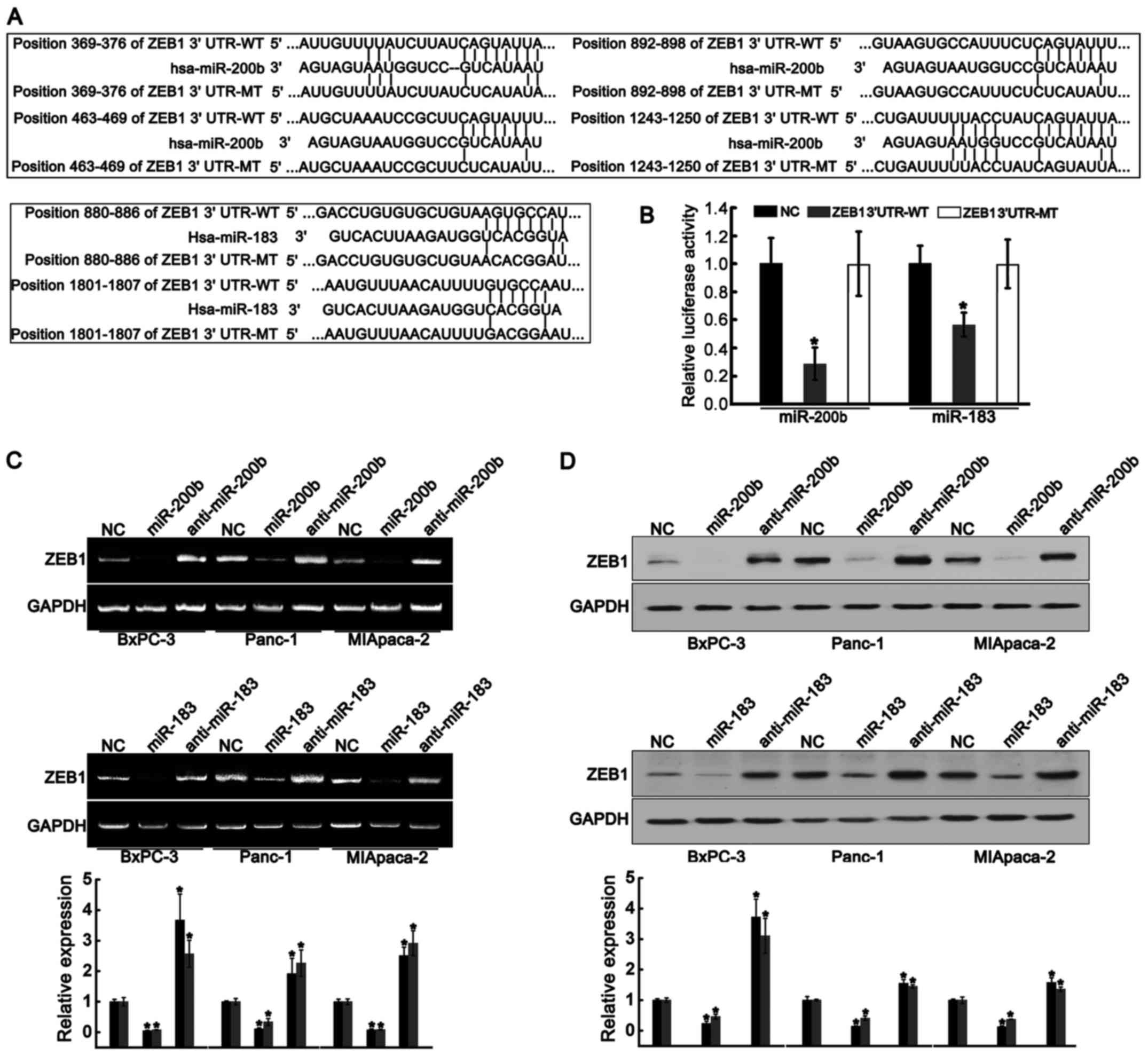
miR-200b and miR-183 directly targets
ZEB1 in PDAC cells
We further explored whether miR-200b and miR-183
directly targeted the-3′UTR region of ZEB1. Targetscan (http://www.targetscan.org/) was used to predict the
recognition sites of miR-200b and miR-183. As shown in Fig. 4A, four putative sites for miR-200b
and two putative sites for miR-183 were found. The sequences
containing these sites were cloned into the luciferase reporter
plasmid to obtain the ZEB1-3′UTR-WT-luc plasmid (wild-type), and
the sequence containing the mutant sites was cloned to construct
ZEB1-3′UTR-MT-luc plasmid (mutant type). Subsequently, the Panc-1
cells were co-transfected with ZEB1-3′UTR-WT or ZEB1-3′UTR-MT and
miR-200b or miR-183. Both miR-200b and miR-183 significantly
inhibited luciferase activities in the cells carrying ZEB1-3′UTR-WT
compared to those carrying negative control (NC) and ZEB1-3′UTR-MT
(Fig. 4B). Furthermore, both
miR-200b and miR-183 notably reduced mRNA and protein levels of
ZEB1 in BxPC-3, Panc-1 and MIApaca-2 cells (Fig. 4C and D). Conversely, both
anti-miR-200b and anti-miR-183 elevated mRNA and protein levels of
ZEB1 (Fig. 4C and D). These
results indicated that miR-200b and miR-183 directly targeted
ZEB1.
ZEB1 knockdown contributes to gemcitabine
sensitivity of PDAC cells
As shown in Fig.
5A, gemcitabine treatment promoted ZEB1 expression, which was
consistent with the results in Fig.
1D. ZEB1 knockdown mildly activated caspase 3. A combination of
ZEB1 knockdown and gemcitabine significantly increased cleaved
caspase 3, suggesting that ZEB1 knockdown facilitated gemcitabine
sensitivity of PDAC cells. Fig. 5B
illustrated that gemcitabine treatment induced cell apoptosis in
ZEB1-depleted cells. These data indicated that ZEB1 knockdown
contributed to gemcitabine sensitivity of PDAC cells.
Negative association between KLF4 and
ZEB1 regulates gemcitabine resistance of PDAC in vivo
We established xenograft tumors of PDAC to determine
the effect of KLF4 overexpression on the ZEB1 expression and
gemcitabine resistance of PDAC in vivo. The results showed
that KLF4 overexpression reduced ZEB1 expression, consistent with
the data in vitro (Fig.
6A–C). Linear regression analysis showed that KLF4 expression
was negatively correlated to ZEB1 expression (Fig. 6D). Compared to the control group,
KLF4 overexpression did not significantly affect tumor growth,
whereas it enhanced gemcitabine sensitivity (Fig. 6E and F). These results suggested
that KLF4 overexpression attenuated gemcitabine resistance of PDAC
in vivo by negatively regulating ZEB1 expression.
Discussion
Chemotherapy resistance has been a major challenge
in improving the overall survival of patients with PDAC.
Gemcitabine is a standard clinical chemotherapeutic drug for
advanced PDAC, but results in a progression-free survival interval
ranging from 0.9 to 4.2 months only (12). Thus, understanding of the mechanism
by which chemotherapy resistance occurs contributes to the
development of novel therapeutic strategies for overcoming advanced
PDAC. KLF4 has been identified as a tumor suppressor in diverse
types of cancer (13–15), including PDAC (5). In the pancreas, KLF4 is predominantly
expressed in PDAC and regulates the expression of cytokeratin-19, a
specific marker for PDAC (16). In
this study, we observed a higher level of KLF4 in BxPC-3 cells
compared to those in Panc-1 and MIApaca-2 cells. Intriguingly,
BxPC-3 cells were more sensitive to gemcitabine than Panc-1 and
MIApaca-2. Fig. 1D illustrated
that gemcitabine treatment led to a dose-dependent decrease in the
expression of KLF4 in BxPC-3, Panc-1 and MIApaca-2 cells. These
results suggested that downregulation of KLF4 might contribute to
gemcitabine resistance of PDAC. A previous study demonstrated that
82.4% of patients with PDAC lack KLF4 expression, which indicates
the implication of KLF4 loss in PDAC progression (17). Our results showed that KLF4
knockdown enhanced gemcitabine resistance of PDAC cells while KLF4
overexpression reduced gemcitabine resistance. These results
confirmed that the low level of KLF4 promoted gemcitabine
resistance of PDAC cells.
How does KLF4 regulate gemcitabine resistance of
PDAC cells? Our results showed that gemcitabine treatment
simultaneously triggered the downregulation of KLF4, miR-200b and
miR-183 in PDAC cells. KLF4 silencing inhibited the expression of
miR-200b and miR-183. In contrast, KLF4 overexpression promoted the
expression of miR-200b and miR-183. ChIP assays revealed that KLF4
positively regulated the expression of miR-200b and miR-183 by
directly binding to their promoters. These results suggested that
lower expression of KLF4 conferred PDAC cells with greater
gemcitabine resistance by upregulation of miR-200b and miR-183.
Consistently, the expression of miR-200b has been found to be
significantly downregulated in gemcitabine-resistant cells
(18). The expression of miR-183
is related to expression of miR-200 family members (19). miR-183 and miR-200 family members
cooperate to affect stemness properties in PDAC cells by
suppressing Bmi1 (19), function
as EMT inhibitors and accelerate epithelial differentiation by
targeting the Wnt/β-catenin signaling pathway (20,21).
In this study, overexpression of miR-183 and miR-200b reduced the
expression of ZEB1 at both mRNA and protein levels in PDAC cells,
even in KLF4-depleted cells that ZEB1 was strongly increased.
Conversely, anti-miR-200b and anti-miR-183 increased the mRNA and
protein levels of ZEB1, even in KLF4-overexpressing cells in which
ZEB1 was significantly inhibited. Combined with the data from
luciferase reporter assays, we confirmed that miR-200b and miR-183
targeted ZEB1 in PDAC cells. Congruously, ZEB1 has been found to be
direct target of miR-200b in hepatocellular carcinoma, osteosarcoma
and non-small cell lung cancer (22–24).
Although it has been confirmed that ZEB1 represses the
transcription of miR-183 in lung cancer (25,26),
it is unclear whether miR-183 directly targets ZEB1. Our results
indicated that KLF4 knockdown enhanced gemcitabine resistance by
suppressing the miR-200b/miR-183/ZEB1 signaling pathway.
ZEB1 is an EMT-activator that is correlated to tumor
metastasis, drug resistance and poor prognosis in different tumor
types (27). A series of studies
have suggested that EMT promotes early-stage dissemination,
invasion and metastasis of PDAC (28,29).
A recent study demonstrates that EMT is dispensable for metastasis
but increases chemoresistance in pancreatic cancer (30). Our results also demonstrated that
ZEB1 knockdown promoted gemcitabine sensitivity of PDAC cells.
In conclusion, we demonstrated that gemcitabine
inhibited the expression of KLF4, blocking KLF4-mediated downstream
signals. Further investigation confirmed that KLF4 induced the
expression of miR-200b and miR-183 by directly binding to the
promoters of miR-200b and miR-183. Elevated miR-200b and miR-183
subsequently inhibited ZEB1 expression by directly targeting
the-3′UTR of ZEB1. Inhibition of ZEB1 attenuated gemcitabine
resistance. In addition, KLF4 overexpression enhanced gemcitabine
sensitivity of PDAC in vivo by inhibiting ZEB1 expression.
Taken together, our results suggested that novel crosstalk between
KLF4 and ZEB1 regulates gemcitabine resistance in PDAC cells
(Fig. 7).
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant nos. 81572272 and 81201778),
Shandong Provincial Natural Science Foundation (grant no.
ZR2013HQ026) and the Science and Technology Development Plan
Project of Shandong Province (grant nos. 2013G0021810 and 2016
GSF201127).
References
|
1
|
Mohammed A, Janakiram NB, Brewer M,
Ritchie RL, Marya A, Lightfoot S, Steele VE and Rao CV:
Antidiabetic drug metformin prevents progression of pancreatic
cancer by targeting in part cancer stem cells and mTOR signaling.
Transl Oncol. 6:649–659. 2013. View Article : Google Scholar
|
|
2
|
Olive KP, Jacobetz MA, Davidson CJ,
Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA,
Caldwell ME, Allard D, et al: Inhibition of Hedgehog signaling
enhances delivery of chemotherapy in a mouse model of pancreatic
cancer. Science. 324:1457–1461. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tetreault MP, Yang Y and Katz JP:
Krüppel-like factors in cancer. Nat Rev Cancer. 13:701–713. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wei D, Wang L, Kanai M, Jia Z, Le X, Li Q,
Wang H and Xie K: KLF4α up-regulation promotes cell cycle
progression and reduces survival time of patients with pancreatic
cancer. Gastroenterology. 139:2135–2145. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yan Y, Li Z, Kong X, Jia Z, Zuo X, Gagea
M, Huang S, Wei D and Xie K: KLF4-Mediated suppression of CD44
signaling negatively impacts pancreatic cancer stemness and
metastasis. Cancer Res. 76:2419–2431. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rowland BD, Bernards R and Peeper DS: The
KLF4 tumour suppressor is a transcriptional repressor of p53 that
acts as a context-dependent oncogene. Nat Cell Biol. 7:1074–1082.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meidhof S, Brabletz S, Lehmann W, Preca
BT, Mock K, Ruh M, Schüler J, Berthold M, Weber A, Burk U, et al:
ZEB1-associated drug resistance in cancer cells is reversed by the
class I HDAC inhibitor mocetinostat. EMBO Mol Med. 7:831–847. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Truesdell P, Ahn J, Chander H, Meens J,
Watt K, Yang X and Craig AW: CIP4 promotes lung adenocarcinoma
metastasis and is associated with poor prognosis. Oncogene.
34:3527–3535. 2015. View Article : Google Scholar
|
|
9
|
Xiong H, Hong J, Du W, Lin YW, Ren LL,
Wang YC, Su WY, Wang JL, Cui Y, Wang ZH, et al: Roles of STAT3 and
ZEB1 proteins in E-cadherin down-regulation and human colorectal
cancer epithelial-mesenchymal transition. J Biol Chem.
287:5819–5832. 2012. View Article : Google Scholar :
|
|
10
|
Rhodes LV, Tate CR, Segar HC, Burks HE,
Phamduy TB, Hoang V, Elliott S, Gilliam D, Pounder FN, Anbalagan M,
et al: Suppression of triple-negative breast cancer metastasis by
pan-DAC inhibitor panobinostat via inhibition of ZEB family of EMT
master regulators. Breast Cancer Res Treat. 145:593–604. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma J, Fu G, Wu J, Han S, Zhang L, Yang M,
Yu Y, Zhang M, Lin Y and Wang Y: 4-cholesten-3-one suppresses lung
adeno-carcinoma metastasis by regulating translocation of HMGB1,
HIF1α and Caveolin-1. Cell Death Dis. 7:e23722016. View Article : Google Scholar
|
|
12
|
Xian G, Zhao J, Qin C, Zhang Z, Lin Y and
Su Z: Simvastatin attenuates macrophage-mediated gemcitabine
resistance of pancreatic ductal adenocarcinoma by regulating the
TGF-β1/Gfi-1 axis. Cancer Lett. 385:65–74. 2017. View Article : Google Scholar
|
|
13
|
Wang Z, Li Y, Kong D, Banerjee S, Ahmad A,
Azmi AS, Ali S, Abbruzzese JL, Gallick GE and Sarkar FH:
Acquisition of epithelial-mesenchymal transition phenotype of
gemcitabine-resistant pancreatic cancer cells is linked with
activation of the notch signaling pathway. Cancer Res.
69:2400–2407. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu T, Chen X, Lin T, Liu J, Li M, Zhang W,
Xu X, Zhao W, Liu M, Napier DL, et al: KLF4 deletion alters gastric
cell lineage and induces MUC2 expression. Cell Death Dis.
7:e22552016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shi M, Cui J, Du J, Wei D, Jia Z, Zhang J,
Zhu Z, Gao Y and Xie K: A novel KLF4/LDHA signaling pathway
regulates aerobic glycolysis in and progression of pancreatic
cancer. Clin Cancer Res. 20:4370–4380. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li H, Wang J, Xiao W, Xia D, Lang B, Yu G,
Guo X, Guan W, Wang Z, Hu Z, et al: Epigenetic alterations of
Krüppel-like factor 4 and its tumor suppressor function in renal
cell carcinoma. Carcinogenesis. 34:2262–2270. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wei D, Wang L, Yan Y, Jia Z, Gagea M, Li
Z, Zuo X, Kong X, Huang S and Xie K: KLF4 is essential for
induction of cellular identity change and acinar-to-ductal
reprogramming during early pancreatic carcinogenesis. Cancer Cell.
29:324–338. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Funel N, Morelli M, Giovannetti E, Del
Chiaro M, Pollina LE, Mosca F, Boggi U, Cavazzana A and Campani D:
Loss of heterozygosity status of D9S105 marker is associated with
downregulation of Kruppel-like factor 4 expression in pancreatic
ductal adenocarcinoma and pancreatic intraepithelial lesions.
Pancreatology. 11:30–42. 2011. View Article : Google Scholar
|
|
19
|
Li Y, VandenBoom TG II, Kong D, Wang Z,
Ali S, Philip PA and Sarkar FH: Up-regulation of miR-200 and let-7
by natural agents leads to the reversal of
epithelial-to-mesenchymal transition in gemcitabine-resistant
pancreatic cancer cells. Cancer Res. 69:6704–6712. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wellner U, Schubert J, Burk UC,
Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D,
zur Hausen A, et al: The EMT-activator ZEB1 promotes tumorigenicity
by repressing stemness-inhibiting microRNAs. Nat Cell Biol.
11:1487–1495. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xia H, Cheung WK, Sze J, Lu G, Jiang S,
Yao H, Bian XW, Poon WS, Kung HF and Lin MC: miR-200a regulates
epithelial-mesenchymal to stem-like transition via ZEB2 and
beta-catenin signaling. J Biol Chem. 285:36995–37004. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen C, Xiang H, Peng YL, Peng J and Jiang
SW: Mature miR-183, negatively regulated by transcription factor
GATA3, promotes 3T3-L1 adipogenesis through inhibition of the
canonical Wnt/β-catenin signaling pathway by targeting LRP6. Cell
Signal. 26:1155–1165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li Q, Song W, Wang W, Yao S, Tian C, Cai X
and Wang L: Suppression of epithelial-mesenchymal transition in
hepatocellular carcinoma cells by Krüppel-like factor 4.
Oncotarget. 7:29749–29760. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li Y, Zeng C, Tu M, Jiang W, Dai Z, Hu Y,
Deng Z and Xiao W: MicroRNA-200b acts as a tumor suppressor in
osteosarcoma via targeting ZEB1. Onco Targets Ther. 9:3101–3111.
2016.PubMed/NCBI
|
|
25
|
Nishijima N, Seike M, Soeno C, Chiba M,
Miyanaga A, Noro R, Sugano T, Matsumoto M, Kubota K and Gemma A:
miR-200/ZEB axis regulates sensitivity to nintedanib in non-small
cell lung cancer cells. Int J Oncol. 48:937–944. 2016.PubMed/NCBI
|
|
26
|
Yang Y, Ahn YH, Chen Y, Tan X, Guo L,
Gibbons DL, Ungewiss C, Peng DH, Liu X, Lin SH, et al: ZEB1
sensitizes lung adenocarcinoma to metastasis suppression by PI3K
antagonism. J Clin Invest. 124:2696–2708. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kundu ST, Byers LA, Peng DH, Roybal JD,
Diao L, Wang J, Tong P, Creighton CJ and Gibbons DL: The miR-200
family and the miR-183~96~182 cluster target Foxf2 to inhibit
invasion and metastasis in lung cancers. Oncogene. 35:173–186.
2016. View Article : Google Scholar
|
|
28
|
Lehmann W, Mossmann D, Kleemann J, Mock K,
Meisinger C, Brummer T, Herr R, Brabletz S, Stemmler MP and
Brabletz T: ZEB1 turns into a transcriptional activator by
interacting with YAP1 in aggressive cancer types. Nat Commun.
7:104982016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Galván JA, Zlobec I, Wartenberg M, Lugli
A, Gloor B, Perren A and Karamitopoulou E: Expression of E-cadherin
repressors SNAIL, ZEB1 and ZEB2 by tumour and stromal cells
influences tumour-budding phenotype and suggests heterogeneity of
stromal cells in pancreatic cancer. Br J Cancer. 112:1944–1950.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zheng X, Carstens JL, Kim J, Scheible M,
Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R:
Epithelial-to-mesenchymal transition is dispensable for metastasis
but induces chemoresistance in pancreatic cancer. Nature.
527:525–530. 2015. View Article : Google Scholar : PubMed/NCBI
|